

Abstract
Development of vaccines capable of eliciting broadly neutralizing antibodies (bNAbs) is a key goal to controlling the global AIDS epidemic. To be effective, bNAbs must block the capture of HIV-1 to prevent viral acquisition and establishment of reservoirs. However, the role of bNAbs, particularly during initial exposure of primary viruses to host cells, has not been fully examined. Using a sensitive, quantitative, and high-throughput qRT-PCR assay, we found that primary viruses were captured by host cells and converted into a trypsin-resistant form in less than five minutes. We discovered, unexpectedly, that bNAbs did not block primary virus capture, although they inhibited the capture of pseudoviruses/IMCs and production of progeny viruses at 48 hours. Further, viruses escaped bNAb inhibition unless the bNAbs were present in the initial minutes of exposure of virus to host cells. These findings will have important implications for HIV-1 vaccine design and determination of vaccine efficacy.
Keywords: HIV-1 transmission, A3R5.7 cells, broadly neutralizing antibodies, viral capture, qRT-PCR, virus entry, HIV-1 vaccine
Introduction
HIV/AIDS is a serious global epidemic the world is facing today. While anti-retroviral drugs have been highly effective in controlling viremia, there is no cure. And, despite over 30 years of concerted efforts worldwide, there still is no HIV vaccine. A preventative vaccine that can block HIV transmission is considered to be the best option to control and ultimately eradicate this disease.
HIV presents numerous challenges for the design of an effective prophylactic vaccine. These include extensive viral diversity; very early establishment of latent viral reservoirs; evolution of mutations in the trimeric envelope protein (Env) in response to the host immune response; complex glycosylation that forms a shield over the Env ectodomain hindering the antibodies from reaching the critical epitopes; and lack of a suitable animal model for determining the correlates of protection and vaccine efficacy. Despite these challenges, several human phase III clinical trials using a variety of HIV-1 immunogen formulations have been conducted. All have thus far failed except for the RV144 Thai trial which showed a modest efficacy of ∼31% at 42 months and 60% at 12 months after initiation of vaccination (Rerks-Ngarm et al., 2009; Robb et al., 2012). Binding antibodies specific to the V2 domain of the envelope HIV-1 glycoprotein, but not neutralizing antibodies, correlated with protection in the RV144 phase III trial (Haynes et al., 2012). Further, vaccine induced polyfunctional CD4+ T-cell subsets were shown to be associated with decreased risk of HIV infection in the same trial (Lin et al., 2015).
One of the major strategies to prevent acquisition of HIV has been to elicit broadly neutralizing antibodies (bNAbs). These antibodies arise in individuals during the chronic phase of HIV-1; however, these antibodies are not able to control the disease at that late stage. Whether they can prevent infection if administered passively is being tested in high risk volunteers in an ongoing HVTN 703 (AMP) trial. These bNAbs can also be generated in vitro from B cells of individuals with chronic infection and have been shown to neutralize several different strains and subtypes of HIV-1 (Chuang et al., 2013; Falkowska et al., 2014; Georgiev et al., 2013; Huang et al., 2014; Huang et al., 2012; Liao et al., 2013; Walker et al., 2011; Walker et al., 2009; Wu et al., 2010). However, in the current neutralization assays, the read-out of virus infectivity is days (in vitro) or weeks (in vivo) after infection. Hence, these time-points are far removed from the initial virus entry. Moreover, replication-incompetent, in vitro-cell line produced, pseudoviruses, which are used for assessing neutralization may not be the same as the viral inoculum humans are exposed to during HIV-1 transmission. Therefore, a critical gap exists in the current paradigm due to the lack of quantitative data on virus capture and entry at timescales relative to natural primary virus infection. Such data are essential to directly measure the efficiency of virus capture and entry; determine the effectiveness of bNAbs or immune sera to block virus capture; and to design efficacious preventative vaccines that can block HIV-1 infection.
In this study, we developed a quantitative high-throughput assay to examine early events after exposure of primary HIV-1 to host cells. We found that the primary virus was captured by the host cells and converted into a trypsin-resistant form in less than five minutes. Unexpectedly, we observed that this capture was not inhibited by potent bNAbs, although the bNAbs inhibited subsequent steps of entry and viral replication. Furthermore, we found that the bNAbs must be present in the initial minutes of exposure of the virus to the host cells. Otherwise, a significant portion of the primary viruses escaped even if bNAbs were present for the rest of the infection period. On the other hand, capture of pseudoviruses and infectious molecular clones (IMCs) was inhibited by the bNAbs implicating fundamental differences between the envelopes of primary viruses and pseudoviruses and their recognition by host cells. These findings raise the possibility that at least some of the primary viruses that the humans are exposed to might escape bNAbs and establish early reservoirs. Therefore, it might be necessary to induce capture-blocking antibodies in addition to bNAbs to completely prevent HIV-1 infection during sexual transmission. Our studies thus provide new insights on the mechanisms of HIV-1 transmission and have implications for the design of more effective HIV-1 vaccines.
Materials and Methods
Ethics Statement
RV229B (WRAIR Protocol #1386)
This protocol “Apheresis of blood components from healthy volunteers for in vitro research” and all related documents were approved by the following independent Institutional Review Boards (IRBs): The Division of Human Subject Protection, Walter Reed Army Institute of Research, and the Ethical Review Committee for Research in Human Subjects. All volunteers provided written informed consent following discussion and counseling by the clinical study team prior to enrollment and before any blood draws were performed.
Monoclonal Antibodies and Inhibitors
Monoclonal antibodies 2G12 and b12 were purchased from Polymun Scientific (Austria). The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (PG9), Anti-HIV-1 gp120 Monoclonal (PGT121), Anti-HIV-1 gp41 Monoclonal (4E10) from Dr. Hermann Katinger, Anti-HIV-1 gp120 Monoclonal (3BNC117) from Dr. Michel C. Nussenzweig, and Anti-HIV-1 gp120 Monoclonal (2191, 2219, 2442) from Dr. Susan Zolla-Pazner. CAP256-VRC26.25 and VRC01 were a gift from Dr. John Mascola, VRC, NIH. Anti-CD4 purified antibody, SK3 clone was purchased from BD Biosciences. Synagis was purchased from MedImmune and T20 was purchased from AnaSpec.
Cells and Media
A3R5.7 cells were provided by Dr. Robert McLinden, USMHRP. Media components and reagents were obtained as follows: RPMI-1640, L-glutamine, penicillin/streptomycin (Quality Biologicals Inc.), Accutase (eBiosciences), Trypsin (Sigma-Aldrich), fetal bovine serum (Gemini Bio Products), and G418 (Life Technologies). A3R5.7 cells were grown in RPMI-1640 containing 15% FBS, 1% L-Glutamine, 1% Pen/Strep, and 1 mg/mL G418 at 37°C and 5% CO2. Cells were grown to confluency and used for experiments starting at day three. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: 8E5/LAV from Dr. Thomas Folks. The 8E5 cells were grown in RPMI-1640 containing 05% FBS, 1% L-Glutamine, and 1% Pen/Strep at 37°C and 5% CO2. The cells were harvested and then processed for DNA isolation using the DNeasy Blood and Tissue kit (Qiagen) for cellular and viral DNA standards for qRT-PCR.
HIV-1 capture assay using A3R5.7 cells
A3R5.7 cells grown in G418 media were harvested and then plated. Purified primary virus (preparation and purification as described in Supplemental Information) was added to the wells and the plates were incubated in a CO2 incubator at 37°C for 5 or 30 minutes. Cells and virus were then centrifuged at 1200 rpm for 5 minutes at 4°C. The supernatant containing the virus was removed and the cells were washed and treated with trypsin. Cell pellets were frozen at −20°C, and then later processed for RNA extraction. For details see Supplemental Information.
HIV-1 capture assay using A3R5.7 cells and bNAbs
A3R5.7 cells were plated as above. The virus was pre-incubated at 37°C for 15 minutes in the absence or presence of mAbs and then added to the cells. The cells were incubated at 37°C for 5 or 30 minutes and processed as described above. For details see Supplemental Information.
Addition of exogenous gag RNA in the capture assay
A3R5.7 cells were plated as stated above. Purified primary BaL was mixed with media or with 10 million gag RNA copies (gag RNA transcripts were provided by Dr. Mary Kearney, NCI, NIH), added to cells, and incubated at 37°C for 5 minutes. The cells were washed, trypsin treated, and processed for RNA isolation as described in Supplemental Information.
Expansion experiments for A3R5.7 cells
The cells were seeded and infected with virus as described above for the capture assay. Followed by washing and trypsin treatment, cells were resuspended in media, and incubated for 1 hour or 48 hours at 37°C, after which the cells were harvested, and washed as described in the Supplemental Information. The supernatants were analyzed using p24 ELISA kit (ABL, Inc). The final cell pellets were split into 2 aliquots and each pellet was processed for RNA and DNA extraction.
Virus production in A3R5.7 cells after 48 hours
Assay details are in Supplemental Information. Cells were prepared as described above for the HIV-1 capture assay with bNAbs. Cells were washed, trypsin treated, resuspended in media containing either mAbs or T20, incubated at 37°C for 48 hours, and then harvested. The viral supernatant was collected and the cells pellets were washed with ice cold PBS. A portion of the viral supernatant and cell pellets was processed for nucleic acid extraction.
Experiments to further understand the mechanism of inhibition of viral replication by VRC01 was performed in a similar way as noted above. However, the cells were infected with the virus (pre-incubated in the absence or presence of mAb) for 30 minutes, washed, trypsin treated, and after further washes, resuspended in media. There were three conditions: (i) the virus and cells were incubated together for 30 minutes in the absence of VRC01. Following trypsin treatment and washes as noted above, VRC01 was added and present during the 48 hour incubation; (ii) the antibody and virus were pre-incubated for 15 minutes and then added to the cells for 30 minutes. Following trypsin treatment and washes, the antibody was added and was also present during the 48-hour incubation; (iii) the antibody and virus was pre-incubated for 15 minutes and then the mixture was added to the cells for 30 minutes. Following trypsin treatment and washes, cells were incubated for 48 hours in media in the absence of VRC01, see Fig. 5A.
Fig 5. VRC01 inhibits cell free infection and is required early for effective inhibition.
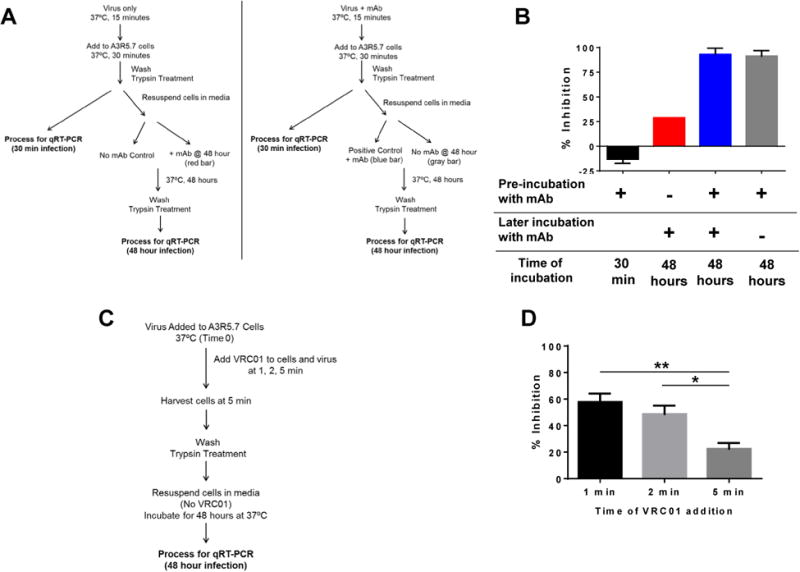
(A) Flow diagram of the experimental set up to examine the requirements of VRC01 for inhibiting viral infection at 48 hours. (B) The bNAb did not inhibit viral capture at the 30-minute time point (black bar). Addition of VRC01 after infection resulted in only a modest inhibition (red bar). Presence of VRC01 during the infection process and also during the 48-hour time point resulted in greater than 90% inhibition (blue bar). Interestingly, VRC01 was not required during the 48-hour infection time for inhibition if present during the capture phase (gray bar). (C) Flow diagram of the experimental set up to examine the time frame for the capture and inhibition by bNAbs. (D) Primary BaLwas added to A3R5.7 cells (time 0) and VRC01 was added at 1, 2, or 5 minutes, washed, trypsin treated, and cells were incubated in media for 48 hours in the absence of VRC01. The ability of VRC01 to inhibit viral capture and subsequent infection measured at the 48-hour time point progressively decreased (60%, 50%, 20%) and was dependent on the time of addition of the bNAb. The data shown are mean ± SEM (*P<0.01, **P<0.001) of at least 2 independent experiments performed in triplicate.
CD4 Receptor blocking experiments
A3R5.7 cells were washed with ice cold PBS before being resuspended in 10% goat sera. Anti-CD4 antibody was added to the cells in goat sera at a final concentration of 20 μg/mL and incubated at 4°C for 15 minutes. The cells were then washed, resuspended in media, and infected with purified primary BaL, BaL.ec1 pseudovirus, BaL IMC, CM235 pseudovirus, 40061.LucR and C6980V0C72 IMCs as described above in the capture assay experiments. For infections that occurred for 48 hours, the samples were processed as described above for the 48-hour infection assay. For details see Supplemental Information.
Timing of addition of bNAb after cell and virus contact
A3R5.7 cells were placed in a 37°C heat block in Eppendorf tubes. The virus was added to the cells (time 0) followed by the addition of VRC01 at 1, 2, or 5 minutes. The cells and virus were incubated for a total time of 5 minutes before they were processed as described above, washed with ice cold PBS, and then trypsin treated. After the trypsin treatment, the cells were resuspended in media and incubated for 48 hours at 37°C. The cells were then harvested, washed and RNA extracted from the cell pellets. For details, see Figure 5C and Supplemental Information.
Real-time qRT-PCR and qPCR
RNA was extracted from the infected cell pellets using the RNeasy Mini Kit (Qiagen). Viral RNA present in the supernatant was extracted using the QIAamp Viral RNA Mini Kit (Qiagen). DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen). The two-step real-time RT-PCR assay was performed with a Viia7 (Applied Biosystems) using the Taqman RNA-to-Ct kit (Applied Biosystems). DNA detection qPCR assay was performed using the Taqman Universal Master Mix II (Applied Biosystems), Taqman primers, and probes as detailed in the Supplemental Information. Assay acceptability was contingent on the R2 value for the HIV-1 and cell linear regressions were >0.95.
Results
A quantitative assay to examine virus capture and replication of primary HIV-1 in host cells
We utilized purified primary HIV-1 propagated in PBMCs (Jobe et al., 2012) and the human T lymphoblastoid cell line, A3R5.7, to quantify virus capture. This T cell line expresses CD4, CXCR4, CCR5, and the integrin homing receptor α4β7, in copy numbers similar to that observed on human PBMCs (McLinden et al., 2013). Viral infection with this cell line has been shown to be efficient, productive, and envelope dependent (Marechal et al., 1998).
The amount of virus was quantified by measuring viral gag RNA or long-terminal-repeat (LTR) RNA copies present inside the cells by quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR) (Fig 1). We determined the number of viral RNA copies or the number of virus particles associated with 1 million cells after infection for 5 minutes based on the TCID50 and p24 concentration (Fig 2A, B). Due to the high sensitivity of qRT-PCR, we performed infection in the absence of reagents used to enhance viral infection, such as DEAE Dextran and polybrene. Moreover, this would be closer to natural infection. Based on these data, we decided to use an amount of virus (∼1 ng of p24 = 1×107 viral particles) that infected less than 1% of the cell population such that the amount of viral RNA quantified from these cells was within the linear range. Furthermore, these conditions more closely represented the low multiplicity of infection (MOI) and efficiency of infection observed based on the estimated peract HIV transmission risk (Patel et al., 2014). In order to eliminate a majority of the virus particles that might be loosely bound to the cell surface, the cells were treated with trypsin, as has been described in previous studies (Marechal et al., 1998; Wang et al., 2007). A3R5.7 cells are sensitive to trypsin treatment and were treated under gentle conditions (0.05% trypsin, 5 min, 25°C). The remaining viral genomes associated with the cells thus represent captured, and presumably internalized, viruses that are trypsin-resistant (Fig. 1).
Fig 1. Illustration of examination of virus capture at early time points of infection.

Cells were incubated with HIV-1 for varying time periods at 37°C to allow for binding of the virus. Cells were then washed and trypsin treated to remove surface bound virus. Subsequently the cells were lysed, RNA isolated, and then analyzed for gag or LTR RNA copies by qRT-PCR.
Fig 2. Primary HIV-1 is captured by A3R5.7 cells within 5 minutes and is replication competent.
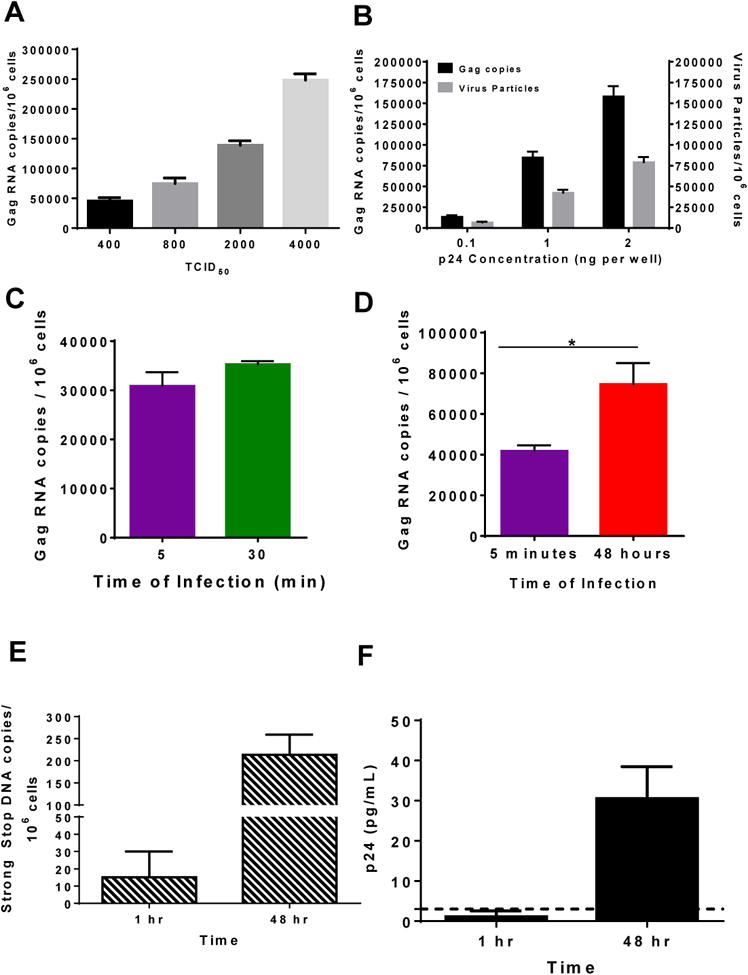
(A, B) Concentration dependent capture of HIV-1 with A3R5 cells. A3R5.7 cells were infected with different amounts of purified primary BaL for 5 minutes at 37° C based on the TCID50 (A) or p24 concentration (B). The amount of gag RNA copies/106 cells (black bar) as well as the equivalent amount of virus particles (gray bar) in (B) was determined assuming that 2 RNA copies are present inside one virus particle. (C) Cells were infected for 5 (purple) or 30 minutes (green) with purified primary BaL. Thirty minutes of infection did not significantly increase the number of viral RNA copies associated with the cells. (D) A3R5.7 cells were infected for 5 minutes (purple), washed, trypsin treated, resuspended in media, and cultured for 48 hours, after which the cells were processed as described above. There was a significant increase in the amount of gag RNA present inside the cells at 48 hours (red) indicating that the virus that was captured by the cells in 5 minutes was replication competent. (E) DNA was extracted and the number of gag DNA copies was determined. Viral DNA was detected at the 1 hour time point with a significant increase in the number of gag DNA copies at 48 hours, demonstrating that the virus particles that entered the cells were replication competent. (F) Supernatants were harvested and p24 concentrations were determined using a p24 ELISA kit (ABL). The data shown are mean ± SEM (*P < 0.01) of at least 3 independent experiments performed in triplicate.
A series of control experiments were performed to determine that the HIV-1 gag RNA detected was due to virus capture and entry into A3R5.7 cells and not due to nonspecific transfer of free RNA present in the purified HIV-1 preparations. Incubation of A3R5.7 cells with purified BaL along with the exogenous addition of 10 million gag copies did not significantly increase the number of RNA copies observed inside the cells (Fig S1A). To rule out any nonspecific uptake of the virus, we utilized a purified IMC lacking the env protein (delta env) as another control. Despite the presence of 48 million viral RNA copies in the delta env IMC (see inset in Fig S1B), there was no viral RNA associated with the cells after 30 minutes of infection (Fig S1B). These results demonstrated that the HIV-1 gag RNA copies quantified by qRT-PCR was due to specific envelope-dependent viral capture but not due to nonspecific uptake of free RNA or virus.
When A3R5.7 cells were infected with purified subtype B primary BaL for 5 minutes, ∼30,000 copies of gag RNA copies/106 cells were detected, representing the number of cell-associated viral genomes that are trypsin resistant. Increasing the time of infection to 30 minutes only slightly increased the number of gag RNA copies (Fig. 2C). Thus, 5 minutes was sufficient for HIV-1 capture by these T cells. The lack of increase in the amount of gag RNA copies observed after a longer time of infection indicates that there is a saturating amount of virus that can be captured by the cells, as well as that the virus that is captured is specific. If non-specific virus uptake was to occur, an increase in gag RNA copies after a longer time of infection would have been observed. Importantly, the virus that was captured was replication competent as shown by i) a 1.8-fold increase in viral RNA copy number after 48 hours compared to the 5-minute infection (Fig 2D), ii) the appearance of strong stop viral DNA reverse transcripts (Fig 2E) and iii) the release of progeny virus into the culture supernatant as determined by p24 ELISA (Fig 2F), after the subsequent 48-hour incubation.
bNAbs Inhibit Viral Replication of Primary Viruses
Having established a quantitative assay to measure the capture of primary HIV-1 at early time points, we utilized this method to examine the ability of various bNAbs to inhibit HIV-1 infection of A3R5.7 cells with R5 and X4 tropic viruses BaL, CAM0002BBY, and 92UG029 (subtypes B, CRF01_AG, and A respectively). The virus was pre-incubated with the bNAb for 15 minutes at 37° C prior to infection of A3R5.7 cells (Fig 3A). The amount of bNAbs used was 2.5, 5, or 10 μg/mL, which equates to 1.5×1012, 3×1012, or 6×1012 molecules of antibody. Since the amount of virus used was 0.5-1×107 viral particles and each particle contains ∼7-14 envelope protein trimers (Chertova et al., 2002; Zhu et al., 2006), the amount of mAb used was in great excess compared to the number of trimers present on the virus. The same procedure as above was used except that fresh medium containing the bNAb was added to the infected cells after trypsin-treatment during the 48-hour incubation (see Fig 3A for details). The percentage inhibition by the bNAb was determined by comparing the viral RNA copies associated with the cells in the presence of bNAbs to that in their absence. Synagis, a mAb against Respiratory Syncytia Virus (RSV), and T20, an HIV-1 fusion inhibitor were used as negative and positive controls, respectively.
Fig 3. Broadly neutralizing antibodies (bNAbs) inhibit viral infection but not viral capture of primary virus by A3R5.7 cells.
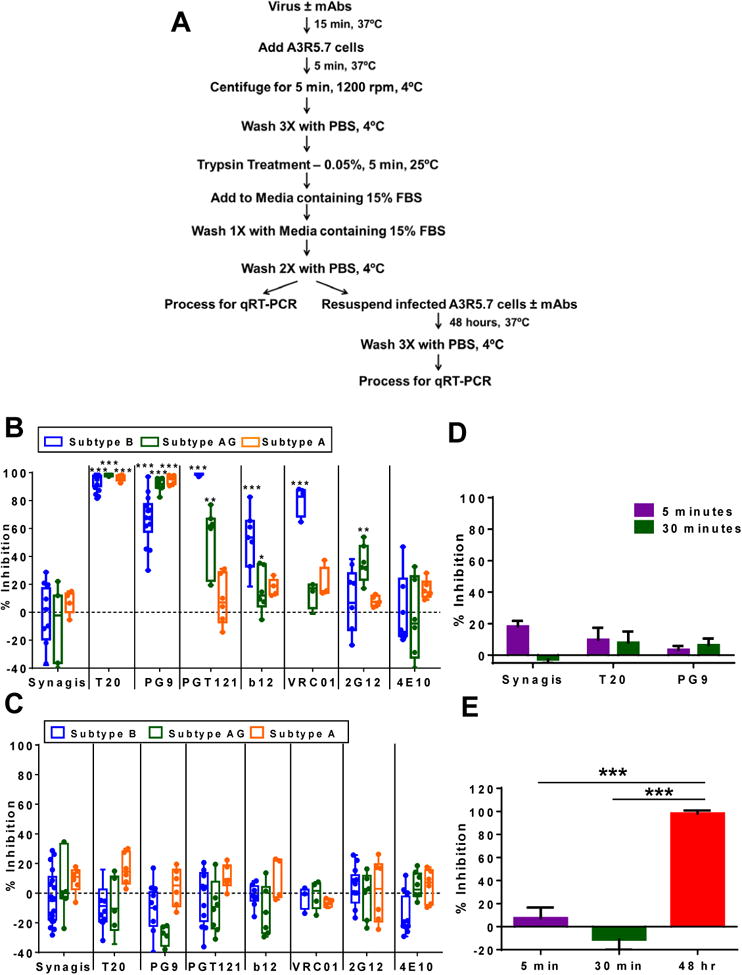
(A) Scheme used to examine bNAbs during viral capture and infection. (B) Inhibition of viral infection at 48 hours was observed for bNAbs with different primary virus isolates. Subtype B BaL (blue) was most susceptible to PG9, PGT121, VRC01, and b12; subtype CRF02_AG CAM0002BBY (green) was inhibited by PG9 and PGT121; subtype A 92UG029, (orange) was inhibited only by PG9. Almost 100% inhibition was observed with T20. (C) None of the bNAbs as well as T20 inhibited viral capture at the 5-minute time point. (D) Increasing the amount of time for viral capture from 5 (purple) to 30 (green) minutes did not affect the inhibition of T20 or PG9. (E) A3R5.7 cells were infected with BaL in the presence of b12, 4E10, 2G12, PG9 and PGT121 (2.5 μg/mL each) for 5 (purple) or 30 (green) minutes, or for 5 minutes followed by a culture period of 48 hours (red). While viral capture was not inhibited, the combination of bNAbs inhibited viral infection to >95% at the 48- hour time point. The data are shown as box-whisker plots (the box extends from the 25th to 75th percentiles, the whiskers contain the min to max showing all points) or as bar graphs. The data shown are mean ± SEM (*P<0.01, **P<0.001, ***P<0.0001) of at least 3 independent experiments performed in triplicate.
After 48 hours of infection, no significant inhibition was observed with Synagis, whereas >90% inhibition was observed with T20 against all 3 subtypes tested (Fig 3B). The bNAbs directed against different regions of the env protein structure; the V1V2 domain (PG9), V3 loop (PGT121), CD4 binding site (VRC01, b12), glycans (2G12), or MPER (4E10), inhibited the different subtypes to varying degrees (Fig 3B). PG9 potently inhibited all 3 viruses (70% to 90%), whereas PGT121, VRC01, and b12 inhibited BaL (99%, 85%, and 52%, respectively), and to a lesser extent CAM0002BBY (53% to 16%) and 92UG029 (18% to 9%). The glycan antibody 2G12 and the MPER region antibody 4E10 showed minimal inhibition against all the viruses tested (Fig 3B). The inhibition observed with PGT121, PG9 and T20 based on the viral RNA copies present inside the cells was mirrored in the gag RNA copies present in the supernatant, indicating that viral production was greatly diminished as represented by the % inhibition (Fig S2A). Overall, these data obtained at the 48-hour time point are in agreement with the previously published work with bNAbs using either luminescence or measurement of p24 as the readout (Chuang et al., 2013; Falkowska et al., 2014; Georgiev et al., 2013; Huang et al., 2014; Huang et al., 2012; Liao et al., 2013; Walker et al., 2011; Walker et al., 2009; Wu et al., 2010).
bNAbs Do Not Inhibit Capture of Primary Viruses
We next analyzed the ability of bNAbs to prevent virus capture at the 5-minute time point under the same conditions. Surprisingly, and in contrast to the 48-hour time point, neither the bNAbs nor the T20 inhibited viral capture by the A3R5.7 cells. The lack of inhibition of capture was observed with all the viruses examined and was independent of the subtype (Fig 3C) or the infection time (Fig. 3D). Particularly informative is the lack of inhibition by VRC01, which, due to its high affinity interaction with the CD4 binding site should have blocked virus capture by the CD4 receptor, the first step of the virus entry pathway. The lack of capture inhibition at the 5 minute time point was not due to limiting amounts of the antibody since concentrations of even 10 μg/mL of VRC01 showed no significant inhibition when compared to the control Ab (Fig S2B). At the same concentration, VRC01 inhibited BaL by 98% at the 48-hour time point (Fig S2C).
We examined several other antibodies, including the newer class of more potent antibodies such as 3BNC117 (Shingai et al., 2013) and CAP256-VRC26 (Doria-Rose et al., 2014), at 10 μg/mL concentration. 3BNC117 was highly effective at inhibiting viral replication of subtype B primary virus BaL at 48 hours (>90% inhibition) but it was unable to inhibit viral capture at the early time points. Similar results were observed with the CAP256-VRC26 mAb with subtype C primary virus, TZBD9/11 (Supplementary Table S1).
It has been previously demonstrated that neutralization potency is enhanced when a combination of bNAbs is used (Kong et al., 2015). Therefore, we utilized a cocktail of five potent bNAbs (b12, 4E10, 2G12, PG9 and PGT121, 2.5 μg/mL each) to determine if this could block viral capture during early time points. The bNAb cocktail showed no significant inhibition (<10%) of capture of the primary virus BaL by A3R5.7 cells at both 5 and 30 minutes of infection (Fig 3E). In contrast, >95% inhibition was observed at 48 hours of infection (Fig 3E).
bNAbs Inhibit Capture of Pseudoviruses and Infectious Molecular Clones
To determine if purification of the primary virus somehow selected a particular class of HIV-1 particles or virions with altered envelope structures, we performed the 5-minute capture/infection experiments with both unpurified and purified viruses from the same batch of primary virus grown in PBMCs. No significant differences were observed; neither the unpurified nor the purified BaL viruses were inhibited by PG9 at the 5-minute time point (Fig 4A).
Fig 4. PG9 and anti-CD4 mAbs inhibit capture of pseudoviruses and infectious molecular clones with A3R5.7 cells.
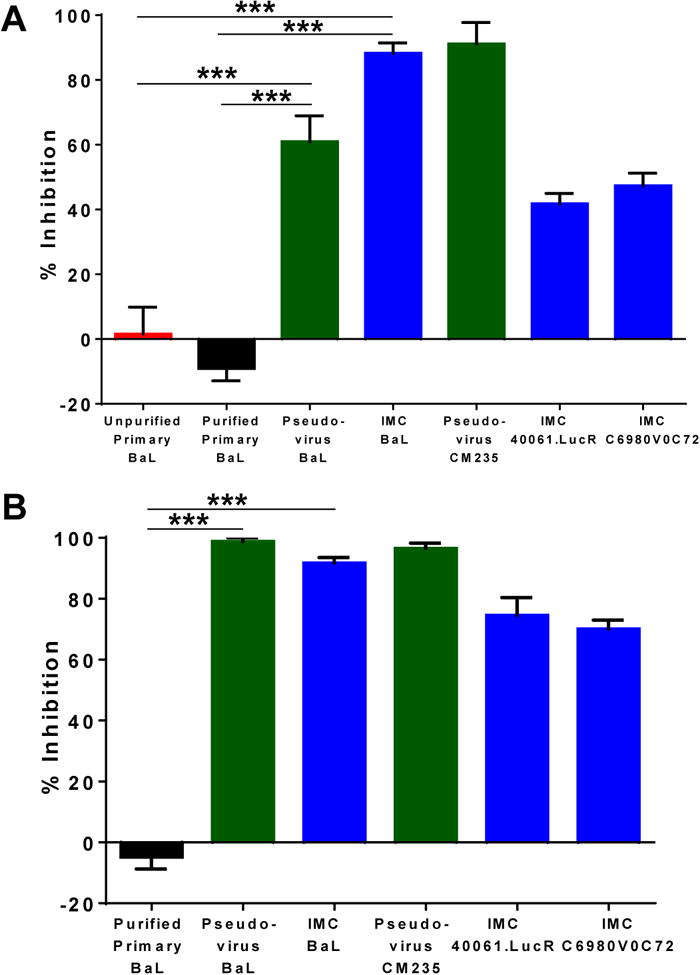
(A) A3R5.7 cells were infected with unpurified and purified primary BaL as well as the equivalent pseudovirus, BaL.ec1, CM235 Pseudovirus, and IMCs 40061.LucR and C6980V0C72 in the presence of PG9 for 5 minutes. PG9 inhibited the capture of the pseudoviruses (60-85%) and IMCs (40-85%) but not primary BaL. (B) The same viruses were used to infect A3R5.7 cells in the presence of the anti-CD4 mAb (SK3) for 5 minutes. The mAb did not inhibit the capture of the primary virus, but did inhibit the capture of the pseudoviruses and IMCs. The data shown are mean ± SEM (***P<0.0001) of at least 3 independent experiments performed in triplicate.
We then examined the ability of PG9 to inhibit the capture of the pseudovirus, BaL.ec1 and BaL IMC. Pseudoviruses and IMCs produced from the 293T cell line have been the viruses of choice in the current TZM-bl neutralization assays. Strikingly, in contrast to the primary BaL, the capture of pseudovirus BaL.ec1 and the BaL IMC was greatly inhibited, up to 74% and 88%, respectively (Fig 4A). Similar results were also observed with subtype AE pseudovirus (CM235), the transmitted/founder subtype AE IMC (40061.LucR), and subtype C IMC (C6980V0C72) (Fig. 4A). These results demonstrate that the pseudoviruses and IMCs produced in 293T cells exhibit a distinct capture phenotype from the primary viruses produced in PBMCs.
To further investigate this distinction, we utilized the anti-CD4 mAb SK3 to block HIV-1 capture through CD4, its primary receptor. It was expected that blocking this interaction would block capture of primary viruses as well as pseudoviruses and IMCs. However, the data mirrored that obtained with the bNAbs. Blocking of the CD4 receptor with excess SK3 did not inhibit the capture of the primary BaL (Fig 4B). In contrast, SK3 greatly inhibited the capture of both the pseudovirus and the IMCs (70-100%) (Fig 4B). It is important to note that although the anti-CD4 receptor antibody did not block the capture of primary BaL, later processes of viral infection and replication were greatly inhibited (75-88%), at the 48 hour time point and similar results were obtained with both unpurified and purified BaL (Fig S3A). This indicates that although the primary virus may not utilize CD4 for its initial interactions with the T cell, it does require CD4 for later processes including fusion and entry. We also examined the effect of soluble CD4 on the capture of primary and IMC BaL by A3R5.7 cells. Even at a concentration of 5 μg/mL, the protein was unable to inhibit the capture of primary BaL, but was able to inhibit the capture of the BaL IMC (Fig S3B). These results demonstrate that the capture of pseudoviruses and IMCs by A3R5.7 cells required CD4-interaction, while the capture of primary viruses was independent of CD4 interaction.
Early Exposure of Primary Virus to bNAbs is Essential for Inhibition of Viral Replication
To evaluate whether the bNAbs need to be present throughout the 48-hour incubation period to inhibit virus replication, or if there is a critical time period for the bNAbs to be effective, we performed a series of experiments utilizing three bNAbs, VRC01, PG9, and PGT121 by adding them individually at different time points during the capture and/or infection process. Primary BaL was pre-incubated with media or with the mAb for 15 minutes followed by the addition to A3R5.7 cells for 30 minutes for the capture/infection process. Cells from each of the two conditions were then washed, trypsin treated, and either analyzed for gag RNA (30 min time point), or were resuspended in media alone or in media containing the mAb for 48 hours and then analyzed for gag RNA (Fig 5A). The data with VRC01 is shown in Fig 5B.
The controls behaved as described in the earlier experiments (Fig. 3) in that pre-incubation of the virus with VRC01 followed by infection did not result in inhibition of virus capture at the 30-minute time point (black bar; Fig 5B). If however, in addition, VRC01 was also present throughout the 48-hour incubation period (positive control; blue bar, Fig 5B) over 90% inhibition was observed. Interestingly, when VRC01 was pre-incubated with the virus, but was absent during the 48-hour incubation (gray bar, Fig 5B), over 90% inhibition was observed, which was similar to the positive control. On the other hand, if VRC01 was not pre-incubated with the virus, but was only added during the subsequent 48-hour incubation (red bar, Fig 5B), a modest inhibition of only 30% was observed. In this latter scenario, since the mAb was added after the capture, it was not effective in completely blocking the subsequent entry process and generation of productive virus. It is also possible that the mAb inhibited cell-to-cell virus transmission, hence partially effective, in agreement with previous work (Abela et al., 2012; Malbec et al., 2013). These results demonstrate that although the bNAbs do not inhibit primary virus capture, their presence before capture was essential, and sufficient to inhibit viral replication. Similar patterns of inhibition were observed for both PG9 and PGT121 (Fig S4).
To further define the window of time for capture and inhibition by bNAbs, HIV-1 BaL was added to A3R5.7 cells at time 0, followed by the addition of VRC01 after 1, 2, or 5 minutes (Fig 5C). Cells were harvested at 5 minutes, washed, trypsin treated, resuspended in media, and then incubated for 48 hours at 37° C in the absence of VRC01. The data showed that capture occurred within one to two minutes post exposure of the virus with the cells. Addition of VRC01 one or two minutes after virus exposure allowed the escape of ∼50% of the viral genomes (Fig 5D) from inhibition by the bNAb. The escape was up to ∼80% when the bNAb was added 5 minutes after exposure of the virus to cells. These data demonstrate that primary virus is captured by the cells at a fast timescale, in as little as 1 minute, after which the virus is no longer fully susceptible to the inhibitory effects of the bNAb. This means that the bNAb must be present before the virus comes into contact with the cells during the capture phase, in order to exert its effect in the subsequent steps of virus entry. It is also important to note that when the bNAb is pre-incubated with the virus before exposure to cells at a saturating concentration, ∼7% of the captured viral genomes apparently escaped the inhibitory effect of VRC01 (Fig 5B, blue bars).
Conclusions
Induction of bNAbs by vaccination has been a major driving force in the HIV vaccine field and this is yet to be achieved. However, the basic mechanisms by which bNAbs block infection by diverse viruses are still poorly understood. Furthermore, the dynamics of initial interactions when a primary virus encounters a host cell and the effect of different bNAbs on these interactions have not been thoroughly investigated. These interactions might be important for locating and engaging with the primary CD4 receptor that then leads to membrane fusion and entry, and also for cell-to-cell transmission, an important facet of the viral transmission process. Antibodies which can interfere with these initial interactions might therefore be effective in preventing HIV-1 transmission and establishment of early reservoirs. To address these questions, we developed a quantitative assay and examined the very early time points after exposure of primary HIV-1 to host cells. Our results generated new and some unexpected insights.
First, our results demonstrated that primary HIV-1 was captured by the host cells within minutes after exposure. We define “capture” as the attachment of the virus to the host cell and subsequent conversion of the virus into a trypsin-resistant form, probably by internalization (Fig 6). The efficiency of capture was on the order of 1:100 – 1:300 viruses, based on the capture of about 30,000 viral genomes (15,000 viruses) per one million A3R5.7 cells in a typical experiment. The captured virus was replication competent as shown by the appearance of strong-stop DNA reverse transcripts in about an hour after capture, an increase in viral genome copy number and the release of progeny viruses into the culture medium during the subsequent 48 hours of incubation. Furthermore, our results demonstrated that capture was envelope dependent since neither free RNA nor the virus particles lacking the envelope showed significant capture.
Fig 6. Proposed schematic representations of virus capture.
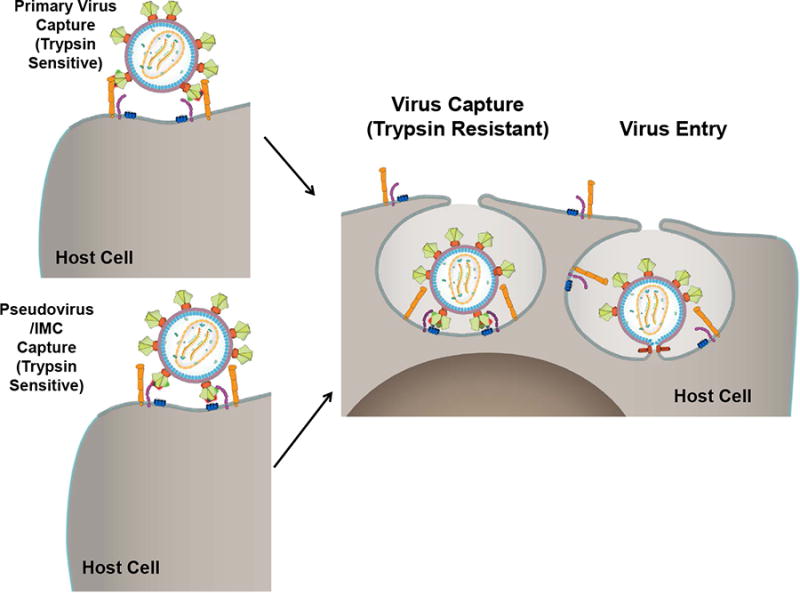
Within minutes of exposure of HIV-1, the virus attaches to the cell through either the CD4 receptor (purple) (pseudovirus/IMC) or some other accessory molecule on the cell surface (orange) (primary virus) that facilitates subsequent interaction with the CD4 receptor. At this stage, the virus is trypsin sensitive if the interactions are of low affinity or if they have not fully stabilized. The captured virus immediately undergoes internalization and the virus becomes trypsin resistant. This is followed by fusion of the viral and cell membranes and entry of the capsid into the host cell.
The bNAbs inhibited virus production at 48 hours in all of our assays. These data are in agreement with numerous previous studies using the TZM-bl and similar neutralization assays (Chuang et al., 2013; Falkowska et al., 2014; Georgiev et al., 2013; Huang et al., 2014; Huang et al., 2012; Liao et al., 2013; Walker et al., 2011; Walker et al., 2009; Wu et al., 2010). Unexpectedly, however, the potent bNAbs did not block the capture of primary virus at the 5-minute (or 30-minute) time point. The fusion inhibitor T20 also behaved in a similar fashion. This inability to inhibit primary virus capture was consistently observed with all the bNAbs tested, including the highly potent 3BNC117 and CAP256 antibodies, as well as a cocktail of five potent bNAbs. These antibodies recognize various linear and/or conformational epitopes located at different sites on the envelope protein structure. The question therefore is, if the bNAbs do not inhibit primary virus capture, how do they inhibit viral replication at the 48-hour time point? Our data demonstrated that viral capture was extremely fast and the virus became trypsin-resistant in less than 5 minutes irrespective of the presence or absence of bNAbs. Therefore, the bNAbs must have exerted their inhibitory activity subsequent to viral capture. Future studies examining the mechanism of activity on viral replication of primary viruses could further our understanding of these bNAbs.
Our studies further showed that although the bNAbs do not inhibit capture, their presence during capture is essential for subsequent inhibition of viral entry and replication. In fact, for maximum inhibition, the bNAb must be in contact with the virus before exposure to cells. If the bNAb was added just 2 minutes after the addition of virus to cells, only partial inhibition (50%) of viral replication was observed at 48 hours (Fig 5D), compared to over 90% inhibition if the bNAb was added before addition of virus to cells (Figs 3B, 5B). If however, the bNAb was added 5 minutes after exposure of the virus to the cells, then only ∼20% inhibition was observed (Fig 5D). Thus, once the virus was captured, if it was not already associated with the bNAb, the virus became refractory to the antibody and escaped inhibition even if the bNAbs were present subsequently in the external medium.
The second unexpected finding was that pseudoviruses and IMCs exhibited a distinct capture phenotype. Unlike the primary viruses, the capture of pseudoviruses and IMCs was significantly inhibited by bNAbs. This might be due to differences in the structure and composition of the envelopes of these viruses. The primary viruses produced from PBMCs through multiple rounds of viral infection consist of a pool of heterogenous quasispecies. On the other hand, pseudoviruses and IMCs produced by a single round of virus assembly from 293T cells are a relatively homogenous population. In addition, there might be differences in the glycosylation patterns of the envelopes produced by the two different cell systems. Finally, differences in the presence of other host proteins on the surface of the virion due to budding from the host cell cannot be excluded.
We hypothesize a bimodal mechanism that explains our findings and some unexpected findings reported recently (Geijtenbeek et al., 2000) (Fig 6). The ability of HIV-1 to infect different cell types, some with extremely low levels of CD4, and to remain attached to host cells lacking CD4 receptors during cell-to-cell transmission raises the question if one of the first interactions between the HIV-1 Env trimer and the cell might be a surface molecule other than the CD4 receptor. There is evidence that primary viruses could be captured through cell surface molecules such as α4β7 (Cicala et al., 2011; Guzzo et al., 2017; McKinnon et al., 2011), DC-SIGN (Geijtenbeek et al., 2000), tetraspanins (Connell and Lortat-Jacob, 2013; von Lindern et al., 2003), and Siglec-1 (Jobe et al., 2016). Capture facilitates movement of the virus on the surface and engagement with the CD4 and CCR5 receptors which then leads to internalization, possibly by encapsulation of the complex into an endocytic-type vesicle. The virus is no longer exposed on the cell surface and hence, trypsin-resistant, and undergoes fusion with the host cell membrane and delivery of nucleocapsid into the cytoplasmic compartment (Fig. 6). If the virus is in complex with the bNAb, then one or more steps of the subsequent entry and fusion process will be blocked, consequently inhibiting viral replication. On the other hand, pseudoviruses and IMCs are captured on the cell surface primarily through interaction with the CD4 receptor. Extensive and complex glycosylation of these relatively homogeneous viruses produced by 293T cells (Croset et al., 2012) may render them less prone to be captured by other attachment molecules. Indeed, differences in glycosylation patterns and varying degrees of infectivity have been noted. The Env of transmitted-founder viruses of subtypes A, C, and D have fewer N-linked glycosylation sites (Derdeyn et al., 2004; Joseph et al., 2015; Sagar et al., 2009), and a bias towards transmission of poorly glycosylated subtype C viruses in female to male transmission has been reported (Ping et al., 2013). In addition, there might be differences in the structure and conformation of the envelope trimer. The trimers of a pseudovirus or IMC might exist in a closed state, which is CD4 dependent (White et al., 2010), whereas those in a primary virus are in a dynamic state sampling different conformations. These various aspects of primary viruses versus pseudoviruses and IMCs require further investigation.
Our findings will have important implications to mechanisms of HIV-1 transmission and design of effective HIV-1 vaccines: First, our results show that rapid capture might be one of the mechanisms used by primary HIV-1 during sexual transmission. Based on our results, this would limit the window of time for the availability of the virus to antibodies to ∼2 minutes. This means that, even if potent bNAbs were present at the site of exposure, a small but significant portion of viruses might escape inhibition. These might be sufficient to establish early reservoirs (Whitney et al., 2014). Indeed, in preliminary experiments using the highly potent 3BNC117 antibody at saturating concentration (10 μg/mL) (90% inhibition), we found that at 96 hours after infection, ∼12,000 gag RNA copies were present in the cells (Fig S5A) and ∼40,000 gag RNA copies in the supernatant (Fig S5B), roughly double the numbers present at 48 hours (Figs S5A and S5B). These may represent viruses that escaped the inhibition by the mAb, some of which were replication competent and subsequently released progeny viruses into the supernatant. Recent data from in vivo challenge experiments are also consistent with our analyses. NHPs inoculated with a “sterilizing” concentration of PGT121 (2 mg/kg) (Moldt et al., 2012) followed by challenge with SHIV-SF162P3 did not show any viremia in the blood, as predicted. However, various organs including the female reproductive tract, bone marrow, and spleen were positive for viral RNA and DNA at 1, 3, and 7 days post infection. Transfer of spleen cells 7 days after challenge into naive hosts resulted in replication competent infectious virus production in the new host demonstrating that the bNAb did not completely prevent virus transmission at the mucosal port of entry (Liu et al., 2016). These unexpected in vivo findings seem to show that although bNAbs were effective in blocking transmission and viral replication, they did not do so completely, which in part might have been due to escape during the capture phase and subsequent replication.
Second, we infer from our results that it might be necessary to elicit capture-blocking antibodies in addition to bNAbs in order to completely block HIV-1 transmission. Capture-blocking antibodies, by interfering with the initial attachment of the virus with the host cell, would minimize bNAb escape. In the RV144 vaccine trial, the only trial that showed 31% vaccine efficacy, elicitation of certain V2 domain-specific binding antibodies correlated with protection (Haynes et al., 2012). This correlation of protection was also recapitulated in the macaque SHIV challenge model (Pegu et al., 2013). The V1V2 domain positioned at the tips of the HIV-1 envelope glycoprotein spikes has been implicated in binding to the cell surface molecules such as α4β7 (Cicala et al., 2011; Guzzo et al., 2017; McKinnon et al., 2011), DC-SIGN (Geijtenbeek et al., 2000), and Siglec-1 (Jobe et al., 2016). Furthermore, a primatized anti-α4β7 mAb provided significant protection against SHIV challenge in macaque models (Byrareddy et al., 2016). SIV-infected macaques on anti-retroviral therapy (ART) did not show viral rebound and maintained normal CD4+ T cell counts after ART was withdrawn if the animals were previously administered the mAb (Byrareddy et al., 2016). Thus, collectively, the evidence is compelling to suggest that antibodies that block capture may have a significant impact on blunting HIV-1 infection. A combination of capture blocking and bNAbs could therefore completely prevent HIV-1 transmission and establishment of early reservoirs.
Finally, in view of the promiscuity of HIV-1 tropism and its ability for cell-to-cell transmission, our studies highlight the importance of a quantitative, kinetically controlled, high throughput assay using primary virus isolates to complement traditional neutralization assays. It would allow evaluation of the effectiveness of antibodies to prevent initial viral capture as well as subsequent viral entry and replication, both of which might be essential in order for a prophylactic vaccine to completely block HIV-1 acquisition during sexual transmission.
Supplementary Material
Highlights.
-
-
Capture of HIV-1 by host cells occurs within minutes after exposure
-
-
Potent bNAbs do not inhibit capture of primary HIV-1
-
-
bNAbs inhibit HIV replication but need to be present before exposure to host cells
-
-
Primary viruses, pseudoviruses, and IMCs differ in their susceptibility to bNAbs
Acknowledgments
We would like to thank Drs. Mary Kearney, Claudia Cicala, Victoria Polonis, Sodsai Tovanabutra, Agnes Chenine, and Eric Freed for providing critical comments and for scientific discussion. We would like to thank Dr. John Mascola for providing the following reagents for our study: VRC01 and CAP256-VRC26.25 antibodies. We thank Dr. Wadad Alsalmi for help in preparing Figure 6. This work was supported by a grant from the NIAID, NIH (AI102725) to Venigalla B. Rao. Dr. Jiae Kim was also funded from the above grant. The work was also supported by a cooperative agreement (W81XWH-11-2-0174) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense (DoD).
Footnotes
Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Army, Department of Defense, nor the U.S. Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS pathogens. 2012;8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrareddy SN, Arthos J, Cicala C, Villinger F, Ortiz KT, Little D, Sidell N, Kane MA, Yu J, Jones JW, Santangelo PJ, Zurla C, McKinnon LR, Arnold KB, Woody CE, Walter L, Roos C, Noll A, Van Ryk D, Jelicic K, Cimbro R, Gumber S, Reid MD, Adsay V, Amancha PK, Mayne AE, Parslow TG, Fauci AS, Ansari AA. Sustained virologic control in SIV+ macaques after antiretroviral and alpha4beta7 antibody therapy. Science. 2016;354:197–202. doi: 10.1126/science.aag1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertova E, Bess JW, Jr, Crise BJ, Sowder IR, Schaden TM, Hilburn JM, Hoxie JA, Benveniste RE, Lifson JD, Henderson LE, Arthur LO. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), Is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. Journal of virology. 2002;76:5315–5325. doi: 10.1128/JVI.76.11.5315-5325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang GY, Acharya P, Schmidt SD, Yang Y, Louder MK, Zhou T, Kwon YD, Pancera M, Bailer RT, Doria-Rose NA, Nussenzweig MC, Mascola JR, Kwong PD, Georgiev IS. Residue-level prediction of HIV-1 antibody epitopes based on neutralization of diverse viral strains. Journal of virology. 2013;87:10047–10058. doi: 10.1128/JVI.00984-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicala C, Arthos J, Fauci AS. HIV-1 envelope, integrins and co-receptor use in mucosal transmission of HIV. Journal of translational medicine. 2011;9(Suppl 1):S2. doi: 10.1186/1479-5876-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell BJ, Lortat-Jacob H. Human immunodeficiency virus and heparan sulfate: from attachment to entry inhibition. Frontiers in immunology. 2013;4:385. doi: 10.3389/fimmu.2013.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset A, Delafosse L, Gaudry JP, Arod C, Glez L, Losberger C, Begue D, Krstanovic A, Robert F, Vilbois F, Chevalet L, Antonsson B. Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. Journal of biotechnology. 2012;161:336–348. doi: 10.1016/j.jbiotec.2012.06.038. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, Staupe RP, Altae-Tran HR, Bailer RT, Crooks ET, Cupo A, Druz A, Garrett NJ, Hoi KH, Kong R, Louder MK, Longo NS, McKee K, Nonyane M, O'Dell S, Roark RS, Rudicell RS, Schmidt SD, Sheward DJ, Soto C, Wibmer CK, Yang Y, Zhang Z, Program NCS, Mullikin JC, Binley JM, Sanders RW, Wilson IA, Moore JP, Ward AB, Georgiou G, Williamson C, Abdool Karim SS, Morris L, Kwong PD, Shapiro L, Mascola JR. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 2014;509:55–62. doi: 10.1038/nature13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowska E, Le KM, Ramos A, Doores KJ, Lee JH, Blattner C, Ramirez A, Derking R, van Gils MJ, Liang CH, McBride R, von Bredow B, Shivatare SS, Wu CY, Chan-Hui PY, Liu Y, Feizi T, Zwick MB, Koff WC, Seaman MS, Swiderek K, Moore JP, Evans D, Paulson JC, Wong CH, Ward AB, Wilson IA, Sanders RW, Poignard P, Burton DR. Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity. 2014;40:657–668. doi: 10.1016/j.immuni.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Georgiev IS, Doria-Rose NA, Zhou T, Kwon YD, Staupe RP, Moquin S, Chuang GY, Louder MK, Schmidt SD, Altae-Tran HR, Bailer RT, McKee K, Nason M, O'Dell S, Ofek G, Pancera M, Srivatsan S, Shapiro L, Connors M, Migueles SA, Morris L, Nishimura Y, Martin MA, Mascola JR, Kwong PD. Delineating antibody recognition in polyclonal sera from patterns of HIV-1 isolate neutralization. Science. 2013;340:751–756. doi: 10.1126/science.1233989. [DOI] [PubMed] [Google Scholar]
- Guzzo C, Ichikawa D, Park CG, Phillips D, Liu Q, Zhang P, Kwon A, Miao H, Lu J, Rehm C, Arthos J, Cicala C, Cohen MS, Fauci AS, Kehrl JH, Lusso P. Virion incorporation of integrin a4b7 facilitates HIV-1 infection and intestinal homing. Science Immunology. 2017;2 doi: 10.1126/sciimmunol.aam7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. The New England journal of medicine. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang GY, Druz A, Doria-Rose NA, Laub L, Sliepen K, van Gils MJ, de la Pena AT, Derking R, Klasse PJ, Migueles SA, Bailer RT, Alam M, Pugach P, Haynes BF, Wyatt RT, Sanders RW, Binley JM, Ward AB, Mascola JR, Kwong PD, Connors M. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature. 2014;515:138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe O, Peachman KK, Matyas GR, Asher LV, Alving CR, Rao M. An anti-phosphoinositide-specific monoclonal antibody that neutralizes HIV-1 infection of human monocyte-derived macrophages. Virology. 2012;430:110–119. doi: 10.1016/j.virol.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Jobe O, Trinh HV, Kim J, Alsalmi W, Tovanabutra S, Ehrenberg PK, Peachman KK, Gao G, Thomas R, Kim JH, Michael NL, Alving CR, Rao VB, Rao M. Effect of cytokines on Siglec-1 and HIV-1 entry in monocyte-derived macrophages: the importance of HIV-1 envelope V1V2 region. Journal of leukocyte biology. 2016;99:1089–1106. doi: 10.1189/jlb.2A0815-361R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Swanstrom R, Kashuba AD, Cohen MS. Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nature reviews. Microbiology. 2015;13:414–425. doi: 10.1038/nrmicro3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R, Louder MK, Wagh K, Bailer RT, deCamp A, Greene K, Gao H, Taft JD, Gazumyan A, Liu C, Nussenzweig MC, Korber B, Montefiori DC, Mascola JR. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. Journal of virology. 2015;89:2659–2671. doi: 10.1128/JVI.03136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, Zhu J, Shapiro L, Program NCS, Mullikin JC, Gnanakaran S, Hraber P, Wiehe K, Kelsoe G, Yang G, Xia SM, Montefiori DC, Parks R, Lloyd KE, Scearce RM, Soderberg KA, Cohen M, Kamanga G, Louder MK, Tran LM, Chen Y, Cai F, Chen S, Moquin S, Du X, Joyce MG, Srivatsan S, Zhang B, Zheng A, Shaw GM, Hahn BH, Kepler TB, Korber BT, Kwong PD, Mascola JR, Haynes BF. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Finak G, Ushey K, Seshadri C, Hawn TR, Frahm N, Scriba TJ, Mahomed H, Hanekom W, Bart PA, Pantaleo G, Tomaras GD, Rerks-Ngarm S, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Michael NL, Kim JH, Robb ML, O'Connell RJ, Karasavvas N, Gilbert P, S CDR, McElrath MJ, Gottardo R. COMPASS identifies T-cell subsets correlated with clinical outcomes. Nature biotechnology. 2015;33:610–616. doi: 10.1038/nbt.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ghneim K, Sok D, Bosche WJ, Li Y, Chipriano E, Berkemeier B, Oswald K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Jimenez J, Mondesir J, Lee B, Giglio P, Chandrashekar A, Abbink P, Colantonio A, Gittens C, Baker C, Wagner W, Lewis MG, Li W, Sekaly RP, Lifson JD, Burton DR, Barouch DH. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science. 2016;353:1045–1049. doi: 10.1126/science.aag0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malbec M, Porrot F, Rua R, Horwitz J, Klein F, Halper-Stromberg A, Scheid JF, Eden C, Mouquet H, Nussenzweig MC, Schwartz O. Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. The Journal of experimental medicine. 2013;210:2813–2821. doi: 10.1084/jem.20131244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal V, Clavel F, Heard JM, Schwartz O. Cytosolic Gag p24 as an index of productive entry of human immunodeficiency virus type 1. Journal of virology. 1998;72:2208–2212. doi: 10.1128/jvi.72.3.2208-2212.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon LR, Nyanga B, Chege D, Izulla P, Kimani M, Huibner S, Gelmon L, Block KE, Cicala C, Anzala AO, Arthos J, Kimani J, Kaul R. Characterization of a human cervical CD4+ T cell subset coexpressing multiple markers of HIV susceptibility. Journal of immunology. 2011;187:6032–6042. doi: 10.4049/jimmunol.1101836. [DOI] [PubMed] [Google Scholar]
- McLinden RJ, Labranche CC, Chenine AL, Polonis VR, Eller MA, Wieczorek L, Ochsenbauer C, Kappes JC, Perfetto S, Montefiori DC, Michael NL, Kim JH. Detection of HIV-1 neutralizing antibodies in a human CD4(+)/CXCR4(+)/CCR5(+) T-lymphoblastoid cell assay system. PloS one. 2013;8:e77756. doi: 10.1371/journal.pone.0077756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldt B, Rakasz EG, Schultz N, Chan-Hui PY, Swiderek K, Weisgrau KL, Piaskowski SM, Bergman Z, Watkins DI, Poignard P, Burton DR. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18921–18925. doi: 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, Borkowf CB, Brooks JT, Lasry A, Lansky A, Mermin J. Estimating per-act HIV transmission risk: a systematic review. Aids. 2014;28:1509–1519. doi: 10.1097/QAD.0000000000000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIV(mac251) acquisition in an immunization regimen that mimics the RV-144 Thai trial. Journal of virology. 2013;87:1708–1719. doi: 10.1128/JVI.02544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping LH, Joseph SB, Anderson JA, Abrahams MR, Salazar-Gonzalez JF, Kincer LP, Treurnicht FK, Arney L, Ojeda S, Zhang M, Keys J, Potter EL, Chu H, Moore P, Salazar MG, Iyer S, Jabara C, Kirchherr J, Mapanje C, Ngandu N, Seoighe C, Hoffman I, Gao F, Tang Y, Labranche C, Lee B, Saville A, Vermeulen M, Fiscus S, Morris L, Karim SA, Haynes BF, Shaw GM, Korber BT, Hahn BH, Cohen MS, Montefiori D, Williamson C, Swanstrom R, Study CAI, the Center for, H.I.V.A.V.I.C Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. Journal of virology. 2013;87:7218–7233. doi: 10.1128/JVI.03577-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH, Investigators MT. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. The New England journal of medicine. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- Robb ML, Rerks-Ngarm S, Nitayaphan S, Pitisuttithum P, Kaewkungwal J, Kunasol P, Khamboonruang C, Thongcharoen P, Morgan P, Benenson M, Paris RM, Chiu J, Adams E, Francis D, Gurunathan S, Tartaglia J, Gilbert P, Stablein D, Michael NL, Kim JH. Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC-HIV (vCP1521) and AIDSVAX B/E: a post-hoc analysis of the Thai phase 3 efficacy trial RV 144. The Lancet Infectious diseases. 2012;12:531–537. doi: 10.1016/S1473-3099(12)70088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar M, Laeyendecker O, Lee S, Gamiel J, Wawer MJ, Gray RH, Serwadda D, Sewankambo NK, Shepherd JC, Toma J, Huang W, Quinn TC. Selection of HIV variants with signature genotypic characteristics during heterosexual transmission. The Journal of infectious diseases. 2009;199:580–589. doi: 10.1086/596557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingai M, Nishimura Y, Klein F, Mouquet H, Donau OK, Plishka R, Buckler-White A, Seaman M, Piatak M, Jr, Lifson JD, Dimitrov DS, Nussenzweig MC, Martin MA. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature. 2013;503:277–280. doi: 10.1038/nature12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lindern JJ, Rojo D, Grovit-Ferbas K, Yeramian C, Deng C, Herbein G, Ferguson MR, Pappas TC, Decker JM, Singh A, Collman RG, O'Brien WA. Potential role for CD63 in CCR5-mediated human immunodeficiency virus type 1 infection of macrophages. Journal of virology. 2003;77:3624–3633. doi: 10.1128/JVI.77.6.3624-3633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Protocol GPI, Koff WC, Wilson IA, Burton DR, Poignard P. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Protocol GPI, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Janas AM, Olson WJ, Wu L. Functionally distinct transmission of human immunodeficiency virus type 1 mediated by immature and mature dendritic cells. Journal of virology. 2007;81:8933–8943. doi: 10.1128/JVI.00878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, Bartesaghi A, Borgnia MJ, Meyerson JR, de la Cruz MJ, Bess JW, Nandwani R, Hoxie JA, Lifson JD, Milne JL, Subramaniam S. Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS pathogens. 2010;6:e1001249. doi: 10.1371/journal.ppat.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014;512:74–77. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.