

Abstract
A group of papers investigates functional regulatory elements in genomes from human tissue samples and cell lines. What can neuroscientists learn from the gigantic data set and how will it affect the direction of neuroepigenetics?
A striking and unexpected conclusion that emerged from the completion of the Human Genome Project (Figure 1A) was that the number of genes in the human genome is no more than that of C. elegans (Lander et al., 2001; Venter et al., 2001). Further, we learned that over 95% of the human genome exhibits no protein-coding information. These discoveries shifted our focus from genomes to epigenomes to explain how the complex variegation of human cells and tissues may arise from a precise orchestration of this limited number of genes. The modern definition of epigenome is reversibly encoded information to the genome without altering the underlying DNA sequences. The epigenome works as landmarks for each cell to correctly interpret the invariable scriptio continua of DNA-based genome. There are ever-expanding facets of the epigenome, each of which requires specific experimental modalities to assess—DNaseseq for open chromatin, Hi-C for chromatin long-range interaction, ChIP-seq for transcription factor binding or his-tone post-translational modifications, and bisulfite-seq for 5-methylcytosine modifications (Figure 1B). All of the epigenomic information is marked on a single string of genomic DNA, but most epigenetic studies often pick a subset of epigenomic features due to the economical reason. Thus, the interdisciplinary Encyclopedia of DNA Elements (ENCODE) project was launched to achieve holistic and comprehensive understanding of human epigenomes (Figure 1A) (ENCODE Project Consortium, 2004). Over the last decade, the ENCODE project has successfully generated a large amount of epigenomic data from over a hundred cell or tissue types (ENCODE Project Consortium, 2012 ENCODE Project Consortium, 2007). Very recently, the Roadmap Epigenomics Mapping Consortium had their first large-scale release, which focused on epigenomes of human primary cells and tissue samples, including nine human adult brain structures (Figure 1C) (Roadmap Epigenomics Consortium et al., 2015). Now there is substantial evidence that epigenetic regulation is especially crucial for brain function and the mammalian brain exhibits a particularly plastic epigenetic landscape (Guan et al., 2009; Guo et al., 2011). Here we highlight the value, application, limitation, and future of large-scale epigenomic studies from the viewpoint of the emerging field of neuroepigenetics.
Figure 1. The Past, Present, and Future of Epigenomic Studies in the Nervous System.
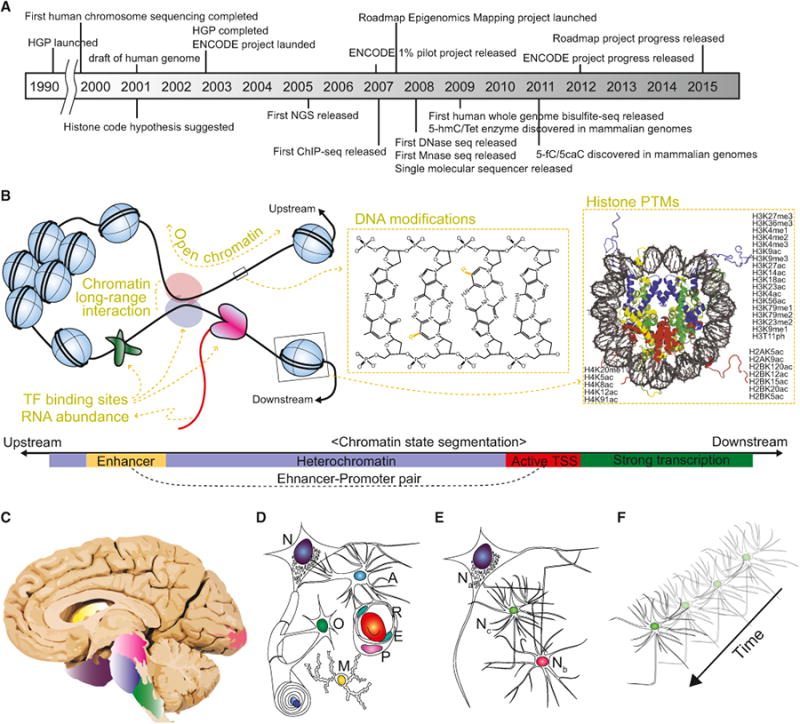
(A) The timeline of key discoveries and technical advances in the field of epigenetics. HGP, Human Genome Project; ENCODE, Encyclopedia of DNA Elements; NGS, Next Generation Sequencer; ChIP, chromatin immunoprecipitation; hmC, 5-hydroxymethylcytosine; fC, 5-formylcytosine; caC, 5-carboxylcytosine; Mnase, micrococcal nuclease; Tet, Ten-eleven translocation. (B) Key epigenomic features that current large-scale epigenomic projects uncover. Open chromatin can be assayed by DNase-seq, FAIRE-seq, or ATAC-seq; chromatin long-range interactions can be assayed by Hi-C; transcription factor (TF) binding sites can be assayed by TF ChIP-seq; RNA abundance can be assayed by RNA-seq; DNA modifications can be assayed by whole-genome bisulfite sequencing; and histone post-translational modifications can be assayed by histone ChIP-seq (top). Using general principles achieved by large epigenome data sets, chromatin states can be defined based on epigenomic features, such as chromatin accessibility or histone PTMs (bottom). As such, although each genomic region can have hundreds of epigenomic features requiring hundreds of epigenetic assays, we only need a minimal set of epigenomic features to define chromatin states. (C-F) The future of neuroepigenetics. The Roadmap Epigenomics Project distinguished nine brain structures of human brain: angular gyrus, head of caudate nucleus, inferior parietal lobule, inferior temporal gyrus, middle frontal gyrus, midbrain, occipital pole, pons, and medulla oblongata (C). Each brain region comprises a variety of distinct cell types, including neurons (N), astrocytes (A), oligodendrocytes (O), microglia (M), blood cells (R), endocytes (E), and pericytes (P) (D). All these cell types with distinct functions and epigenomes collectively contribute to the epigenome data generated. Thus, it is difficult to know which cell type is the major contributor for an observed epigenomic transition, or whether the observed epigenomic transition is the result of the epigenome change within each cell type or the result of changes in the cellular composition of the sample. Each cell type also contains multiple subtypes (E). For example, there are a number of different neuronal subtypes (denoted as Na, Nb, and Nc) with distinct functions, morphologies, and probably epigenomes. Furthermore, each neuronal subtype can exhibit differential epigenome over time during development or upon physiological or pathological perturbations (F).
Since the ENCODE consortium was launched in 2003, we have experienced an exponential growth in the epigenome database. Several major releases have significantly advanced our understanding of the human epigenome (Figure 1A). First, the ENCODE consortium revealed 1% of the human epigenome and transcriptome using microarray technologies in 2007. This pilot release provided a number of groundbreaking discoveries and insights, including the identification of pervasive genome-wide transcriptional activity and characterization of distal regulatory elements and megabase-scale chromatin domains (ENCODE Project Consortium, 2007). Second, the ENCODE consortium released 30 publications with massive data sets in 2012, which provided a far more comprehensive view of the epigenetic landscape and additional insights (ENCODE Project Consortium, 2012). Using 147 cell types from mostly cultured cell lines, these studies sought to establish generally applicable principles of the human epigenome, which included a hierarchical network of transcription factor (TF) binding, predicting TF binding using known TF motifs and DNase I footprinting, and predicting functional chromatin states by integrating histone modifications and chromatin accessibility (Figure 1B) (ENCODE Project Consortium, 2012; Ernst and Kellis, 2010; Ernst et al., 2011; Thurman et al., 2012). Third, the Roadmap Epigenomics Mapping Consortium released an even greater amount of data focused on human primary cells and tissues (Roadmap Epigenomics Consortium et al., 2015), which revealed more comprehensive and fundamental epigenetic principles with potential for widespread biological relevance. For example, Ziller et al. attributed key transcription factors to regional epigenomic transition throughout consecutive stages of in vitro neural differentiation (Ziller et al., 2015). Gjoneska et al. showed that epigenome changes in a mouse Alzheimer disease model occur at conserved regulatory elements for immune cells (Gjoneska et al., 2015).
Currently, the growth of the epigenome database derived from these consortiums has three immediate benefits. First, these large data sets can be probed for epigenetic marks of interest at specific loci by individual researchers to guide their own studies. Second, it allows targeted approaches for genome-wide analyses. Recent works by Lunnon et al. and De Jager et al. are excellent examples of how epigenetic perturbations in human brain disorders can be identified using targeted approaches (De Jager et al., 2014; Lunnon et al., 2014). These studies employed microarray-based methylation analysis, which covers only 1.5% of CpGs in the genome but included most identified promoters and functional genomic elements based on previous genome-wide studies. Designing assays that could specifically target only functional elements would be a very powerful tool for large-scale screening of brain-specific epigenomic perturbations. Third, we can infer chromatin state information using only key epigenomic features (Figure 1B, bottom). The roadmap project consortium reported that a combination of a subset of histone modifications is sufficient to define the nearby transcription levels and even to impute the chromatin accessibility and local DNA methylation levels (Roadmap Epigenomics Consortium et al., 2015). Therefore, if such an assumption holds true for other tissues of interest, we can achieve a similar level of precision as large-scale epigenomic projects using a fraction of epigenomic assays.
Is there an endpoint to the trend of massive data accumulation in epigenetics? Clearly, epigenome projects would not achieve “completion” in the same way as the Human Genome Project. First, unlike the largely invariable genome, the epigenome varies along multiple and interacting dimensions that include different cell and tissue types, specific developmental stages, and physiological or pathological perturbations (Figures 1C–1F). Second, there has been a rapid expansion of identified epigenetic features, such as different DNA base modifications, hundreds of histone post-translational modifications and binding of transcription factors, nucleosome occupancy, and 3-dimensional chromatin architectures (Figures 1A and 1B). If the variability of the epigenome among tissue and cell types with different developmental, physiological, or pathological conditions, combined with the myriad facets of epigenetic features, precludes the identification of all possible reference epigenomes, then what is the future of the epigenome project, especially from the neuroepigenetics point-of-view?
First, we expect to identify generally applicable rules defining chromatin states using a minimal number of epigenetic features. There are a few tools defining chromatin features using a subset of histone modifications without bias. Moreover, the Roadmap Epigenomics project showed the possibility of computing the distribution of missing epigenetic marks with precision based on other observed epigenetic data sets (Ernst and Kellis, 2015). It suggests that certain epigenetic marks are dependent variables, and thus not necessary to be assayed for all biological conditions. Eventually, utilizing the simplest rules to distinguish the distribution and rearrangement of underlying chromatin functional segments would become possible. Second, we expect to see the emergence of single-cell epigenomics to address the heterogeneity issue (Figures 1C–1F). Heterogeneity is indeed a very imminent challenge for neuroepigenetics (Shin et al., 2014). There are two types of heterogeneity: cell-type/static heterogeneity and temporal/dynamic heterogeneity. Static heterogeneity indicates cells with distinct functions, such as neurons and astrocytes, which are therefore expected to exhibit distinct basal epigenomes. Dynamic heterogeneity indicates a change of the epigenome within the same cell population resulting from changes in activity, a critical property of dynamic brain circuits and function (Guo et al., 2011). Static heterogeneity can be partially addressed by cell selection through methods such as fluorescence-activated cell sorting, yet these approaches may preclude identification of unappreciated heterogeneity among the seemingly homogenous population. Dynamic heterogeneity can only be tackled by single-cell epigenetic assays because of the continuous nature of the process. Third, we expect to achieve allelic information of various epigenomic features via sequential profiling of different epigenetic marks in the same sample, such as Chip-bisulfite sequencing (Guo et al., 2014). Fourth, we need to know the causal and temporal relationships between different epigenetic marks. These mechanistic insights will be tremendously helpful in understanding which epigenomic assay to use when we study more specific developmental or disease conditions.
Epigenetic mechanisms are emerging as an important component in human brain function and dysfunction (De Jager et al., 2014; Guan et al., 2009; Lunnon et al., 2014). The mammalian brain also exhibits unique epigenetic features, such as high levels of hydroxymethylcytosine (Kriaucionis and Heintz, 2009) and nonCpG methylation (Guo et al., 2014). Moreover, over 90% of disease alleles are located at non-coding distal regulatory elements, suggesting the importance of epigenetic mechanisms in the brain. The immense complexity of neural structures and circuitry, in conjunction with both static and dynamic heterogeneity of cellular populations, make it challenging to understand the mechanisms and consequences of epigenetic modifications. However, considering the relatively short history of the large-scale epigenomic projects and the rapid advancement of supporting technologies that include assays for novel epigenomic features, next-generation sequencing technology, and bioinformatics makes us optimistic for the future of neuroepigenetics.
References
- De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, et al. Nat Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Nat Biotechnol. 2015:3157. doi: 10.1038/nbt.3157. Published online February 18, 2015 http://dx.doi.org/10.1038/nbt. [DOI] [PMC free article] [PubMed]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, et al. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, et al. Nat Neurosci. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. Nat Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. International Human Genome Sequencing Consortium. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, Troakes C, Al-Sarraj S, Burrage J, Macdonald R, et al. Nat Neurosci. 2014;17:1164–1170. doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Ming GL, Song H. Nat Neurosci. 2014;17:1463–1475. doi: 10.1038/nn.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, Issner R, Gifford CA, Goren A, Xing J, et al. Nature. 2015;518:355–359. doi: 10.1038/nature13990. [DOI] [PMC free article] [PubMed] [Google Scholar]