

Significance
Apoptosis is crucial for immune system function and limiting tumor development. Because BAK and BAX are essential effectors of apoptosis, understanding how they are activated to form the oligomeric mitochondrial pores that kill cells is a major goal of the field. We define a requirement for two sites on mitochondrial BAK for its interaction with, and activation by, BCL-2 homology 3 (BH3)-only proteins during apoptosis and determine that binding of BH3-only proteins at a distal site promotes exposure of a canonical site to allow terminal BAK activation and homooligomerization. Additionally, we provide insight into how BAK and BAX kill cells, identifying that the oligomeric pore is limited to interactions between the BH3 domain and canonical groove and does not involve additional protein interfaces.
Keywords: apoptosis, BAK, BAX, BH3-only protein, mitochondria
Abstract
BAK and BAX are the essential effectors of apoptosis because without them a cell is resistant to most apoptotic stimuli. BAK and BAX undergo conformation changes to homooligomerize then permeabilize the mitochondrial outer membrane during apoptosis. How BCL-2 homology 3 (BH3)-only proteins bind to activate BAK and BAX is unclear. We report that BH3-only proteins bind inactive full-length BAK at mitochondria and then dissociate following exposure of the BAK BH3 and BH4 domains before BAK homodimerization. Using a functional obstructive labeling approach, we show that activation of BAK involves important interactions of BH3-only proteins with both the canonical hydrophobic binding groove (α2–5) and α6 at the rear of BAK, with interaction at α6 promoting an open groove to receive a BH3-only protein. Once activated, how BAK homodimers multimerize to form the putative apoptotic pore is unknown. Obstructive labeling of BAK beyond the BH3 domain and hydrophobic groove did not inhibit multimerization and mitochondrial damage, indicating that critical protein–protein interfaces in BAK self-association are limited to the α2–5 homodimerization domain.
BAK and BAX are the pivotal effectors of intrinsic apoptosis, with one or the other being required for mitochondrial damage and cell death (1, 2). They are activated by interaction with BCL-2 homology 3 (BH3)-only proteins including BID and BIM (3). Structures show that peptides based on the BH3 domains of activator BH3-only proteins can bind directly to BAK and BAX via a hydrophobic groove (comprising α-helices 2–5) that is also shared with their prosurvival homologs (4–8). BAX has been proposed to have an additional activation site, distinct from the hydrophobic groove, at the rear of the molecule comprising α-helices 1 and 6 (9). Binding of stapled BH3 peptides at this site induced the dissociation of the BAX C-terminal transmembrane domain from the hydrophobic groove to facilitate mitochondrial localization (10, 11). Interaction of BH3-only proteins at this noncanonical site is not thought to be necessary for BAK activity because BAK is constitutively anchored in the mitochondrial outer membrane (MOM) via its transmembrane domain (12). However, recent reports that both BAX (13, 14) and BAK (15) are constantly “retrotranslocated” from the mitochondria to the cytosol suggest conserved mechanisms of activation for BAK and BAX.
Characterizing the interaction of BH3-only proteins with full-length BAK at mitochondria has proven difficult because the bound BH3-only protein dissociates during consequent BAK conformation changes, including exposure of the N-terminus (amino terminus) and BH3-domain (16–19) and dissociation of their α2–5 helices (“core”) from their α6–8 helices (“latch”) (6, 7) to facilitate self-association. Additionally, structural studies have been largely confined to truncated protein or peptides in the absence of a membrane where interactions between BH3-only proteins and BAK occur.
Once activated BAK and BAX self-associate to form pores that damage the MOM to release cytochrome c and other apoptotic factors. A key step is the formation of symmetrical BH3:groove homodimers (17, 20), which then form higher order oligomers including ring-like pores and aggregates (21, 22). However, how BAK and BAX homodimers multimerize is unknown. From linkage or proximity measurements based on electroparamagnetic resonance and double electron–electron resonance various secondary interfaces have been implicated to allow homodimers to associate (23–28). Alternatively, based on linkage analysis and mathematical modeling, random aggregation of homodimers has also been proposed (29).
Understanding the molecular details of BAK and BAX activation and oligomerization may facilitate the rational design of small molecules that can inhibit them and so inhibit pathological apoptosis. We find that cBID interacts with inactive BAK at mitochondria and dissociates following exposure of the BAK α2 (BH3) and α1 (BH4) domains. We also show that cBID promotes BAK activation by binding a site involving the BAK α6 to promote opening of the canonical hydrophobic groove. Blocking experiments also indicate that a stable protein–protein interface outside of the core BH3:groove dimerization domain is not essential for pore formation and MOM damage.
Results
BAK Conformation Change Destabilizes the cBID:BAK Interaction at Mitochondria.
BH3-only proteins are thought to dissociate from BAK at mitochondria due to the induced conformation change and/or self-association of BAK. Thus, we hypothesized that constraining BAK in its inactive conformation, thereby preventing conformation change and subsequent homooligomerization, would stabilize the cBID:BAK interaction at mitochondria. We thus induced disulphide linkage between the two native cysteines in BAK (C14 and C166, Fig. 1A). This tether impaired BAK conformation change, evidenced by decreased exposure of N-terminal and BH3 epitopes (Fig. 1B) and MOM permeabilization (Fig. 1C). As predicted, the tether stabilized the interaction with cBID at mitochondria (Fig. 1D). As controls, cBID failed to coimmunoprecipitate with either single-cysteine BAK mutant (C14S or C166S) that was not constrained by intramolecular disulphide linkage (Fig. 1D).
Fig. 1.

Constraining events in BAK activation stabilize interaction with cBID. (A) Model of BAK C14:C166-induced disulphide linkage. N-terminal 20 amino acids are represented as a hashed line because the residues are absent from the crystal structure [Protein Data Bank (PDB) ID code 2IMS]. (B) An intramolecular tether constrains cBID-induced BAK conformation change. Membrane fractions from Bak−/−Bax−/− mouse embryonic fibroblasts (MEFs) expressing BAK, BAK C14S, or BAK C166S were treated with bismaleimidoethane (BMOE, 0.5 mM) followed by recombinant cBID (100 nM, 30 °C, 30 min) before immunoprecipitation with conformation-specific BAK antibodies that recognize the N-terminus (amino acids 23–38) or BH3 domain (4B5). C14:C166 tether is indicated (arrowhead). (C) C14:C166 tether impairs cBID-induced BAK apoptotic function. Membrane fractions as in B were treated with the cysteine cross-linker BMOE (0.5 mM) then recombinant cBID (100 nM, 30 °C, 30 min) before fractionation into supernatant (S) and pellet (P) and immunoblotting for cytochrome c. (D) Constraining BAK conformation change stabilizes interaction with cBID. Membrane fractions as in B were treated with CuPhe to induce disulphide linkage followed by recombinant cBID (100 nM, 30 °C, 30 min) before immunoprecipitation with an antibody that recognizes both active and inactive BAK (7D10). (E) Cartoon representation of BAK (PDB ID code 2IMS) (4). The hydrophobic groove (green) and the residues mutated to cysteine (yellow) are indicated. (F) Intramolecular tethers constrain cBID interaction. Membrane fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were treated and analyzed as in D. (G) Schematic of the stepwise conformation change of BAK and BID interaction. All data are representative of at least three independent experiments.
To define the step in BAK activation at which cBID dissociates we used additional constraints between the α1–α2, α1–α6, and α5–α6 that we have shown impair defined steps in BAK activation to block cytochrome c release (7, 19) (Fig. 1E). We first confirmed that the induced intramolecular tethers did not alter the conformation (Fig. S1A) or oligomerization state (Fig. S1B) of BAK. Like the C14:C166 tether, tethering Y41 (α1) to A79 (α2) or A28C (α1/2loop) to L163C (α6) stabilized the interaction with cBID (Fig. 1F). We have previously shown that tethering V142C (α5) to F150C (α6) to constrain “core/latch” dissociation still shows evidence of cBID-induced N-terminal epitope exposure, indicating that cBID still interacts with the tethered form to induce early steps in BAK conformation change (19). However, in contrast with the α1:α2 and α1/2 loop:α6 tethers, tethering α5:6 failed to stabilize the cBID:BAK complex (Fig. 1F), placing α5/6 dissociation downstream of both cBID dissociation and separation of α1 and α2 (BH3). Because core/latch dissociation precedes dimerization, our data suggest that cBID dissociates from BAK as a consequence of activating conformation change, likely due to α1 (BH4) and/or α2 (BH3) exposure, rather than due to displacement of BH3-only proteins from the groove by a BAK BH3 domain during BAK homodimerization (Fig. 1G).
Fig. S1.

Disulphide tethers do not induce BAK conformation change or oligomerization. (A and B) Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were treated or not with oxidant (CuPhe) to induce intramolecular disulphide linkage before treatment with proteinase K and reducing SDS/PAGE (A) or solubilization with digitonin and BN-PAGE (B).
cBID Interacts with Soluble and Mitochondrial BAK at Both the Canonical Hydrophobic Groove and at α6.
Whereas interactions of BAX with BH3-only proteins also occur at a distal site involving the α1 helix (30) and the α6 helix (9), interactions of BAK with BH3-only proteins seem restricted to the hydrophobic groove (11, 12). To investigate how cBID engages BAK at mitochondria we used an intermolecular disulphide-linkage approach to test whether cBID could link to regions in BAK other than the groove. To establish disulphide-linkage conditions we first used a functional semicytosolic mutant of BAK (BAK/BAXCS, where CS indicates C-segment) (31), because it could be at least partially activated by cBID in cytosolic fractions (Fig. S2 A and B), but its failure to undergo full activation and hence homooligomerize (Fig. S2A) allowed a stable interaction with cBID (Fig. S2C).
Fig. S2.

cBID and Bim directly activate a cytosolic BAK variant. (A) A cytosolic BAK variant is activated by cBID. Membrane or cytosolic fractions from Bak−/−Bax−/− MEFs expressing BAK or a BAK variant that is significantly cytosolic (BAK/BAXCS) were treated with recombinant cBID (100 nM, 30 °C, 30 min) before induction of disulphide linkage (CuPhe) and nonreducing SDS/PAGE. Note that BAK/BAXCS in cytosolic fractions becomes activated, as indicated by the loss of an intramolecular disulphide link between its two endogenous cysteines, C14 (N-segment) and C166 (α6–7 loop), that are known to be proximal in the inactive conformer but distal in the active form (20, 39) (Fig. 1A), but does not oligomerize. (B) Cytosolic BAK is activated by “activator” BH3-only proteins. Membrane (P) or cytosolic (S) fractions from Bak−/−Bax−/− MEFs expressing BAK or BAK/BAXCS were treated with recombinant cBID variants that contain the BH3 domains of BID, BIM, or BAD (100 nM, 30 °C, 30 min) before immunoprecipitation with a conformation-specific BAK antibody (Ab-1) or an antibody that recognizes both active and inactive BAK (7D10). (C) Failure to oligomerize stabilizes BAK interaction with cBID. Membrane or cytosolic fractions from Bak−/−Bax−/− MEFs expressing BAK or BAK/BAXCS were treated with recombinant cBID (100 nM, 30 °C, 30 min) before solubilization of samples with digitonin (dig) or Triton X-100 (TX), immunoprecipitation of inactive and active BAK, and immunoblotting for cBID. Note that the interaction was disrupted by Triton X-100, which induces BAK conformation change (40).
We introduced cysteine in the BID BH3 domain at position I83 or R84 of recombinant BID and confirmed they retained apoptotic activity (Fig. S3A). WT cBID (which lacks cysteine in the activating p15 fragment) and cBID I83C and R84C variants were incubated with cytosolic extracts from cells stably expressing BAK/BAXCS with cysteines engineered in either the groove (H99C or K113C) or α6 (R156C, D160C, or H164C). Disulphide linkage was induced with oxidant [copper (II) (1,10-phenanthroline)3, CuPhe] and assessed on nonreducing SDS/PAGE following coimmunoprecipitation. Disulphide-linked cBID:BAK heterodimers were evident between the cBID R84C variant with cysteine at H99 in the BAK groove (Fig. 2A) and also at R156 and H164 in the BAK α6 (Fig. 2B). A G94E mutation in BID reduces its interaction with BCL-2 proteins (32) and, consistent with this, cBID G94E showed reduced ability to link with BAK/BAXCS at both sites (Fig. 2C and Fig. S3B).
Fig. S3.

cBID interacts with BAK to induce its activation and cytochrome c release. (A) Cysteine was engineered in the BH3-domain of cBID at two positions that we have previously shown to cross-link in BAK (red). Important hydrophobic residues are underlined. Mitochondrial fractions from WT MEFs were incubated with the indicated concentrations of cBID, cBID I83C, or cBID R83C and cytochrome c release assessed. (B–D) Nonreducing SDS/PAGE and immunoblotting of lysates corresponding to Fig. 2 C–E, respectively. cBID:BAK is indicated (arrowhead). (E) Cys null cBID G94E inhibits linkage at both the groove and α6. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were preincubated with equimolar (1×) or threefold (3×) molar excess cBID ∆Cys/G94E for 15 min before incubation with cBID R84C/G94E for 15 min and induction of disulphide linkage and immunoprecipitation as in D. All data are representative of at least two independent experiments. cBID:BAK linkages (arrowhead); asterisk indicates IgG.
Fig. 2.

cBID interacts with the α6 as well as the hydrophobic groove of BAK. (A and B) cBID binds the hydrophobic groove and α6 of BAK/BAXCS. Cytosolic fractions from Bak−/−Bax−/− MEFs expressing BAK/BAXCS mutants with cysteines at the indicated positions in the groove (A) or α6 (B) were incubated with the indicated HA-cBID variants (100 nM, 30 °C, 30 min) and disulphide-linked. BAK immunoprecipitations were run under nonreducing conditions and immunoblotted for BAK or BID. Note that the p15 fragment of cBID cysteine variants cross-linked to other proteins in the cytosol with likely linkage to cBID p7 fragment the most abundant. Note that in subsequent experiments in this figure we detected recombinant BID with an HA antibody. (C) Mutation in cBID BH3 reduces interaction at the groove and α6 of BAK/BAXCS. Cytosolic fractions as in A and B were incubated with the indicated HA-cBID variants (100 nM, 30 °C, 30 min), disulphide-linked, and analyzed as in A. (D) cBID interacts with the hydrophobic groove and α6 of mitochondrial BAK. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were incubated with HA-cBID variants (100 nM, 30 °C, 15 min) and disulphide-linked. Membranes were solubilized with digitonin and BAK 7D10 immunoprecipitations were run under nonreducing conditions and immunoblotted for BAK or HA.
This approach was then used to map the transient interaction of cBID with BAK at mitochondria. cBID R84C did not detectably link at either the hydrophobic groove or α6 of BAK resident at mitochondria, because cBID R84C efficiently promoted BAK activation and homodimerization (Fig. 2D and Fig. S3C, BAK:BAK). However, hypomorphic cBID R84C/G94E, which failed to induce BAK homodimers, linked readily to both the groove (H99C) and α6 (R156C) of mitochondrial BAK (Fig. 2D and Fig. S3C). No linkage was observed with WT BAK (C14 and C166), BAK∆Cys, or BAK with a cysteine at N124 (Fig. 2D). The ability of cBID R84C/G94E to link at both the groove and α6 was competitively inhibited by preincubation with a cBID G94E/Cys null variant (Fig. S3E), supporting that the linkage profile reported genuine protein–protein interactions.
Thus, cBID binds BAK at mitochondria to promote its conformation change and consequently dissociates to facilitate BAK homooligomerization. In contrast, the hypomorphic cBID G94E variant binds BAK but does not induce activating conformation change and homooligomerization, and consequently its interaction with BAK at mitochondria persists, allowing it to disulphide-link at both the groove and α6 (Fig. 2D and Fig. S3C). The inability of cBID G94E to promote BAK activation was potentially due to its limited affinity. Alternatively, interactions between the C-terminal end of the BID BH3 domain with the BAK hydrophobic groove may be key in initiating BAK conformation change.
Obstructing the Hydrophobic Groove or α6 Inhibits BAK Activation.
To map the functionally relevant activation site(s) on mitochondrial BAK we labeled single cysteines engineered at various positions throughout BAK with cysteine-reactive 5-kDa PEG- maleimide and tested the effect on BAK-mediated cytochrome c release. Labeling of BAK cysteine variants was confirmed by retarded migration on reducing SDS/PAGE (Fig. S4 A and B). Ability to label each variant before activation correlated with the solvent exposure of residues in the structure of BAK (4). For example, residues in the buried face of the α2 (M71C) and in the core α5 helix (e.g., A130C, L132C, R137C, L138C, and A139C) were resistant to labeling (Fig. 3B, Figs. S4C and S5, and Table S1). Additionally, labeling was consistent with known changes in BAK conformation during apoptosis as indicated by the ability of M71C in the BAK BH3 domain to label only following its cBID-induced exposure (Fig. S4C). PEG-maleimide treatment had no impact on cytochrome c release nonspecifically or by labeling an alternative target, becasue PEG-maleimide treatment of WT BAK (labeled on both C14 and C166) and BAK ∆Cys did not impair MOM permeabilization (Fig. 3A). Labeling of the majority of positions throughout BAK did not impair cytochrome c release and, additionally, labeling did not induce cytochrome c release (Fig. 3 A and B and Fig. S5). Labeling at residue Y89, which would be expected to obscure the hydrophobic groove (4), significantly impaired BAK-mediated cytochrome c release in response to 100 nM cBID (Fig. 3 A and B, Fig. S5A, and Table S1), confirming the groove’s important role in BAK apoptotic function (20, 33).
Fig. S4.

PEG-maleimide labeling of BAK cysteine residues. (A and B) Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing BAK or BAK ∆Cys were treated with the indicated concentration of PEG-maleimide for 30 min (A) or with 0.5 mM PEG-maleimide for the indicated time (B) at room temperature before analysis on reducing SDS/PAGE and immunoblotting for BAK. (C) Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing BAK M71C were pretreated with 0.5 mM PEG-maleimide before activation with cBID (pre), or membrane fractions were coincubated with PEG-maleimide and cBID (during) before analysis on reducing SDS/PAGE and immunoblotting for BAK.
Fig. 3.

BAK hydrophobic groove and α6 site are important for BAK apoptotic activity. (A and B) Obstructive labeling of the groove abrogates BAK-mediated MOM permeabilization. Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were labeled with PEG-maleimide (5 kDa) before treatment with cBID (100 nM, 30 °C, 30 min) and supernatant (S) and membrane (P) fractions were immunoblotted for cytochrome c and membrane fractions immunoblotted for BAK. PEG-maleimide labeling and its effect on BAK-mediated cytochrome c release is shown in B. A labeled residue was defined as showing ≥50% labeled form on SDS/PAGE. Asterisks indicate residues that do not label in inactive BAK but label after activation with cBID. Endogenous cysteines (C14 and C166) in BAK are indicated in bold. The α2/BH3 domain (red), α3–5/hydrophobic groove (green), and α-helices (underlined) are indicated. (C) Obstructive labeling of the BAK α6 impairs apoptotic function in response to limiting cBID concentration. Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were treated as in A except they were treated with 1 nM cBID. (D) Double labeling of the BAK α6 impairs apoptotic function in response to high concentrations of cBID. Membranes from Bak−/−Bax−/− MEFs expressing FLAG-BAK D160C/H164C were treated with cBID (5 or 100 nM) or heated at 43 °C and assessed for cytochrome c release. (E) Obstructive labeling of the groove and α6 inhibits cBID-induced conformation change. Membrane fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were incubated with cBID and PEG-maleimide as indicated, followed by incubation with the conformation-specific antibody BAK aa23-38 and BN-PAGE. (F) Labeling of groove and α6 residues inhibits BAK activity. Cartoon representation of the structure of inactive BAK (PDB ID code 2IMS) showing the groove (green) and α2 (BH3 domain, raspberry). Residues that when labeled with PEG-maleimide inhibited cBID-induced BAK activation at 1 nM (yellow) or 100 nM (red) or did not inhibit BAK activation (blue) are indicated. All data are representative of at least two independent experiments.
Fig. S5.

PEG-maleimide labeling commonly does not abrogate BAK apoptotic function. (A) Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing BAK or the indicated BAK Cys mutants were treated with PEG-maleimide before treatment with cBID (100 nM, 30 °C, 30 min) or heated at 43 °C for 30 min. Supernatant (S) and membrane (P) fractions were immunoblotted for cytochrome c and membrane fractions immunoblotted for BAK. Note that the blots for BAK and BAK ∆Cys are the same as those presented in Fig. 5A. Asterisk indicates a nonspecific band when cytochrome c blots for G72C, Q73C, I80C, R88C, E92C, and H164C were reprobed for BAK. (B) Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK Cys mutants were treated or not with PEG-maleimide and immunoblotted for BAK. Note that BAK L132C was expressed at relatively low levels so a longer exposure of this variant from the same blot is shown. (C) Profile of PEG-maleimide labeling of exposed BAK residues. Cartoon representation of the structure of inactive BAK (PDB 2IMS) showing the groove (green) and α2 (BH3 domain, raspberry). Residues that were labeled with PEG-maleimide (blue) or not (black) are indicated.
Table S1.
Summary of PEG-maleimide labeling on BAK
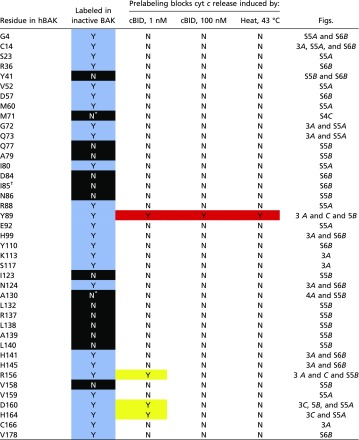 |
Colouring (black, blue, red, and yellow) corresponds to Fig. 3B. N, no and Y, yes.
Labels following activation.
Cysteine mutant is loss of function.
Given the flexibility of the linear PEG-maleimide molecule, we reasoned that an inhibitory effect of the label on cBID-induced BAK activation might be overcome by the high concentrations (100 nM) of cBID used in these initial experiments. Thus, we tested whether an inhibitory effect of PEG-maleimide label might be revealed by a lower (threshold) concentration of cBID (1 nM, Fig. 3C and Fig. S6A). As expected, labeling Y89C inhibited BAK function (Fig. 3C). Labeling at most other positions did not impair BAK activity in response to 1 nM cBID (Fig. S6B), but labeling at three positions in the BAK α6 reduced BAK apoptotic function (Fig. 3C and Table S1). Simultaneous labeling at two positions in α6 (D160 and H164) blocked BAK activity even when induced by high concentrations of cBID (Fig. 3D). BAK activity induced by the BH3 domain of either BIM or PUMA was also impaired by α6 labeling (Fig. S6C), suggesting a conserved mechanism.
Fig. S6.

PEG-maleimide labeling commonly does not inhibit BAK activation induced by limiting concentrations of cBID. (A) Dose–response of cBID-induction of cytochrome c release. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing BAK were treated with the indicated concentration of cBID (30 °C, 30 min). Supernatant (S) and membrane (P) fractions were immunoblotted for cytochrome c. (B) Labeling does not inhibit BAK function in response to limiting dose of cBID. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were treated with PEG-maleimide before treatment with cBID (1 nM, 30 °C, 30 min) for 30 min. Supernatant (S) and membrane (P) fractions were immunoblotted for cytochrome c and membrane fractions immunoblotted for BAK. (C) BAK activation induced by BIM and PUMA is inhibited by labeling the α6. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing BAK R156C were pretreated with PEG-maleimide before incubation with recombinant cBID or chimeras of cBID bearing the BH3 domain from BIM or PUMA and cytochrome c release was assessed as in A.
To test whether labeling impaired cBID-induced conformation change we performed blue native (BN)-PAGE in combination with gel shift with a conformation-specific antibody (amino acids 23–38). As expected, unlabeled BAK gel-shifted with the antibody only after activation with cBID (Fig. 3E, indicated by the loss of the dimer in lane 5 compared with lane 2), and treatment with PEG-maleimide did not perturb complete antibody gel shift following activation of BAK ∆Cys or G4C (Fig. 3E, compare lanes 5 and 6). Labeling of Y89C with PEG-maleimide to obscure the hydrophobic groove reduced, but did not completely block, gel shift with the conformation-specific antibody in response to both 1 and 100 nM cBID (Fig. 3E, compare lanes 5 and 6). Labeling of R156C and H164C in α6 likewise reduced gel shift of BAK induced by 1 nM cBID (Fig. 3 E and F). Labeling of BAK G4C did not impair dimerization, as evidenced by labeled dimers and the lack of labeled monomer after cBID treatment (Fig. 3E, lane 3). In contrast, clear retention of labeled monomeric BAK R156C and BAK H164C variants further supports that labeling of α6 impairs cBID-induced activation. That obstructing the α6 perturbs BAK activation rather than homooligomerization is consistent with the effect of α6 labeling’s being overcome by high concentrations of cBID.
Labeling of the BAK α6 Inhibits cBID-Induced Opening of the BAK Groove.
The crystal structure of inactive BAK revealed that the side chains from R88 and Y89 partially occlude the hydrophobic groove and suggested that the groove needed to open to present a binding site for BH3-only proteins (4). Consistent with this, in the structure of BAK with a BID BH3 peptide bound in its hydrophobic groove, the groove is more open and thereby increases the solvent exposure of residues at the base of the groove including A130 (4, 33) (Fig. S7). Accordingly, we found that although BAK A130C was not efficiently labeled before activation, its labeling was encouraged by cBID (Fig. 4 A and B, during). Labeling of A130C was reduced after cBID had induced BAK homooligomerization (post, Fig. 4A), consistent with its burial beneath a bound BAK BH3 domain in the BAK BH3:groove homodimer (7). We hypothesized that binding of cBID to the α6 activation site may promote opening of the hydrophobic groove to facilitate BID BH3 domain binding. Consistent with this, labeling α6 at either R156 or H164 inhibited the PEG-maleimide labeling of A130C induced by cBID (Fig. 4C). In contrast, both cysteines could be efficiently labeled when BAK was activated by heat (Fig. 4C). This suggests that binding of cBID to the α6 site promotes exposure or opening of the groove to allow BH3-only protein binding and BAK activation (Fig. 4D).
Fig. S7.

Activation of BAK involves opening of the hydrophobic groove to expose A130. (A) Inactive BAK has an occluded groove. Cartoon (Left) or surface (Right) representation of the X-ray structure of inactive BAK (orange) [PDB ID code 2IMS (4)], showing the side chains of Y89 and R88 (yellow) that occlude the hydrophobic groove (α3–5, green) as a site to receive the BH3-domain from BH3-only proteins. A130 (red) is buried in the inactive conformer. (B) BAK groove adopts a more open configuration to expose A130 when a BID BH3 peptide occupies the groove. Surface representation of the lowest-energy NMR structure of BAK:BID BH3 peptide [PDB ID code 2M5B (35)] with BID BH3 peptide atoms shown (Left) or removed to show the solvent exposed A130 (Right). Hydrophobic groove (α3–5, green), hydrocarbon stapled BID BH3 peptide (raspberry), and exposed A130 (red) are indicated.
Fig. 4.

Labeling the α6 inhibits cBID-induced exposure of the hydrophobic groove. (A) Labeling reveals transient exposure of the α5 groove residue A130 during activation. Membranes from Bak−/−Bax−/− MEFs expressing the BAK A130C were treated with PEG-maleimide before treatment with cBID (100 nM, 30 °C, 30 min) (pre), in the presence of cBID (during), or after cBID treatment (post) and immunoblotted for BAK. (B) A130 localizes to the base of the hydrophobic groove in inactive BAK. A130, R156, and H164 are indicated on a cartoon representation of the structure of inactive BAK (PDB 2IMS). (C) Labeling of α6 residues impairs induced exposure of A130C. Membranes from Bak−/−Bax−/− MEFs expressing BAK variants were pretreated with PEG-maleimide without quenching before treatment with cBID (30 °C, 30 min) or heating at 43 °C (30 min) and immunoblotted for BAK. (D) Obstructive labeling informs the stepwise activation of BAK at mitochondria.
Labeling of BAK Homodimers Does Not Interfere with Apoptotic Activity.
Obstructive labeling at the groove could affect BAK-mediated cytochrome c release either by inhibiting cBID-induced conformation change or by blocking downstream BAK homooligomerization (Fig. 5A). To assess the influence of labeling on BAK oligomerization downstream of cBID-induced activation, we tested the effect of labeling on heat-induced cytochrome c release that is independent of BH3-only proteins (34). Labeling in the canonical hydrophobic groove (Y89) impaired BAK-mediated MOM permeabilization in response to heat, supporting the important role of the groove not only in BAK activation but also in homodimerization (20) (Fig. 5B). As expected, those mutants whose labeling did not inhibit cytochrome c release induced by cBID also did not inhibit heat-induced cytochrome c release (Fig. 5B and Fig. S5). Additionally, double labeling of the α6 did not impair BAK activity when induced by heat (Fig. 3D).
Fig. 5.

Labeling of the groove impairs BAK homooligomerization and apoptotic function. (A) Obstructive labeling can impair distinct steps in BAK activation and oligomerization. (B) Labeling of the groove impairs BAK apoptotic function independent of interaction with BH3-only proteins. Membranes from Bak−/−Bax−/− MEFs expressing the indicated BAK mutants were treated with PEG-maleimide before treatment with cBID (100 nM, 30 °C, 30 min) or heated at 43 °C for 30 min. Supernatant (S) and membrane (P) fractions were immunoblotted for cytochrome c and membrane fractions for BAK. (C) Labeling of the groove impairs BAK homooligomerization. Mitochondrial fractions from Bak−/−Bax−/− MEFs expressing the indicated BAK variants were treated with PEG-maleimide before treatment with cBID (100 nM, 30 °C, 30 min), in the presence of cBID (during), or after cBID treatment (post) or alternatively heated at 43 °C for 30 min and assessed on BN-PAGE. (D) Labeling of residues on the periphery of the BAK core homodimer does not impair apoptotic function. Cartoon representation of the BAK α2–5 core BH3:groove homodimer (PDB ID code 4U2V) (7). Residues that label and impair BAK function (red) and residues that label but do not affect BAK function (blue) are indicated. Data are representative of at least three independent experiments.
Consistent with the proposed important role for the hydrophobic groove in the homodimerization, BN-PAGE revealed that labeling of Y89C and A130C prevented homodimerization in response to heat as well as in response to cBID (Figs. 3E and 5C). The effect of labeling on dimerization of BAK Y89C was more pronounced than on dimerization of R156C and H164C (Fig. 3E, lane 3; note the labeled dimers in R156C and H164C compared with their absence in Y89C). This likely reflects the role of the hydrophobic groove in both activation and homodimerization, whereas the role of α6 is restricted to activation.
When PEG-maleimide labeling was performed after cBID-induced activation, dimers of BAK Y89C were largely resistant to labeling (Fig. 5C), indicating that Y89C is buried in dimerized BAK (Fig. 5D). Both C14 and C166 were efficiently labeled in WT BAK but did not impair oligomerization (Fig. 5C). However, once activated and oligomerized these positions in BAK became more resistant to labeling, as indicated by the marked increase in 1× and 2× labeled dimer species when labeled after oligomerization in comparison with the predominant 4× prelabeled species (Fig. 5C, compare lanes 5 and 8). This indicates that both the N termini (C14) and α6 (C166) become buried once BAK is oligomerized. Thus, taken together, labeling a single position in the groove with PEG-maleimide was sufficient to block homodimerization and MOM permeabilization, yet labeling of each position tested outside of the groove failed to do so (Fig. 5D and Fig. S5).
Discussion
Our data indicate that BH3-only proteins like cBID interact with full-length BAK at mitochondria via the canonical hydrophobic groove comprising α2–5 and also at a site on the opposite face of BAK comprising α6, indicating that the BAK and BAX activation mechanisms are conserved (11, 33). A previous report failed to find evidence of an interaction between BH3 peptides and the putative rear site in a recombinant mutant BAK protein in solution (12). This difference led to the proposition that this noncanonical site was restricted to BAX to promote its translocation to the MOM, with the mitochondrial localization of BAK precluding the need for activation via the noncanonical site. Our findings with full-length BAK at mitochondria suggest that the consequence of BH3-only protein binding at the noncanonical site is likely the same for BAK as it is for BAX: it promotes opening or accessibility of the hydrophobic groove. In BAK that resides at mitochondria this exposes a reactive groove for BH3-only protein binding, whereas in cytosolic BAX it additionally serves to encourage dissociation of the transmembrane anchor from the hydrophobic groove and promote mitochondrial localization. The increased accessibility of the groove may be due to induced conformation change to allosterically open the hydrophobic groove. Alternatively, because both BAK and BAX interact with VDAC2 at the MOM (35–37), binding of BH3-only proteins to the α6 site in BAK or BAX may be an important step in dissociating them from VDAC2 during apoptosis. Recent evidence that the distinct subcellular localization of BAK and BAX is a consequence of their relative rates of retrotranslocation to the cytosol argues against qualitatively different modes of BAK and BAX activation (13, 15). Although our linkage and blocking data support a two-site mechanism of BAK activation, it remains possible that labeling the BAK α6 indirectly impairs a key event in BAK activation induced by groove binding that precedes homodimerization.
Structural information regarding regions beyond the core dimerization domain of α2–α5 in dimerized BAK and BAX is lacking. PEG-maleimide labeling was consistent with the known structures of inactive (4) and homodimeric BAK (7). This includes residues of BAK that become exposed during activation such as those in the α2/BH3 domain and base of the hydrophobic groove. Additionally, Y89C’s inability to be labeled in the homodimer form is consistent with its burial in a BH3:groove homodimer (7), providing further support that full-length BAK adopts a symmetric BH3:groove homodimer in the MOM. Thus, PEG-maleimide labeling is an efficient way to interrogate the conformations of full-length BAK and BAX in the MOM while also informing functional relevance.
BAK and BAX homodimers have been proposed to multimerize via secondary interfaces including α1, α3/4, α4/5, and α6 to form the oligomers necessary for MOM permeabilization (7, 23–25, 27, 28, 38, 39). However, labeling of residues in these implicated secondary interfaces failed to significantly affect MOM permeabilization, suggesting that these putative interfaces are not necessary for BAK multimerization and higher order pore formation. We have previously shown that mutation of the BAK α6 abrogated higher order complex formation and apoptotic function downstream of BH3:groove homodimerization, indicating that BAK BH3:groove homodimers are not sufficient to mediate MOM permeabilization and cause cell death (38), while also implicating the α6 in higher-order pore formation. However, even dual labeling of α6 residues failed to significantly inhibit BAK-mediated cytochrome c release in response to heat, suggesting that mutation of α6 may inhibit apoptotic function by impairing events that lead to the aggregation of dimers such as conformation change and/or membrane integration following BH3:groove dimerization. The lack of a putative secondary protein interface that mediates BAK oligomerization suggests that BAK (and by homology likely also BAX) homodimers may aggregate in a disordered fashion, as implicated by our mathematical modeling of BAK oligomers based on disulphide-linkage constraints (29). Additionally, our data are consistent with the notion that homodimers associate with the phospholipid of the MOM to form a proteolipidic pore (21, 40, 41). We have reported that BAX and BAK homodimers expose a hydrophobic face comprising aromatic residues in α4 and α5 that may promote interaction with the lipids of the MOM (6, 7, 42). Biophysical studies showed evidence of proteolipidic pore formation by BAX and BAK (21, 40, 43), and our earlier studies indicating that the higher order oligomeric pore is heterogeneous and unstable once removed from the MOM (23, 38) are consistent with the characteristics of proteolipidic pores.
Experimental Procedures
Cell Culture and Retroviral Infection.
SV40-transformed Bak−/−Bax−/− mouse embryonic fibroblasts were generated, passaged, and retrovirally transduced with BAK expressions constructs as described (20). Details are provided in SI Experimental Procedures.
Subcellular Fractionation, Cytochrome c Release, Cysteine Linkage Analysis and PEG-Maleimide Labeling, Immunoprecipitation, SDS/PAGE, and BN-PAGE.
Procedures were performed essentially as described (20). For PEG-maleimide labeling, mitochondria-enriched membrane fractions were resuspended in fractionation buffer and treated with methoxy PEG-maleimide (5 kDa, PLS-234; Creative PEGworks). Details are provided in SI Experimental Procedures.
SI Experimental Procedures
Recombinant Protein Production and Purification.
The recombinant cBID and BID chimeras with the BH3 domains of BIM or PUMA were cloned into pGEX-4T2 to add a C-terminal HA tag and generated as described (3). Protein was expressed in Escherichia coli BL21 (DE3) induced with 1 mM isopropyl β-d-1-thiogalactopyranoside overnight at 18 °C. Bacteria were lysed in lysis buffer (PBS with 1 mM EDTA, 10 μg/mL aprotinin, and 10 μg/mL leupeptin), with 1 mM DTT, 0.5 mM PMSF, 266 μg/mL lysozyme, and 37 μg/mL DNase I with a homogenizer (EmulsiFlex; Avestin). The lysate was centrifuged and filtered before incubation with glutathione beads (GE Healthcare Bio-Sciences AB). Washed beads were incubated overnight at 4 °C with thrombin in 50 mM Tris, pH 8.0, 150 mM NaCl, and 5 mM MgCl2. For recombinant BID chimeras, BID was engineered with a thrombin cleavage site in place of the caspase 8 cleavage site to allow one-step cleavage and purification. For recombinant cBID, full-length cBID was eluted with glutathione and cleaved with recombinant caspase-8 as described (3). Eluates were further purified by gel filtration (Superdex 75; GE Healthcare Bio-Sciences AB) in Tris-buffered saline (TBS; 20 mM Tris, pH 8.0, and 150 mM NaCl) with 2 mM DTT and recombinant protein was aliquoted and stored at −80 °C.
Cell Culture and Retroviral Infection.
Mouse embryonic fibroblasts from Bak−/−Bax−/− C57BL/6 embryos harvested at embryonic day 14.5 were transformed by transfecting with SV40-large T antigen. MEFs were passaged in Dulbecco’s modified Eagles medium supplemented with 10% FCS, 55 μM 2-mercaptoethanol, and 250 μM asparagine at 37 °C and 10% CO2. BAK constructs were cloned into the retroviral expression vector pMX-IRES-GFP (internal ribosome entry site-green fluorescent protein) and were retrovirally transduced in MEFs using Phoenix ecotropic packaging cells as described (20).
Subcellular Fractionation, Cytochrome c Release, Cysteine Linkage Analysis, PEG-Maleimide Labeling, and Limited Proteolysis.
Cytosol and mitochondria-enriched heavy membrane were fractionated following cell membrane permeabilization with 0.025% wt/vol digitonin in fractionation buffer (20 mM Hepes KOH, pH 7.5, 100 mM sucrose, 100 mM KCl, and 2.5 mM MgCl2) for 10 min on ice. Where indicated membrane fractions were incubated for 30 min at 30 °C in the presence of caspase-8 cleaved recombinant BID (cBID), BID variants with the BH3 domain from either BIM or PUMA, or were incubated at 43 °C for heat-induced BAK activation. Cytosol and membrane fractions were separated by centrifugation at 13,000 × g for 5 min at 4 °C and fractions assessed for cytochrome c release by immunoblotting. For oxidant-induction of disulphide linkage, cytosol or membrane fractions in fractionation buffer were treated with CuPhe (1 mM) for 30 min on ice. Alternatively, membranes were resuspended in cross-linking buffer (20 mM Hepes KOH, pH 7.5, 100 mM sucrose, 100 mM KCl, 2.5 mM MgCl2, and 2 mM EDTA) and treated with the 8-Å cysteine-reactive chemical cross-linker BMOE (0.5 mM) for 30 min at room temperature. Oxidized and chemically cross-linked samples were electrophoresed under nonreducing and reducing conditions, respectively. For PEG-maleimide labeling, mitochondria-enriched membrane fractions were resuspended in fractionation buffer and treated with methoxy PEG-maleimide (5 kDa, PLS-234; Creative PEGworks) at room temperature for 30 min at 0.5 mM unless otherwise indicated, followed by quenching with n-ethylmaleimide (20 mM). In experiments where PEG-maleimide labeling occurred during cBID-induced or heat-induced activation, PEG-maleimide was added shortly before the activating stimulus and was not quenched. Limited proteolysis to investigate BAK protein conformation was performed as previously described (38). Briefly, membrane fractions resuspended in fractionation buffer with 4 μg/mL pepstatin A (Sigma) were prechilled to 0 °C then incubated with 30 μg/mL proteinase K (Roche) on ice for 20 min. The reaction was stopped by the addition of 1 mM PMSF and samples were immunoblotted with antibody to the BAK BH3 domain (4B5).
Immunoprecipitation, SDS/PAGE, BN-PAGE, and Immunoblotting.
For immunoprecipitation, cells were solubilized on ice for 30 min in lysis buffer (20 mM Tris⋅HCl, pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and 10% glycerol) containing either 1% vol/vol Triton X-100 or 1% wt/vol digitonin. Immunoprecipitation was performed with anti-HA (16B12; Covance), anti-BAK 7D10 that recognizes inactive and active BAK (D. C. S. Huang, Walter and Eliza Hall Institute, Parkville, Australia), or the conformation-specific BAK antibodies aa23-38 (B5897; Sigma-Aldrich), Ab-1 (BD Biosciences), or 4B5 (D. C. S. Huang).
For SDS/PAGE, lysates of whole cells, cellular fractions, or immunoprecipitates in nonreducing or reducing Laemmli sample buffer were electrophoresed through Tris-glycine gels (Bio-Rad) and transferred to PVDF membrane.
BN-PAGE was performed essentially as described. Membrane fractions were solubilized in 1% digitonin in 20 mM Bis-Tris (pH 7.4), 50 mM NaCl, 10% glycerol, and 10 mM DTT before centrifugation at 13,000 × g to pellet insoluble debris. BN-PAGE loading dye [5% wt/vol Coomassie Blue R-250 (Bio-Rad) in 500 mM 6-aminohexanoic acid and 100 mM Bis-Tris, pH 7.0] was added to each sample. Gels were electrophoresed in anode buffer (50 mM Bis-Tris, pH 7.0) and blue cathode buffer [50 mM N-2-hydroxy-1,1-bis(hydroxymethyl)ethylglycine and 15 mM Bis-Tris unbuffered containing 0.02% wt/vol Coomassie Blue G-250]. Blue cathode buffer was replaced with clear buffer (without Coomassie Blue) when the dye front was one-third of the way through the resolving gel. Gels were transferred to PVDF in Tris-glycine transfer buffer containing 20% methanol and 0.037% SDS. Blots were destained in 50% methanol and 25% acetic acid and washed with TBS before immunoblotting.
Membranes were blocked in 5% wt/vol nonfat milk in TBS-T before immunoblotting with antibodies raised against BAK (amino acids 23–38 or 7D10) and cytochrome c (clone 7H8.2C12; BD Biosciences). Immunoprecipitates and preimmunoprecipitation cell lysates were immunoblotted for HA (3f10, Roche) or BAK (amino acids 23–38). Secondary antibodies used were horseradish peroxidase-conjugated anti-rabbit IgG, anti-mouse IgG, and anti-rat IgG (Southern Biotech).
Acknowledgments
We thank Kristen Scicluna for technical assistance and Peter Colman, Sweta Iyer, and Peter Czabotar for helpful discussions and advice on the manuscript. This work was supported by National Health and Medical Research Council Grants 1059290 and 1078924, Australian Research Council Fellowship FT100100791 (to G.D.), and operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Support 9000220.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1702453114/-/DCSupplemental.
References
- 1.Lindsten T, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei MC, et al. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hockings C, et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015;6:e1735. doi: 10.1038/cddis.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moldoveanu T, et al. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 6.Czabotar PE, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Brouwer JM, et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci USA. 2013;110:E986–E995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edlich F, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–116. doi: 10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schellenberg B, et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell. 2013;49:959–971. doi: 10.1016/j.molcel.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todt F, et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015;34:67–80. doi: 10.15252/embj.201488806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu Y-T, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 17.Dewson G, et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012;19:661–670. doi: 10.1038/cdd.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffiths GJ, et al. Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene. 2001;20:7668–7676. doi: 10.1038/sj.onc.1204995. [DOI] [PubMed] [Google Scholar]
- 19.Alsop AE, et al. Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis. Nat Commun. 2015;6:6841. doi: 10.1038/ncomms7841. [DOI] [PubMed] [Google Scholar]
- 20.Dewson G, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Salvador-Gallego R, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35:389–401. doi: 10.15252/embj.201593384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Große L, et al. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–413. doi: 10.15252/embj.201592789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewson G, et al. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Iyer S, et al. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015;22:1665–1675. doi: 10.1038/cdd.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, et al. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35:208–236. doi: 10.15252/embj.201591552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subburaj Y, et al. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat Commun. 2015;6:8042. doi: 10.1038/ncomms9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bleicken S, et al. Structural model of active Bax at the membrane. Mol Cell. 2014;56:496–505. doi: 10.1016/j.molcel.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aluvila S, et al. Organization of the mitochondrial apoptotic BAK pore: Oligomerization of the BAK homodimers. J Biol Chem. 2014;289:2537–2551. doi: 10.1074/jbc.M113.526806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uren RT, et al. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife. 2017 doi: 10.7554/eLife.19944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cartron PF, et al. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Ferrer PE, Frederick P, Gulbis JM, Dewson G, Kluck RM. Translocation of a Bak C-terminus mutant from cytosol to mitochondria to mediate cytochrome C release: Implications for Bak and Bax apoptotic function. PLoS One. 2012;7:e31510. doi: 10.1371/journal.pone.0031510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Yin X-M, Chao DT, Milliman CL, Korsmeyer SJ. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 33.Moldoveanu T, et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pagliari LJ, et al. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA. 2005;102:17975–17980. doi: 10.1073/pnas.0506712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazarou M, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285:36876–36883. doi: 10.1074/jbc.M110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma SB, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: A requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014;21:1925–1935. doi: 10.1038/cdd.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 38.Ma S, et al. Assembly of the Bak apoptotic pore: A critical role for the Bak protein α6 helix in the multimerization of homodimers during apoptosis. J Biol Chem. 2013;288:26027–26038. doi: 10.1074/jbc.M113.490094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, et al. Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem. 2010;285:17614–17627. doi: 10.1074/jbc.M110.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terrones O, et al. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem. 2004;279:30081–30091. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- 41.Basañez G, et al. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci USA. 1999;96:5492–5497. doi: 10.1073/pnas.96.10.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westphal D, et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci USA. 2014;111:E4076–E4085. doi: 10.1073/pnas.1415142111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian S, Wang W, Yang L, Huang HW. Structure of transmembrane pore induced by Bax-derived peptide: Evidence for lipidic pores. Proc Natl Acad Sci USA. 2008;105:17379–17383. doi: 10.1073/pnas.0807764105. [DOI] [PMC free article] [PubMed] [Google Scholar]