

Abstract
Intestinal cyclic guanosine monophosphate (cGMP) signaling regulates epithelial homeostasis and has been implicated in the suppression of colitis and colon cancer. In this study, we investigated the cGMP-elevating ability of the phosphodiesterase-5 (PDE5) inhibitor sildenafil to prevent disease in the azoxymethane/dextran sulfate sodium (AOM/DSS) inflammation-driven colorectal cancer model. Treatment of mice with sildenafil activated cGMP signaling in the colon mucosa and protected against DSS-induced barrier dysfunction. In mice treated with AOM/DSS, oral administration of sildenafil throughout the disease course reduced polyp multiplicity by 50% compared to untreated controls. Polyps that did form in sildenafil treated mice were less proliferative and more differentiated compared to polyps from untreated mice, but apoptosis was unaffected. Polyps in sildenafil treated mice were also less inflamed; they exhibited reduced myeloid-cell infiltration, and reduced expression of iNOS, IFNγ, and IL-6 compared to untreated controls. Most of the protection conferred by sildenafil was during the initiation stage of carcinogenesis (38% reduction in multiplicity). Administration of sildenafil during the later promotion stages did not affect multiplicity but had a similar effect on the polyp phenotype, including increased mucus production, and reduced proliferation and inflammation. In summary, the results demonstrate that oral administration of sildenafil suppresses polyp formation and inflammation in mice treated with AOM/DSS. This validation of PDE5 as a target highlights the potential therapeutic value of PDE5 inhibitors for the prevention of colitis-driven colon cancer in humans.
Keywords: cGMP, colon cancer, PDE5 inhibitor, colitis, prevention
Introduction
Inflammation is a driver of carcinogenesis, and patients with inflammatory bowel disease (IBD) have increased risk for developing colorectal cancer (CRC) (1,2). This association provides rationale for the utility of anti-inflammatory therapies for CRC prevention (3). Indeed, epidemiological evidence has shown that long-term non-steroidal anti-inflammatory drug (NSAID) use correlates with reduced incidence of CRC including in patients without IBD (4,5). NSAIDs are widely known to prevent adenoma formation, but might also be an effective treatment for some tumors (6,7). While the effects of NSAIDs are presumed to be due to inhibition of cyclooxygenases (COX) and prostaglandin synthesis, COX-independent anti-tumor effects have also been observed. Exisulind/Aptosyn™ is a sulfone derivative of the NSAID sulindac with no NSAID activity, but it can block colon cancer cell growth by inhibiting phosphodiesterases (PDEs) (8). Exisulind has been reported to induce apoptosis of colon cancer cells in vitro by increasing 3′, 5′-cyclic guanosine monophosphate (cGMP) and activation of type-1 cGMP-dependent protein kinase (PKG1) (9). Clinical trials in human familial adenomatous polyposis (FAP) patients and also sporadic adenomas, demonstrated that exisulind could induce regression of colorectal polyps by promoting apoptosis and mucus differentiation in the glandular epithelium (10,11). Exisulind faltered in a phase 3 clinical trial and ultimately was not approved due to undisclosed safety issues (12). This outcome might be attributed to the fact that exisulind is a weak PDE5 inhibitor, and that therapeutic doses are associated with hepatotoxicity (13).
Interest in PDE5 as a cancer target waned with exisulind, but compelling evidence has accumulated that indicates a critical role for cGMP in the regulation of epithelial homeostasis and protection against colitis and colon cancer in mice. The endogenous peptide hormones guanylin and uroguanylin generate cGMP in the intestinal epithelium by activating receptor guanylyl-cyclase C (GCC) (14). Knockout mice that are defective in cGMP signaling components exhibit crypt hyperplasia, increased luminal apoptosis, and reduced differentiation, particularly of goblet cells (15–17). Treatment of wild type mice with the PDE5 inhibitor vardenafil suppressed proliferation and apoptosis, and increased secretory cell density, indicating that homeostatic regulation of the intestinal epithelium by cGMP is a normal physiological process (18). The anti-proliferative effect of cGMP suggested a tumor suppressive role, and this idea was strengthened by the observation that GCC knockout mice are more susceptible to tumorigenesis in both carcinogen-induced, and in Apcmin mouse models (19,20). Lending further support for a tumor suppressive role of endogenous cGMP, treatment with exogenous uroguanylin was able to reduce tumorigenesis in Apcmin mice (21). The mechanisms underlying the anti-tumor effects of cGMP in the intestine are not fully understood. It is possible that direct inhibition of proliferation by cGMP could suppress tumor progression. However, recent work from several laboratories has highlighted barrier-protective functions of cGMP, which when compromised in GCC-deficient mice lead to increased susceptibility to colitis (22–24). Moreover, increasing intestinal cGMP with either GCC agonists or with PDE5 inhibitors can protect against experimental colitis in mice (18,25). Because inflammation is a central driver of many cancers, it is reasonable that promoting epithelial barrier function could also contribute to the prevention of intestinal tumorigenesis by cGMP.
The molecular mechanisms underlying the effects of cGMP in the intestine are currently an area of active investigation. The upstream cGMP-generating machinery includes guanylin/uroguanylin that bind and activate GCC in the epithelial plasma-membrane. The homeostatic effects of cGMP have been shown to be mediated by PKG2, but the downstream effector pathways are less clear (17,18). In gastric and colon cancer cell lines, cGMP has been reported to inhibit Ras/ERK (26,27) and β-catenin signaling (9,28), either of which could suppress proliferation and promote differentiation. In both cell lines and in mice, cGMP has been shown to inhibit the AKT and JNK pathways (18,20), which regulate homeostasis in the intestinal epithelium. More recently it was reported that by suppressing AKT, cGMP/PKG2 activates Forkhead Box O (FoxO) transcription factors in both colon cancer cell lines and in vivo (29). As tumor suppressors, FoxO can inhibit proliferation in dividing cells and provide antioxidant protection to quiescent cells (30,31). It is therefore likely that FoxO is an important downstream effector of cGMP on proliferation and barrier maintenance in the intestinal epithelium.
Despite growing evidence for the anti-inflammatory role of cGMP signaling in the intestine, the effect of increasing cGMP on inflammation-driven carcinogenesis has not been tested. By increasing epithelial cGMP, PDE5 inhibitors can protect against dextran-sulfate sodium (DSS)-induced colitis in mice and can activate several anti-tumor effector pathways. The objective of the present study was therefore to test the ability of the PDE5 inhibitor sildenafil to suppress tumorigenesis in the azoxymethane/dextran sulfate sodium (AOM/DSS) model of colorectal cancer in mice.
Materials and Methods
Animals
Age-matched, male C57Bl/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) at six weeks of age. Animals were acclimated for two weeks in the animal husbandry facility prior to experimentation. For all studies, the mice were maintained in a controlled environment 20°C and 14-hours light/10-hours dark cycle with free access to food and water. Pharmaceutical grade PDE5 inhibitor sildenafil (Revatio™) was dissolved in water immediately prior to use. During various treatment courses, mice were allowed to drink freely from water bottles, with a calculated sildenafil dose of 5.7 mg/kg daily throughout the disease course. The Augusta University Institutional Animal Care and Use Committee approved all mouse procedures.
Colitis model (DSS)
Inducing disease in this model was performed as described previously (18). Briefly, age-matched male mice were treated with 3% w/v dextran sulfate sodium (DSS) (36 000–50 000 Da; MP Biomedicals) in the drinking water for five days ad libitum, then switched to normal drinking water for five days during the recovery stage. Disease activity index (DAI) included measures of weight loss (0 = 0%, 1 = 1–5%, 2 = 5–10%, 3 = 10–20%, 4 = >20%), diarrhea (0 = normal stool, 1 = soft stool, 2 = very soft stool, 3 = diarrhea, 4 = dysenteric diarrhea) and rectal bleeding (0 = none, 1 = occult bleeding, 2 = blood on anus, 3 = blood on fur/tail, 4 = gross bleeding). Maximal disease activity index and intestinal barrier permeability were observed on day 3 of the DSS recovery stage. To assess intestinal barrier permeability, age-matched male mice were starved overnight and given fluorescein isothiocyanate (FITC)-dextran (0.5 mg/g body weight, 4 kDa; Sigma-Aldrich) by oral gavage. Serum was collected 1.5 hrs after oral gavage and the concentration of FITC-dextran was measured by quantitating fluorescence in a Cytation multiplate reader (Biotek, Winooski, VT).
Colorectal tumorigenesis model
Procedures followed here were carried out as described previously with the following modifications (32). Mice were given a single 7.5 mg/kg intraperitoneal (IP) dose of azoxymethane (13.4M 98%, Sigma Aldrich) and subsequently subjected to three consecutive cycles of DSS treatment (3% w/v DSS for 5 days) at weeks 9, 12 and 15 of age. Recovery periods after first and second cycles of DSS were for 14 days. After the third DSS cycle, mice were allowed to recover for 21 days prior to euthanasia. Throughout the study, body weight was measured to monitor disease progression. All mice were sacrificed at 18 weeks of age and the colorectums were processed for quantitation of polyp number, size, and histological analyses. Briefly, colorectums were flushed with ice cold PBS and longitudinally cut open, flattened, highlighted with 1% Alcian blue dye, and placed on a light box for macroscopic high-resolution image captures. These images were enlarged for identification and marking of suspected polyps, and ImageJ software was used for enumeration and size quantification of the identified polyps. Size classification for polyps are the following, small (less than 4mm2), medium (4–8mm2), and large (greater than 8mm2).
Immunoblot analysis
Mucosal tissues were scraped and polyps were excised with scissors and forceps and processed with lysis buffer containing 1% NP40, 0.1% SDS, protease and phosphatase inhibitors (EMD Millipore). Polyps were dissociated with a short pulse in a tissue grinder. Tissue lysates were prepared and separated on PAGE gels as described previously (17). Protein was quantified using a BCA protein quantification kit (Thermo Scientific). Antibodies used for immunoblotting recognized β-actin (1:2000, Sigma), PKG1 (1:1000, gift from Robert Feil), PKG2 (1:1000, Santa Cruz Biotechnology), Phospho-VASP (Ser239) (1:1000, Cell Signaling), VASP (1:1000, Cell Signaling), PDE5 (1:1000, Cell Signaling), β-catenin (1:500, BD Transduction), Cyclin D1 (1:1000, Cell Signaling), Phospho-Akt (Ser473) (1:1000, Cell Signaling), and Akt (1:1000, Cell Signaling).
Immunohistochemistry (IHC)
Formalin-fixed colorectal tissues were embedded in paraffin and sectioned at a 5μm thickness by the Augusta University Histology core. Sections were subsequently deparaffinized in xylene, rehydrated with decreasing concentrations of ethanol, and boiled in citrate buffer for antigen-retrieval. Sections were either stained directly with hematoxylin-eosin (H&E) or Alcian-Blue/Periodic acid-Schiff (AB/PAS), or immunohistologically with antibodies: Ki67 (1:100, Dako Cytomation) and cleaved caspase-3 (1:500, Cell Signaling Tech). Antibody visualization was with an anti-rat/anti-rabbit ABC staining kit (Santa Cruz) for DAB amplification. Proliferation and apoptosis indices were standardized by counterstaining with hematoxylin and analyzed using ImageJ software. AB/PAS quantitation was done by expressing area of stained tissue as a function of total area. All image analyses used at least 3 animals and at least 5 random fields encompassing a minimum of 50 crypts per mouse. For immunofluorescence, double labeling of paraffin embedded sections, CD11b (1:100 dilution, Novus Biologicals) and GR-1 (1:100 dilution, eBioscience) were used, followed by anti-rabbit/anti-rat Alexa Fluor ® 647 and 488 (1:200 dilution; ThermoFisher Scientific). Sections were mounted in aqueous mounting media with DAPI (ThermoFisher Scientific) and visualized via confocal microscopy (Zeiss LSM 780 Upright Confocal). Quantitation of CD11b and GR-1 double positive staining cells were absolute cell counts from the distal colon per mouse.
Analysis of gene expression
Mucosal tissues were scraped and polyps were excised with scissors and forceps and flash-frozen in TRIzol reagent (Life technologies). RNA was DNAse treated (TURBO DNA-free kit, Life Technologies) and converted to cDNA using M-MLV reverse transcriptase (Invitrogen™). Quantitative PCR (qRT-PCR) analysis of the cDNA was performed using SYBR Green PCR Master Mix (Applied Biosystems). Relative expression levels were calculated using the 2−ΔΔCT method with β-actin (ACTB) as a reference. Amplifications were performed in triplicate wells, and melt curve analysis was done to confirm the specificity of the primers used. Primers were designed using Primer Blast Software (NCBI; Table 1).
Table 1.
Mouse primer sets used for real time quantitative PCR
| Gene Name | Sequence (forward/reverse, 5′→3′) |
|---|---|
| Nos2 | CTATGGCCGCTTTGATGTGC / TTGGGATGCTCCATGGTCAC |
| Tnf | CCCACTCTGACCCCTTTACT / TTTGAGTCCTTGATGGTGGT |
| Il1b | CCCAACTGGTACATCAGCAC / TCTGCTCATTCACGAAAAGG |
| Il6 | CTACCCCAATTTCCAATGCT / ACCACAGTGAGGAATGTCCA |
| Ifng | CCATCAGCAACAACATAAGCGTC / TCTCTTCCCCACCCCGAATCAGCAG |
| Pde5a | GGCAAGCACCATGGAACGAG / TGCGTTGACCATGTCTCTGG |
| Guca2a | TGTCCTGGTAGAAGGGGTCA / CGGGGAGCAAACTTCTTGTG |
| Guca2b | TGGAAGCCATGGTACTTGATG / AAGCGAGGCCATGTCAAGAA |
| Prkg1 | GATGGCCCAGAGTTTCACAT/ ACCATGGGTCCTGGAAAAGT |
| Prkg2 | GGGCATCTGTGATGAGCTTT / CAACCACCCGAACCTATGAC |
| Gucy2c | ACCGACAGTGGAGAACCAAC / TCTTATAGCTGGCCACCCGA |
Statistical Analysis
All data were expressed as mean ± SEM, unless otherwise stated. A Students t test was used for the comparison of two means, unless otherwise stated. The statistical significance was set at p < 0.05.
Results
Sildenafil protects the intestinal epithelium from DSS-induced damage
We have previously reported that IP injection of the PDE5 inhibitor vardenafil can activate cGMP signaling in the intestinal epithelium of mice, leading to changes in epithelial homeostasis and reduced sensitivity to DSS (18). To determine whether orally administered sildenafil could also activate cGMP signaling in the colon epithelium, phosphorylation of the endogenous PKG substrate vasodilator-stimulated phosphoprotein (VASP), and homeostasis were assessed. An acute dose of sildenafil (1.4 mg/kg IP, 4 hr) caused VASP phosphorylation (Fig. 1A), and after seven days led to increased goblet cell density and reduced proliferation in the colon (Fig. 1B, C). When challenged with dextran sulfate sodium (DSS), mice treated with sildenafil exhibited increased barrier function and reduced measures of colitis, including weight loss, bleeding, and diarrhea compared to controls (Fig. 1D, E). Taken together these results demonstrate that different PDE5 inhibitors have similar effects on epithelial homeostasis and that oral delivery of sildenafil can protect the barrier from DSS-induced damage.
Figure 1. Sildenafil protects the intestinal epithelium from DSS-induced damage.
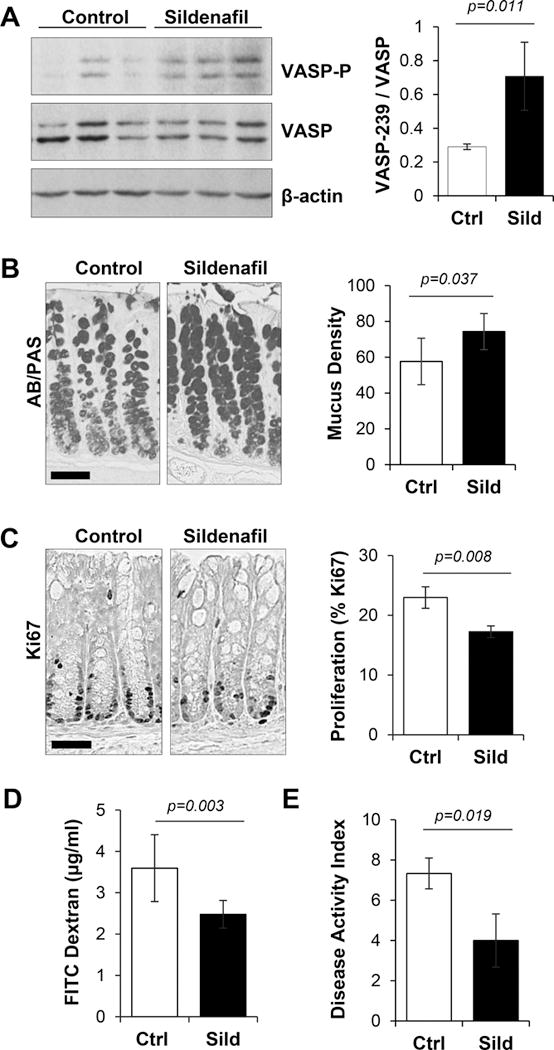
A, Immunoblot shows phosphorylation of the PKG substrate vasodilator stimulated phosphoprotein (VASP-P) in colon mucosa from control and sildenafil treated groups. β-actin is a loading control. Right panel, quantitation of densitometric data from blots shown in the left panel. B, Mucus staining (AB/PAS) of colon sections from untreated (Ctrl) and sildenafil treated mice (Sild) for 7 days. Histogram showing percentage of total AB/PAS-positive staining area. C, Ki67 staining for proliferation in colons from Ctrl and 7 day Sild-treated mice. Histogram shows percent Ki67-positive staining cells per crypt. D, Barrier permeability assessed with FITC-dextran in mice treated with 3% DSS for 5 days with or without sildenafil treatment. E, Disease activity index (DAI) in mice treated with 3% DSS for 5 days with or without sildenafil treatment. A-E, n=3 mice per group. Scale bars in panels B,C are 50 μm.
Sildenafil suppresses polyp formation in the AOM/DSS model of colon cancer
Inflammation resulting from DSS-induced damage to the colon epithelium can promote tumor growth following treatment with the mutagen azoxymethane (AOM). AOM/DSS treatment is widely used because the resulting adenomas show similar genetic mutations and dysplasia-to-carcinoma sequence observed in human colitis-associated colorectal cancer (33). Our demonstration that PDE5 inhibitors can protect mice from DSS prompted us to determine whether sildenafil could also suppress tumorigenesis in the AOM/DSS model. In these experiments, mice were treated with AOM and following a recovery period they were exposed to 3 successive cycles of DSS (Fig. 2A). Sildenafil administered in the drinking water throughout the treatment period conferred protection from body weight loss during the first cycle of DSS, but was not effective during the subsequent DSS cycles (Fig. 2B). Colons from both control and sildenafil treated animals developed numerous polyps in the distal-rectal area with fewer forming proximally (Fig. 2C). Enumeration of the polyps revealed a large range in multiplicity with a median of 10 polyps per mouse in control animals. Sildenafil treatment significantly inhibited polyp formation with a median of 5 polyps per mouse (p=0.0073; Fig. 2D).
Figure 2. Sildenafil suppresses polyp formation in the AOM/DSS model of colon cancer.
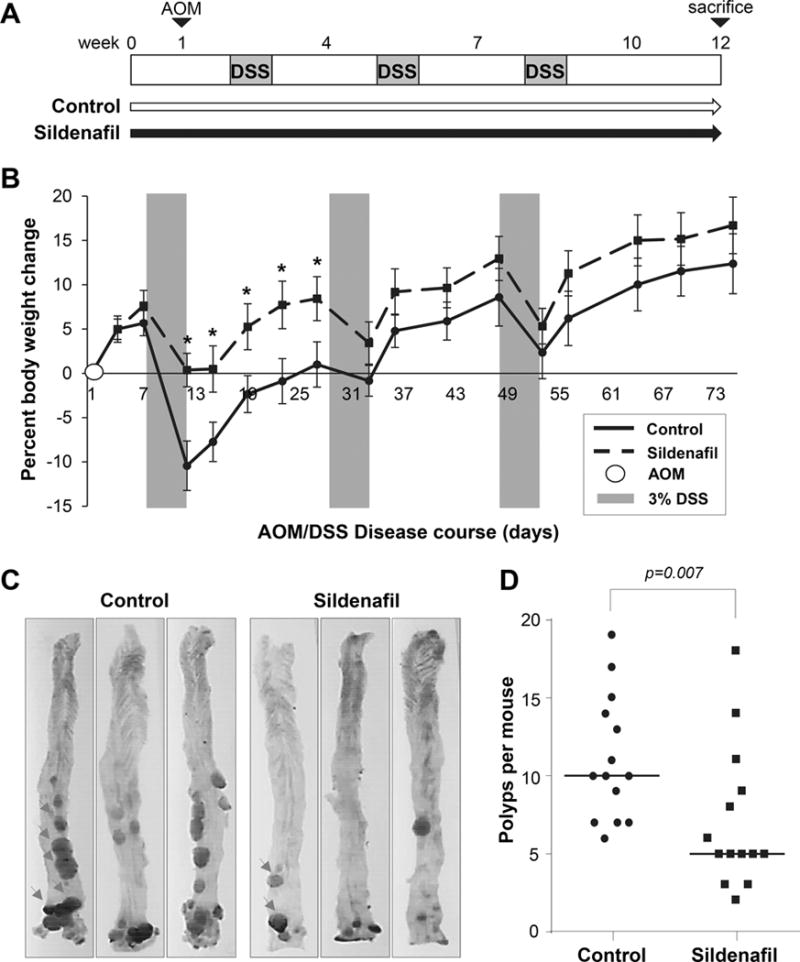
A, Experimental scheme for the AOM/DSS-induced colon carcinogenesis model. C57Bl/6J mice provided water (Control) or water containing sildenafil (5.7mg/kg/day). B, Change in body weight from the initial weight. AOM was administered on day 0, and grey panels depict DSS treatment cycles. C, Representative colons from AOM/DSS treated mice, top-bottom orientation is proximal-rectal and representative polyps are indicated in the first panels by arrows. D, Plot shows the effect of sildenafil on median polyp number per animal (n=14 per group). Asterisks show difference between weight loss between treated and control groups at each day, p < 0.0001; Panel D, p-value generated with Mann–Whitney test.
Polyps from sildenafil treated mice are less inflamed and more differentiated
While a large range of polyp size was observed in the colons of mice treated with AOM/DSS in our studies, histological analysis showed that the majority of the lesions were adenomatous polyps with leukocyte infiltration and varying degrees of dysplasia (Fig. 3A). The suppressive effect of sildenafil on polyp number was more pronounced in the smaller polyps, but the treatment did not affect mean polyp size (Fig. 3B, C). Although there was no difference in polyp size between groups, further analysis showed that there was reduced infiltration of myeloid cells, and reduced expression of inflammatory mediators in polyps from mice treated with sildenafil compared to those from control animals (Fig. 3D, E). This was most significant for iNOS and IFNγ (p=0.012 and 0.038, respectively).
Figure 3. Polyps from sildenafil treated mice are less inflamed.
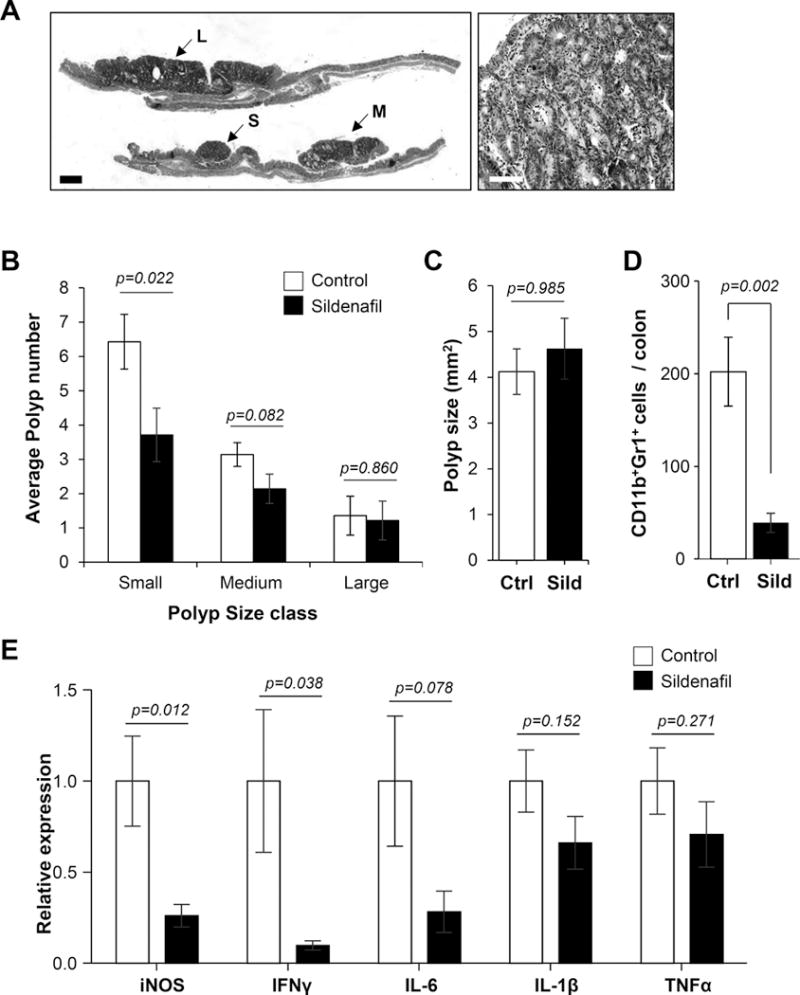
A, Representative image of H&E stained colon section (left panel) and polyp (right panel). Scale bar is 500 μm in the left panel, and 50 μm in the right panel. B, Histogram shows average polyp number in different size classes (small, medium, and large). C, Average polyp size in untreated (Ctrl) and treated (Sild) mice. D, Quantification of CD11b+Gr1+ cells in colorectums of untreated and sildenafil treated mice. E, Real-time qPCR analysis of inflammatory cytokine gene expression in polyps derived from AOM/DSS treated mice. For panels B & C, n=14 mice group; D, n=10 mice per group, E, n=8 polyps per group.
Histological analysis of polyps from AOM/DSS-treated mice showed that the sildenafil treated animals harbored polyps that were less proliferative, and while there was a small increase in apoptotic index in treated mice, the difference was not significant (Fig. 4A, B). There was a notable increase in mucus content in polyps from sildenafil treated animals compared to those from controls (Fig. 4C). Much of the mucus was observed in glandular cavities that were exaggerated in many of the sildenafil treated polyps. These cavities also contained abundant apoptotic cells, indicating a more differentiated epithelium.
Figure 4. Polyps from sildenafil treated mice are less proliferative and more differentiated.
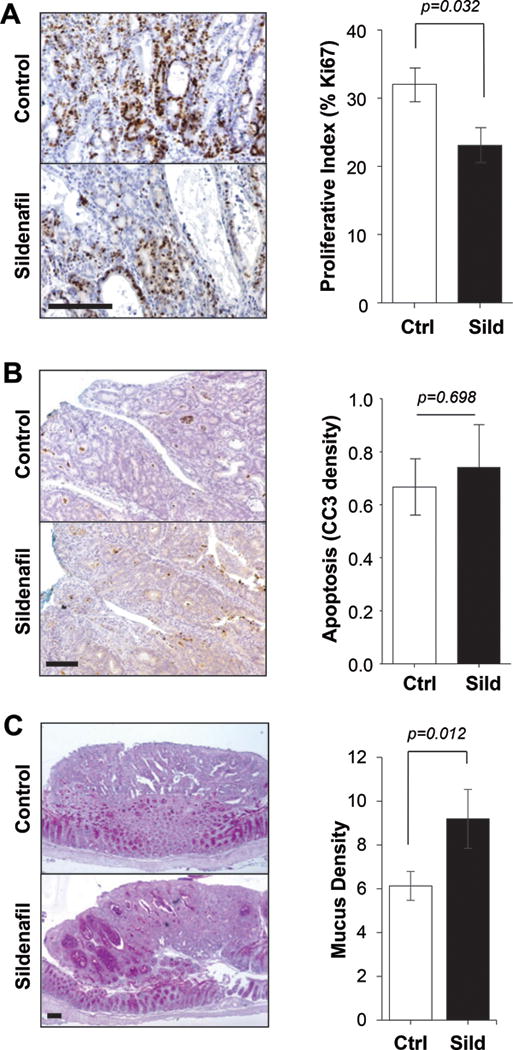
A, IHC staining of Ki67 (left panel) and quantitation (right panel) in polyps of untreated (Ctrl) and treated (Sild) mice. B, IHC staining of cleaved caspase 3 (CC3) (left panel) and quantitation (right panel) in polyps of untreated (Ctrl) and treated (Sild) mice. C, AB/PAS staining of mucus (left panel) and quantitation (right panel) in polyps of untreated (Ctrl) and treated (Sild) mice. In all panels, the scale bar is 100 μm and n=10 mice per group.
Colorectal polyps downregulate the expression of cGMP generating machinery
Components of the cGMP signaling axis have been reported to exhibit altered expression patterns in human colorectal cancer compared to normal mucosa, but this has never been examined in murine colorectal cancer models (34–36). The expression of cGMP signaling components in polyps from AOM/DSS treated mice was measured using qPCR and immunoblotting. The mRNA levels for GCC and guanylin were significantly downregulated in polyps compared to normal colon epithelium (p=0.021 and 0.003, respectively) (Fig. 5A). While uroguanylin did not change, the loss of guanylin expression was dramatic in both control and sildenafil polyps, with a 53 and 35-fold loss, respectively (Fig. 5A). Sildenafil treatment did not significantly affect the expression of these components in the polyps. Signaling downstream of cGMP typically involves PKG1, PKG2, and PDE5 (37). Analysis of mRNA expression for these components showed a small but insignificant increase in PKG1 and a similarly small decrease in PDE5, but a dramatic increase in PKG2 (Fig. 5B) compared to colon mucosa. These changes were not affected by treatment of the mice with sildenafil at either the mRNA or the protein levels (Fig. 5C).
Figure 5. Expression of cGMP signaling components in polyps.
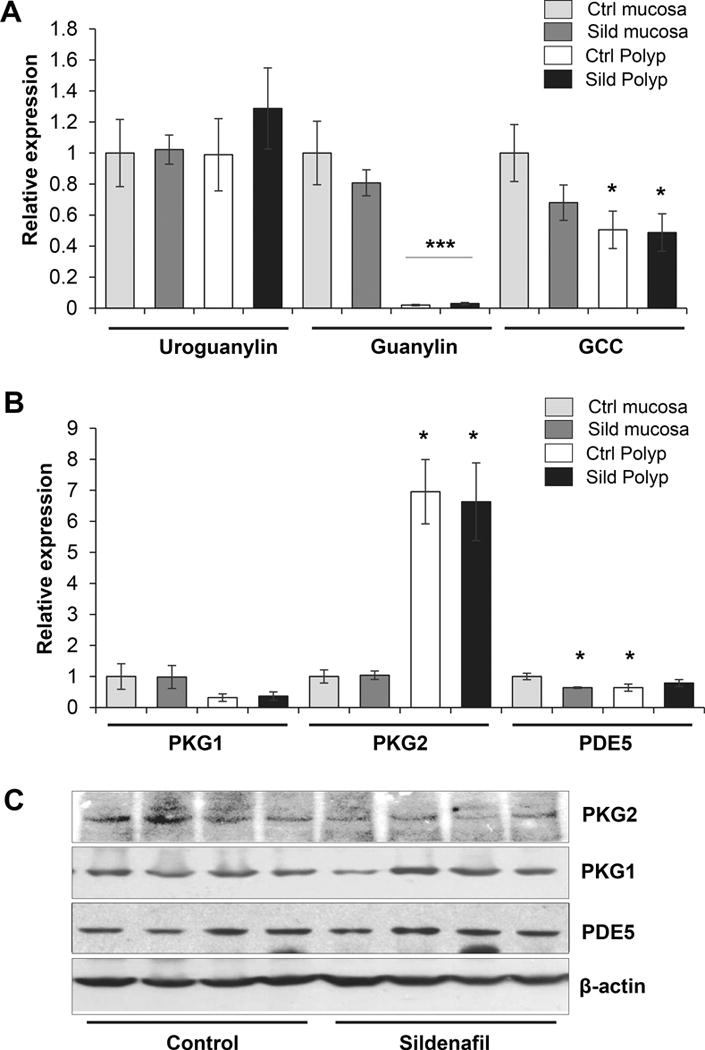
A, Expression of cGMP generators in colon mucosa and polyps measured by real-time qPCR. B, Expression of cGMP effectors in colon mucosa and polyps assayed by real-time CPCR. C, Immunoblot analysis of cGMP effectors in colon polyps of untreated (Control) and treated (Sildenafil) mice, β-actin is a loading control. For panels A–B, n=8 polyps per group, and panel C, n=5 per group. Asterisks, *p < 0.05, ***p < 0.001.
Sildenafil primarily blocks early carcinogenesis in the AOM/DSS model
Administration of sildenafil to mice throughout the AOM/DSS treatment phases resulted in 50% inhibition of polyp multiplicity, but it remains unclear what process of tumorigenesis was affected. To gain additional insight into the mechanism of action of sildenafil, the drug was administered to mice early during the AOM phase prior to exposure to DSS (Fig. 6A). In these experiments the control animals produced a median of 10.5 polyps per mouse, but surprisingly, the sildenafil treatment significantly inhibited polyp formation by 38% with a median of 6.5 polyps per mouse (p=0.039; Fig. 6B). This result suggested that the protracted presence of sildenafil during the later promotion phases did not affect polyp formation despite the marked effect on polyp phenotype. To test this possibility, sildenafil was administered subsequent to the early carcinogenesis phases of AOM treatment and the first DSS cycle. When administered at this later stage, there was a small but insignificant reduction in the number of polyps per mouse (Fig. 6C).
Figure 6. Sildenafil primarily blocks early carcinogenesis in the AOM/DSS model.
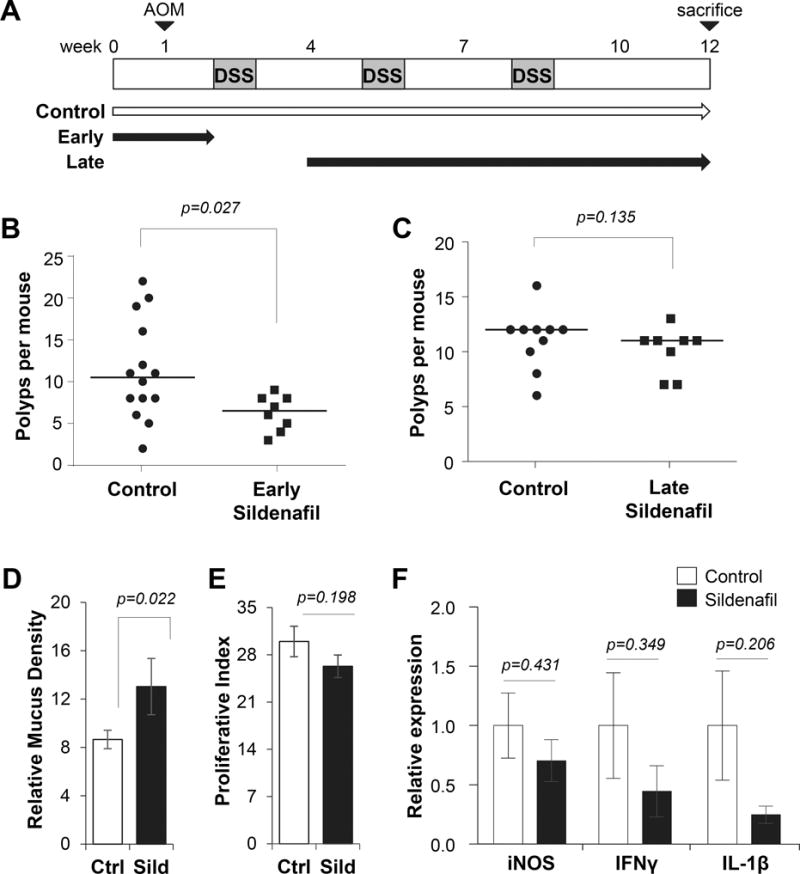
A, Experimental scheme for the AOM/DSS-induced colon carcinogenesis model. Horizontal arrows show control (water only) and early or late administration of sildenafil. B, Plot shows the effect of sildenafil added early on median polyp number per animal. C, Plot shows the effect of sildenafil added later on median polyp number per animal. D, Histogram quantification of proliferative index in polyps of untreated (Ctrl) and late-treated (Sild) mice. E, Histogram quantitation of polyp mucus density. F, Expression of inflammatory cytokine genes in colon tissues assayed by real-time qPCR. For panels B–C, n ≥ 8 mice group and p-values for panel B–C generated with Mann–Whitney test. For Panels D–E, n=11 polyps per group. For panel F, n=8 polyps per group.
The polyps formed in mice treated with sildenafil during later stages had a similar but less pronounced phenotype as those with sildenafil treatment throughout. They were similar to those with sildenafil treatment throughout, in that they showed increased mucus, and reduced proliferation and pro-inflammatory cytokine expression (Fig. 6D–F). The reduced inflammatory leukocytes and cytokines observed in the treated polyps in the present study is consistent with the idea that sildenafil can suppress myeloid-derived suppressor cells, as has been reported using non-colon cancer models (38,39). However, the dramatic effect of sildenafil when added early, and removed before inducing inflammation by DSS treatment suggests that immune regulation is unlikely to play a major role in the suppression of tumorigenesis. This idea is further supported by the lack of effect when sildenafil was administered during the 2nd and 3rd DSS cycles.
Discussion
A large body of evidence derived mostly from knockout animals has identified diverse regulatory roles for cGMP in the intestine, including secretion, epithelial barrier integrity, and proliferation-differentiation along the crypt/villus axis. Activation of cGMP signaling with GCC agonists plecanatide and dolcanatide has recently been reported to reduce symptoms of colitis in mice (25). The ability of GCC agonists to reduce diarrhea in the context of colitis is intriguing as they are established secretagogues, but these observations highlight the translational potential of barrier-protection by elevating cGMP levels. PDE5 inhibitors amplify cGMP generated by the endogenous GCC agonists, and it was shown here that exposing mice to sildenafil in the drinking water elicited similar changes in colon epithelial homeostasis and protection from DSS-induced damage as reported previously using vardenafil (18). This strongly supports the endogenous regulatory role of cGMP in the intestine since the present study used a different mouse strain, a different PDE5 inhibitor, and a different mode of administration.
Based on the ability of PDE5 inhibitors to suppress DSS-induced colitis, we reasoned that increasing cGMP in this manner would also inhibit polyp formation in the AOM/DSS model of colon cancer in mice. This model is widely used because it recapitulates many aspects of human disease (33). The initial genotoxic AOM treatment initiates mutations that are promoted to develop into colorectal adenomas by chronic inflammation that is induced and maintained by cycles of DSS. It was shown here that treating animals with sildenafil throughout AOM/DSS treatment resulted in significantly fewer polyps per mouse relative to control animals on water alone. Surprisingly, the size of the polyps from animals treated with sildenafil did not differ from those from control animals. This result was not anticipated based upon work with human colon cancer cell lines that predicts anti-tumor effects of cGMP signaling that include inhibition of proliferation, increased apoptosis, and inhibition of angiogenesis (9,40,41). Other in vitro studies have demonstrated that cGMP signaling suppresses proliferation without affecting apoptosis, and notably, that it can induce goblet cell differentiation (17,42). While sildenafil did not affect apoptosis, the polyps from treated mice were slightly less proliferative and more differentiated. Intriguingly, this latter finding supports previous observations in polyps from human patients treated with exisulind/Aptosyn (10,11).
Another unexpected observation was reduced tumor inflammation in polyps from sildenafil treated mice as reflected by both cytokine mRNA expression and infiltration of myeloid leukocytes. Inflammatory leukocyte infiltration is a poor prognostic indicator for human colorectal cancer, where it is thought to promote tumor progression and metastasis (43,44). Polyps in the AOM/DSS model typically do not become invasive or metastatic, so the possibility that sildenafil might promote a less aggressive tumor phenotype was not evaluated here. The phenotype of the polyps from sildenafil treated animals ostensibly paralleled that of normal tissue; both showed increased mucus differentiation, and reduced proliferation and inflammation. It is possible that barrier-protection by elevating cGMP can also account for the reduced inflammation of the polyps, particularly smaller and more differentiated ones. The mechanisms underlying the effects of cGMP on epithelial homeostasis and barrier protection in the normal colon epithelium are not fully understood. Suppression of Akt, β-catenin/TCF, and JNK signaling, with subsequent activation of FoxO have all been suggested to occur downstream of cGMP in either normal mouse tissue, in some human colon cancer cell lines, or both (18,20,28,29). In the present study, reduced levels of β-catenin were observed in the polyps from sildenafil treated animals, but this was more likely due to the lower abundance of proliferating cells and inflammation, because there was no evidence of PKG activity such as phosphorylation of vasodilator stimulated phosphoprotein (Supplementary Fig. S1).
It has been reported that several components of the cGMP signaling axis are downregulated either in human colon tumors relative to normal tissue, or by human colon cancer cell lines relative to tumors (17,34,36,45). The lack of detectable cGMP signaling in the polyps of sildenafil treated animals is not surprising considering the striking loss of the cGMP-generating machinery including both guanylin and GCC. The loss of guanylin by the polyps supports previous studies with human specimens, but the loss of GCC does not occur in human tumors (35). We speculated that polyps might increase the expression of alternative PDE’s that might compensate for inhibition of PDE5. While none of the PDE isoforms were affected by sildenafil treatment, we observed a striking increase in the expression of the dual specificity PDE10a (Supplementary Fig. S2). This observation supports a recent study using human specimens (46), indicating that PDE10 upregulation by colon tumors may be a universal phenomenon. Importantly, inhibitors of this isoform can suppress proliferation in colon cancer cell lines, which underscores the potential therapeutic importance of this isoform for colon cancer treatment (46,47). Whether PDE10a upregulation contributed to the suppression of cGMP signaling in the sildenafil treated polyps remains to be determined. The loss of guanylin in the polyps most likely contributed to the lack of detectable PKG activation, and it is therefore difficult to assess the in vivo relevance of anti-tumor signaling pathways previously defined in vitro. The use of exogenous GCC agonists such as linaclotide and plecanatide might elicit more robust intratumoral cGMP signaling. However, future studies will need to determine whether these intraluminal peptide drugs that do not enter circulation are capable of penetrating more dysplastic tumors.
The utility of the AOM/DSS model as an experimental system is that the initiation and promotion phases of carcinogenesis are sufficiently separate to permit testing a drug’s mechanism. The lack of marked cGMP signaling in established polyps suggested that the central anti-tumor effect of sildenafil is early during the initiation process. This idea was confirmed by the observation that sildenafil did not significantly affect polyp multiplicity when administered later during the promotion phase. It was still able to reduce proliferation and inflammation, and promote differentiation, but did not affect the number of polyps formed. In contrast, when sildenafil treatment was restricted to the AOM treatment phase, the polyp multiplicity was dramatically reduced. This unexpected result demonstrated that the important polyp-preventing effect of sildenafil treatment occurred prior to DSS-induced inflammation. While it is presently unclear how increasing cGMP would affect the genotoxic effect of AOM, it is possible that the reduced proliferative compartment in sildenafil treated animals could limit the effect of mutagenic agents. This data supports a previous study with exisulind in rats, where multiplicity was reduced following AOM treatment only if administered during the initiation phase (48). The profound effect of sildenafil early during carcinogenesis does not preclude the importance of its ability to protect against the effects of DSS. This is evident because there was a larger reduction in polyp multiplicity when sildenafil was present throughout all phases of the model compared to treatment during the AOM phase alone. In addition, the DSS dose used in the present study was relatively high, and it is possible that the effect on multiplicity could be even more profound if lower doses were used.
Taken together our results demonstrate that targeting PDE5 with selective inhibitors can inhibit colon tumorigenesis in mice. This contrasts with work suggesting that sildenafil might promote melanoma aggressiveness by potentiating ERK signaling (49). This ERK promoting pathway is not present in colon and gastric cancer cells, where cGMP has been shown to inhibit the ERK pathway (26,27). While the molecular pathways mediating the preventative effect of sildenafil as shown here are not known, the mechanism appears to involve homeostatic changes in the epithelium that confer resistance to both genotoxic and barrier damaging agents. Despite some clinical efficacy of the low-affinity inhibitor exisulind, efforts targeting PDE5 for colon cancer chemoprevention waned due to toxicity at effective doses. However, results shown here using the high-affinity and relatively safe PDE5 inhibitor sildenafil have shown similar effects in mice as were observed by higher levels of exisulind in human FAP patients. This work suggests that the utility of PDE5 as a target for chemoprevention of colorectal cancer might be revisited using currently available high-affinity inhibitors.
Supplementary Material
Acknowledgments
Financial support: This work was supported by the NCI grant #CA172627-01A1
Abbreviations
- AB/PAS
Alcian Blue/ Periodic Acid Schiff
- AOM
azoxymethane
- CC3
cleaved caspase-3
- cGMP
3′,5′cyclic guanosine monophosphate
- DAI
disease activity index
- DSS
dextran sulfate sodium
- GCC
guanylyl cyclase C receptor
- IHC
immunohistochemistry
- IP
intraperitoneal injection
- PDE5
phosphodiesterase-5
- PKG1
type-1 cGMP-dependent protein kinase
- PKG2
type-2 cGMP-dependent protein kinase
- Sild
sildenafil
References
- 1.Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015;372(15):1441–52. doi: 10.1056/NEJMra1403718. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todoric J, Antonucci L, Karin M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev Res (Phila) 2016;9(12):895–905. doi: 10.1158/1940-6207.CAPR-16-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ait Ouakrim D, Dashti SG, Chau R, Buchanan DD, Clendenning M, Rosty C, et al. Aspirin, Ibuprofen, and the Risk of Colorectal Cancer in Lynch Syndrome. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377(9759):31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 6.Elwood PC, Morgan G, Pickering JE, Galante J, Weightman AL, Morris D, et al. Aspirin in the Treatment of Cancer: Reductions in Metastatic Spread and in Mortality: A Systematic Review and Meta-Analyses of Published Studies. PLoS One. 2016;11(4):e0152402. doi: 10.1371/journal.pone.0152402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tougeron D, Sha D, Manthravadi S, Sinicrope FA. Aspirin and colorectal cancer: back to the future. Clin Cancer Res. 2014;20(5):1087–94. doi: 10.1158/1078-0432.CCR-13-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997;57(14):2909–15. [PubMed] [Google Scholar]
- 9.Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, et al. Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000;60(13):3338–42. [PubMed] [Google Scholar]
- 10.Stoner GD, Budd GT, Ganapathi R, DeYoung B, Kresty LA, Nitert M, et al. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv Exp Med Biol. 1999;470:45–53. doi: 10.1007/978-1-4615-4149-3_5. [DOI] [PubMed] [Google Scholar]
- 11.van Stolk R, Stoner G, Hayton WL, Chan K, DeYoung B, Kresty L, et al. Phase I trial of exisulind (sulindac sulfone, FGN-1) as a chemopreventive agent in patients with familial adenomatous polyposis. Clin Cancer Res. 2000;6(1):78–89. [PubMed] [Google Scholar]
- 12.Ratner M. Cancer pathways’ target not validated by clinical results. Nat Biotechnol. 1999;17(3):220. doi: 10.1038/6956. [DOI] [PubMed] [Google Scholar]
- 13.Arber N, Kuwada S, Leshno M, Sjodahl R, Hultcrantz R, Rex D. Sporadic adenomatous polyp regression with exisulind is effective but toxic: a randomised, double blind, placebo controlled, dose-response study. Gut. 2006;55(3):367–73. doi: 10.1136/gut.2004.061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forte LR., Jr Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther. 2004;104(2):137–62. doi: 10.1016/j.pharmthera.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Li P, Lin JE, Chervoneva I, Schulz S, Waldman SA, Pitari GM. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am J Pathol. 2007;171(6):1847–58. doi: 10.2353/ajpath.2007.070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinbrecher KA, Wowk SA, Rudolph JA, Witte DP, Cohen MB. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am J Pathol. 2002;161(6):2169–78. doi: 10.1016/S0002-9440(10)64494-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R, Kwon IK, Thangaraju M, Singh N, Liu K, Jay P, et al. Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am J Physiol Gastrointest Liver Physiol. 2012;303(2):G209–19. doi: 10.1152/ajpgi.00500.2011. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Kwon IK, Singh N, Islam B, Liu K, Sridhar S, et al. Type 2 cGMP-dependent protein kinase regulates homeostasis by blocking c-Jun N-terminal kinase in the colon epithelium. Cell Death Differ. 2014;21(3):427–37. doi: 10.1038/cdd.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li P, Schulz S, Bombonati A, Palazzo JP, Hyslop TM, Xu Y, et al. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133(2):599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 20.Lin JE, Li P, Snook AE, Schulz S, Dasgupta A, Hyslop TM, et al. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology. 2010;138(1):241–54. doi: 10.1053/j.gastro.2009.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shailubhai K, Yu HH, Karunanandaa K, Wang JY, Eber SL, Wang Y, et al. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 2000;60(18):5151–7. [PubMed] [Google Scholar]
- 22.Han X, Mann E, Gilbert S, Guan Y, Steinbrecher KA, Montrose MH, et al. Loss of guanylyl cyclase C (GCC) signaling leads to dysfunctional intestinal barrier. PLoS One. 2011;6(1):e16139. doi: 10.1371/journal.pone.0016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harmel-Laws E, Mann EA, Cohen MB, Steinbrecher KA. Guanylate cyclase C deficiency causes severe inflammation in a murine model of spontaneous colitis. PLoS One. 2013;8(11):e79180. doi: 10.1371/journal.pone.0079180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin JE, Snook AE, Li P, Stoecker BA, Kim GW, Magee MS, et al. GUCY2C opposes systemic genotoxic tumorigenesis by regulating AKT-dependent intestinal barrier integrity. PLoS One. 2012;7(2):e31686. doi: 10.1371/journal.pone.0031686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shailubhai K, Palejwala V, Arjunan KP, Saykhedkar S, Nefsky B, Foss JA, et al. Plecanatide and dolcanatide, novel guanylate cyclase-C agonists, ameliorate gastrointestinal inflammation in experimental models of murine colitis. World J Gastrointest Pharmacol Ther. 2015;6(4):213–22. doi: 10.4292/wjgpt.v6.i4.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu M, et al. PKG II inhibits EGF/EGFR-induced migration of gastric cancer cells. PLoS One. 2013;8(4):e61674. doi: 10.1371/journal.pone.0061674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha S, Chowdhury P, Pal A, Chakrabarti MK. Downregulation of human colon carcinoma cell (COLO-205) proliferation through PKG-MAP kinase mediated signaling cascade by E. coli heat stable enterotoxin (STa), a potent anti-angiogenic and anti-metastatic molecule. J Appl Toxicol. 2008;28(4):475–83. doi: 10.1002/jat.1297. [DOI] [PubMed] [Google Scholar]
- 28.Kwon IK, Wang R, Thangaraju M, Shuang H, Liu K, Dashwood R, et al. PKG inhibits TCF signaling in colon cancer cells by blocking beta-catenin expression and activating FOXO4. Oncogene. 2010;29(23):3423–34. doi: 10.1038/onc.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, Islam B, Bridges A, Sharman S, Hu M, Hou Y, et al. cGMP Signaling Increases Antioxidant Gene Expression by Activating Forkhead Box O 3a in the Colon Epithelium. Am J Pathol. 2016 doi: 10.1016/j.ajpath.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 31.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 32.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protocols. 2007;2(8):1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 33.Robertis MD, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, et al. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. Journal of Carcinogenesis. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou Y, Gupta N, Schoenlein P, Wong E, Martindale R, Ganapathy V, et al. An anti-tumor role for cGMP-dependent protein kinase. Cancer Letters. 2006;240(1):60–8. doi: 10.1016/j.canlet.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 35.Waldman SA, Hyslop T, Schulz S, Barkun A, Nielsen K, Haaf J, et al. Association of GUCY2C expression in lymph nodes with time to recurrence and disease-free survival in pN0 colorectal cancer. JAMA. 2009;301(7):745–52. doi: 10.1001/jama.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson C, Lin JE, Li P, Snook AE, Gong J, Sato T, et al. The paracrine hormone for the GUCY2C tumor suppressor, guanylin, is universally lost in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(11):2328–37. doi: 10.1158/1055-9965.EPI-14-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Browning DD, Kwon IK, Wang R. cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Future Med Chem. 2010;2(1):65–80. doi: 10.4155/fmc.09.142. [DOI] [PubMed] [Google Scholar]
- 38.Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108(41):17111–6. doi: 10.1073/pnas.1108121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon IK, Schoenlein PV, Delk J, Liu K, Thangaraju M, Dulin NO, et al. Expression of cyclic guanosine monophosphate-dependent protein kinase in metastatic colon carcinoma cells blocks tumor angiogenesis. Cancer. 2008;112(7):1462–70. doi: 10.1002/cncr.23334. [DOI] [PubMed] [Google Scholar]
- 41.Soh JW, Mao Y, Kim MG, Pamukcu R, Li H, Piazza GA, et al. Cyclic GMP mediates apoptosis induced by sulindac derivatives via activation of c-Jun NH2-terminal kinase 1. Clin Cancer Res. 2000;6(10):4136–41. [PubMed] [Google Scholar]
- 42.Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc Natl Acad Sci U S A. 2001;98(14):7846–51. doi: 10.1073/pnas.141124698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. 2016;274(1):290–306. doi: 10.1111/imr.12464. [DOI] [PubMed] [Google Scholar]
- 44.Wang K, Karin M. Tumor-Elicited Inflammation and Colorectal Cancer. Adv Cancer Res. 2015;128:173–96. doi: 10.1016/bs.acr.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Steinbrecher KA, Tuohy TM, Heppner Goss K, Scott MC, Witte DP, Groden J, et al. Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem Biophys Res Commun. 2000;273(1):225–30. doi: 10.1006/bbrc.2000.2917. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Lee K, Xi Y, Zhu B, Gary BD, Ramirez-Alcantara V, et al. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and beta-catenin-dependent TCF transcriptional activity. Oncogene. 2015;34(12):1499–509. doi: 10.1038/onc.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K, Lindsey AS, Li N, Gary B, Andrews J, Keeton AB, et al. beta-catenin nuclear translocation in colorectal cancer cells is suppressed by PDE10A inhibition, cGMP elevation, and activation of PKG. Oncotarget. 2016;7(5):5353–65. doi: 10.18632/oncotarget.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy BS, Kawamori T, Lubet RA, Steele VE, Kelloff GJ, Rao CV. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999;59(14):3387–91. [PubMed] [Google Scholar]
- 49.Dhayade S, Kaesler S, Sinnberg T, Dobrowinski H, Peters S, Naumann U, et al. Sildenafil Potentiates a cGMP-Dependent Pathway to Promote Melanoma Growth. Cell Rep. 2016;14(11):2599–610. doi: 10.1016/j.celrep.2016.02.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.