

Key Points
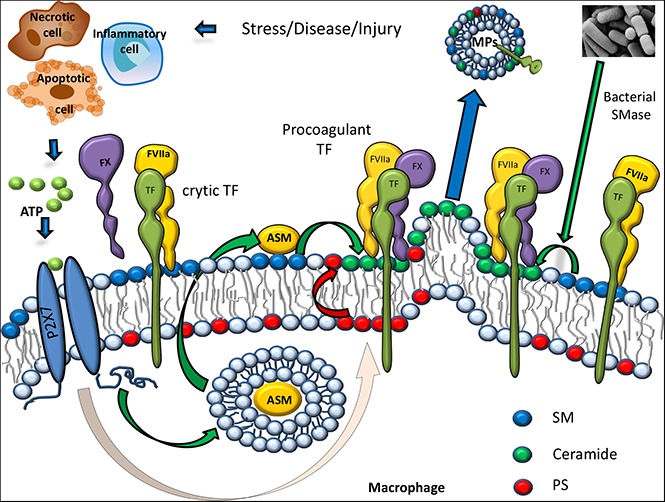
SM in the outer leaflet of the plasma membrane is responsible for TF encryption.
ATP-induced activation of A-SMase leads to hydrolysis of SM in the outer leaflet, which consequently activates TF and releases TF+ MVs.
Abstract
A majority of tissue factor (TF) on cell surfaces exists in an encrypted state with minimal to no procoagulant activity. At present, it is unclear whether limited availability of phosphatidylserine (PS) and/or a specific membrane lipid in the outer leaflet of the plasma membrane contributes to TF encryption. Sphingomyelin (SM) is a major phospholipid in the outer leaflet, and SM metabolism is shown to be altered in many disease settings that cause thrombotic disorders. The present study is carried out to investigate the effect of SM metabolism on TF activity and TF+ microvesicles (MVs) release. In vitro studies using TF reconstituted into liposomes containing varying molar ratios of SM showed that a high molar ratio of SM in the proteoliposomes inhibits TF coagulant activity. Treatment of macrophages with sphingomyelinase (SMase) that hydrolyzes SM in the outer leaflet results in increased TF activity at the cell surface and TF+ MVs release without increasing PS externalization. Adenosine triphosphate (ATP) stimulation of macrophages that activates TF and induces MV shedding also leads to translocation of acid-sphingomyelinase (A-SMase) to the plasma membrane. ATP stimulation increases the hydrolysis of SM in the outer leaflet. Inhibition of A-SMase expression or activity not only attenuates ATP-induced SM hydrolysis, but also inhibits ATP-induced TF decryption and TF+ MVs release. Overall, our novel findings show that SM plays a role in maintaining TF in an encrypted state in resting cells and hydrolysis of SM following cell injury removes the inhibitory effect of SM on TF activity, thus leading to TF decryption.
Visual Abstract
Introduction
Although tissue factor (TF)–mediated coagulation is essential for maintaining hemostasis, the aberrant activation of TF-mediated blood coagulation is a major determinant of thrombotic occlusions, the precipitating event in acute myocardial infarction, unstable angina, and ischemic stroke.1-5 Typically, TF is sequestered from the blood6,7 and exists in a noncoagulant (encrypted) state on hematopoietic cells and other cell types.8-10 A consensus now held in the field is that most of the TF expressed on cell surfaces is maintained in a cryptic, coagulantly inactive state and that an activation step (decryption) is essential for the transformation of cryptic TF to procoagulant active TF.9,11,12 However, molecular differences between cryptic and procoagulant TF and the mechanisms that are responsible for the conversion from one to the other form are poorly understood and often controversial.11,13 Nonetheless, most of the evidence in the literature suggests that externalization of phosphatidylserine (PS) to the outer leaflet of the plasma membrane plays a critical role in regulating TF procoagulant activity at the cell surface.9 However, other pathways, such as the thioredoxin system or thiol-disulfide exchange pathways involving protein-disulfide isomerase (PDI), may also contribute to TF activation by altering TF structure14-16 or through the modulation of PS exposure.17
Although the importance of PS and phosphatidylethanolamine in augmenting TF coagulant activity is well known based on extensive studies performed in reconstituted liposome systems,18-20 it is not entirely clear which and how phospholipids on the plasma membrane influence TF coagulant activity at the cell surface. Plasma membrane phospholipids are organized with mostly phosphatidylcholine (PC) and sphingomyelin (SM) at the outer leaflet, and the aminophospholipids (PS and phosphatidylethanolamine) mostly in the inner leaflet in quiescent cells.21 It is unknown at present whether TF on cell surfaces of native cells exists primarily in the cryptic state because of the limited availability of anionic phospholipids at the outer leaflet of plasma membrane or the existing phospholipids present in the outer leaflet play a critical role in maintaining TF in the cryptic state. In the outer leaflet of mammalian plasma membrane, SM contributes up to 50% of the total phospholipids present on the cell surface.22,23 An experiment comparing the effect of various membrane lipids on TF activity in phospholipid vesicles reconstituted with TF lacking the C-terminal cytoplasmic domain showed slightly lower TF coagulant activity when TF was reconstituted in phospholipid vesicles containing SM compared with vesicles lacking SM.18 This observation was not further pursued. It is possible that a high SM content in the outer leaflet is responsible for keeping TF in its cryptic state at the cell surface in resting cells. Although the importance of SM metabolism is well noted in the generation of bioactive lipids, cholesterol homeostasis, and many biological functions,24,25 knowledge of its effect on coagulation is very limited. SM metabolism is altered in many disease settings, including atherosclerosis, diabetes, sepsis, and cancer,26-34 the same disease settings that also induce aberrant activation of TF.4,35,36 This correlation implicates a possibility that SM metabolism may play a key role in TF encryption and decryption processes. Oxidative stress, chemotherapeutic agents, adenosine triphosphate (ATP), and proinflammatory cytokines are known to activate sphingomyelinases (SMases), which play a central role in SM metabolism in different cell types.31,37-39 Recent studies show that ATP stimulation of the P2X7 receptor decrypts TF and releases TF-bearing microvesicles (MVs).40 Interestingly, Bianco et al37 showed that ATP activation of the P2X7 receptor in glial cells results in rapid activation of acid-sphingomyelinase (A-SMase), translocating A-SMase from intracellular compartments to the outer leaflet of the plasma membrane. It is possible that ATP could activate TF in macrophages through the activation of A-SMase, which hydrolyzes SM in the outer leaflet of the plasma membrane. The present study was conducted to investigate the previous possibility.
Data presented herein show that incorporation of SM into TF proteoliposomes inhibits TF coagulant activity without affecting TF and factor VIIa (FVIIa) interaction or TF-FVIIa active site function. Hydrolysis of SM in the outer leaflet of macrophages by treating them with exogenous SMase enhances cell surface TF coagulant activity and MV shedding. ATP stimulation of macrophages is shown to induce the translocation of A-SMase from cytoplasm to the plasma membrane and the hydrolysis of SM. Inhibition of A-SMase activity by selective inhibitors or short interfering RNA (siRNA) knockdown attenuated the ATP-induced SM hydrolysis, TF decryption, and TF+ MVs shedding. Overall, data presented in the manuscript present a novel mechanism of TF encryption and identify a new mechanism by which ATP signaling activates TF at the cell surface and induces MV shedding. This mechanism could play a major role in intravascular thrombosis in disease settings.
Materials and methods
See supplemental Data for full description of materials and methods.
Human monocyte-derived macrophages (MDMs)
Human peripheral blood mononuclear cells isolated by a density gradient centrifugation were plated into 24-well plates and allowed to adhere for 2 hours in RPMI 1640 medium containing 10% fetal bovine serum. After 2 hours, nonadherent cells were removed, and the adherent monocytes were allowed to mature into MDMs by culturing them in RPMI medium containing serum and supplemented with macrophage colony-stimulating factor (5 ng/mL) for 6 days.
Preparation of TF proteoliposomes
Full-length recombinant human TF was reconstituted into liposomes using PC alone or PC with varying molar ratios of SM or other defined phospholipid compositions as described earlier41 using octyl glucopyranoside as the detergent. The molar ratio of TF:phospholipid in the reconstituted liposomes was 1:10 000.
Isolation of TF+ MVs
Cell supernatants were centrifuged first at 400g for 5 minutes to remove cell debris. Supernatants were transferred into fresh tubes and subjected to centrifugation at 21 000g for 60 minutes at 4°C to sediment MVs. MVs were resuspended in buffer B to the original volume.
Inhibition of A-SMase or PDI by siRNA
MDMs were transfected with a control transfection reagent (mock transfection), scramble siRNA (scRNA) as a control, or siRNA specific for A-SMase or PDI using Lipofectamine RNAiMax transfection reagent (ThermoFisher, Waltham, MA).
Measurement of TF and prothrombinase activities
Cell surface TF and prothrombinase activities were measured as described earlier.42,43 Similar assay protocols were used for measuring TF and prothrombinase activities in liposomes and MVs.
Measurement of SM hydrolysis by [methyl-14C]-choline chloride hydrolysis
To evaluate the hydrolysis of SM in the outer leaflet, the cells were metabolically labeled with [methyl-14C]-choline chloride (0.2 μCi/mL for 48 hours). After removing the unincorporated label and washing cells, the cells were subjected to experimental treatments. The release of the 14C-phosphorylcholine group into the supernatant medium (devoid of MVs) was measured.
Lysenin binding and immunostaining
MDMs were fixed with 4% paraformaldehyde for 1 hour at room temperature. To label SM with lysenin, nonpermeabilized cells were incubated with 0.5 µg/mL lysenin (Peptide Institute Inc., Osaka, Japan) in 2% bovine serum albumin/phosphate-buffered saline for 60 minutes. After removing the unbound lysenin and washing cells, the cells were incubated with rabbit polyclonal anti-lysenin antiserum (Peptide Institute Inc.; 200 × dilution) for 60 minutes. For staining A-SMase, permeabilized MDMs were incubated with rabbit anti-human A-SMase immunoglobulin G (IgG; 0.5 μg/mL) overnight. After removing the unbound primary antibodies and washing the cells, the cells were incubated with 4′,6-diamidino-2-phenylindole (5 μg/mL; to stain nucleus) and AF546 donkey anti-rabbit IgG (2 μg/mL) for 90 minutes before washing and mounting them on a glass slide. A similar protocol was used for TF immunostaining using goat anti-human TF IgG (10 µg/mL) as the primary antibody. To label PS, the fixed cells were incubated with AF488–annexin V for 60 minutes.
Immunofluorescence microscopy and image acquisition
The confocal images were obtained using an LSM 510 Meta confocal system (Carl Zeiss) equipped with an inverted microscope (Axio Observer Z1; Carl Zeiss). Immunostained cells were viewed using Plan-APOCHROMAT 63×/1.4 NA oil objective lens. Zen 2009 software (Carl Zeiss) was used for the image acquisition, quantification of fluorescence signals, and determining colocalization.44
Results
SM inhibits TF coagulant activity in liposomes and on cell surfaces
To investigate the potential effect of SM on TF activity, full-length TF was relipidated into phospholipid vesicles composed of varying molar concentrations of SM with the remainder of the vesicle consisting of PC. As shown in Figure 1A, the presence of 35 mol % SM in TF proteoliposome significantly inhibited TF-FVIIa activation of FX in comparison with TF relipidated in 100% PC. Increasing the SM ratio to 50 mol % in the liposome reduced TF activity markedly. Ceramide, having the similar sphingosine backbone as SM but lacking the phosphorylcholine head group, did not inhibit TF-FVIIa activation of FX (Figure 1B), indicating that the inhibitory effect of SM requires the phosphorylcholine head group. The inhibitory effect of SM on TF-FVIIa activation of FX is not because of impaired FVIIa binding to TF in SM-containing vesicles because increasing FVIIa concentration did not overcome the inhibitory effect of SM (Figure 1C). Furthermore, measurement of TF-FVIIa amidolytic activity showed that SM does not inhibit the amidolytic activity of TF-FVIIa, indicating that SM does not affect FVIIa binding to TF or TF-FVIIa-catalyzed small substrate cleavage (Figure 1D-E). These data also indicate similar levels of TF were incorporated in PC vesicles and vesicles containing SM. SM also significantly inhibited TF coagulant activity of TF reconstituted in PC/PS vesicles, but the inhibition was less pronounced compared with TF relipidated in PC alone (Figure 1F).
Figure 1.

Inhibitory effect of SM on TF coagulant activity. (A) Full-length recombinant human TF was reconstituted in liposomes with PC alone or PC with varying % molar concentrations of SM. TF coagulant activity was measured by its ability to support FVIIa activation of FX using 10 pM of relipidated TF, 1 nM FVIIa, and 175 nM of FX. FXa generation was measured in a chromogenic assay. (B) TF (1.3 nM) relipidated in PC, PC:SM (60:40 mol %), or PC:ceramide (60:40 mol %) was incubated with FVIIa (10 pM) and FX (175 nM), and the activation of FX was measured in a chromogenic assay. (C) TF (10 pM) relipidated in PC (○) or PC: SM (60:40 mol %) (●) was incubated with varying concentrations of FVIIa (1 to 100 pM FVIIa), and TF-FVIIa activation of FX was measured. (D) TF (10 nM) reconstituted in PC or PC with varying % molar concentrations of SM was incubated with FVIIa (100 nM), and TF-FVIIa amidolytic activity was measured using tissue plasminogen activator chromogenic substrate (Chromozym-tPA). (E) TF (10 nM) relipidated in PC (○) or PC: SM (60:40 mol %) (●) was incubated with varying concentrations of FVIIa (1 to 100 nM), and TF-FVIIa amidolytic activity was measured in a chromogenic assay. FVIIa amidolytic activity in the absence of TF was also measured (□). (F) TF (0.5 nM) relipidated in PC alone, PC:SM (60:40 mol %), PC:PS (94:6 mol %), or PC:PS:SM (54:6:40 mol %) was incubated with FVIIa (10 pM) and FX (175 nM), and TF-FVIIa activation of FX was measured. *P < .05; ** P < .01; ns, no statistical significance.
Next, to evaluate whether SM in the outer leaflet of the plasma membrane plays a role in regulating TF coagulant function at the cell surface, human MDMs were treated with exogenous bacterial sphingomyelinase (b-SMase) to hydrolyze SM. The hydrolysis of SM in the outer leaflet was evaluated by monitoring the release of [14C] phosphorylcholine head group from MDMs metabolically labeled with [methyl-14C]-choline chloride. b-SMase treatment increased the hydrolysis of SM in a dose-dependent manner (Figure 2A). Binding of lysenin, a protein that specifically binds SM,45 confirmed that b-SMase treatment markedly reduced SM content on the cell surface (Figure 2B). b-SMase treatment of MDMs increased cell surface TF activity of MDMs in a dose-dependent manner (Figure 2C). A fivefold increase in TF activity was observed in MDMs treated with 1 U/mL b-SMase. Analysis of TF levels on the cell surface by fluorescence-activated cell sorter (FACS) analysis using nonpermeabilized MDMs treated with a control vehicle or b-SMase (1 U/mL for 1 hour) showed no increase in TF levels on the cell surface following b-SMase treatment (Figure 2D). Quantification of TF levels on the cell surface by measuring the fluorescence intensity in the cell membrane of TF immunostained cells by confocal microscopy confirmed that b-SMase treatment did not alter TF levels on the cell surface (control, 72 ± 7; b-SMase-treated, 65 ± 10 arbitrary units, n = 30 to 60 cells). These data rule out the possibility that increase in cell surface TF activity observed in MDMs treated with b-SMase comes from a potential increase in TF levels on the cell surface. SMase treatment of MDMs also enhanced the release of TF-bearing MVs (Figure 2E). SMase treatment had no significant effect on prothrombinase activity at the cell surface and of MVs (Figure 2F-G), suggesting that SMase treatment did not increase PS availability at the cell surface. Consistent with this observation, we found no detectable increase in the binding of AF488–annexin V to b-SMase–treated macrophages compared with MDMs treated with a control vehicle (Figure 2H). Furthermore, annexin V pretreatment failed to attenuate the b-SMase–induced increased cell surface TF activity (Figure 2I). A small decrease in TF activity observed in b-SMase–treated MDMs following annexin V treatment could reflect annexin V inhibition of the basal TF activity. The same concentration of annexin V that had a minimal effect on b-SMase–induced TF activity effectively inhibited most of the cell surface prothrombinase activity in both control and b-SMase–treated cells (Figure 2J). Overall, the previous data indicate that the increased cell surface TF activity following b-SMase treatment comes primarily from decreased SM levels in the outer leaflet of the plasma membrane and not because of a potential increase in PS exposure at the cell surface.
Figure 2.

SM hydrolysis in the outer leaflet of plasma membrane increases cell surface TF activity and releases TF+MVs in macrophages. (A) Human MDMs were metabolically labeled with [methyl-14C]-choline chloride (0.2 μCi/mL) for 48 hours. The labeled cells were treated with a control vehicle or varying concentrations of b-SMase for 60 minutes, and supernatants were collected. The cell supernatants were subjected to centrifugation at 400g for 5 minutes and then 21 000g for 60 minutes to remove cell debris and pellet MVs, respectively. The supernatants devoid of cell debris and MVs were counted for the radioactivity to determine the release of [14C]-phosphorylcholine. (B) MDMs treated with a control vehicle or b-SMase (1 U/mL) for 60 minutes were fixed and incubated with lysenin (0.5 µg/mL), a specific binding protein to SM, for 1 hour. The bound lysenin was detected by using anti-lysenin antiserum followed by secondary antibodies conjugated with AF567 fluorophore. (C) MDMs were treated with varying concentrations of b-SMase for 1 hour. At the end of the treatment, cell supernatants were removed, cells were washed, and cell surface TF activity was measured by adding 10 nM FVIIa and 175 nM FX and determining the rate of FXa generation in a chromogenic assay. (D) MDMs were treated with a control vehicle or b-SMase (1 U/mL) for 1 hour. Intact, nonpermeabilized cells were labeled with control IgG (“C”) or rabbit anti-human TF IgG, followed by fluorophore-conjugated secondary antibodies. The immunostained cells were subjected to FACS analysis. As a positive control, MDMs were stimulated with 1 µg/mL lipopolysaccharides from Escherichia coli O111:B4 (LPS) for 4 hours to enhance TF antigen levels on the cell surface by inducing de novo synthesis of TF. (E) MDMs were treated with varying concentrations of b-SMase for 1 hour. At the end of the treatment, cell supernatants were removed, and MVs were isolated as described in “Materials and methods.” TF activity in MVs was measured as the rate of FXa generated by adding 10 nM FVIIa and 175 nM FX. (F) Cell surface prothrombinase activity of MDMs treated with a control vehicle or varying concentrations of b-SMase for 1 hour was determined by incubating MDMs with FVa (10 nM), FXa (1 nM), and prothrombin (1. 4 µM) and measuring the amount of thrombin generated in a chromogenic assay. (G) Prothrombinase activity in MVs harvested from MDM treated with varying concentrations of b-SMase for 1 hour. The reagent concentrations used for the assay were same as in panel F. (H) AF488–annexin V labeling of MDMs treated with a control vehicle or b-SMase (1 U/mL) for 1 hour. As a positive control, MDMs were treated with calcium ionophore (10 µM) for 10 minutes. Effect of annexin V on cell surface TF (I) and prothrombinase (J) activities. MDMs were pretreated with annexin V (400 nM) for 30 minutes and then treated with a control vehicle or b-SMase (1 U/mL) for 1 hour. *P < .05; **P < .01; ***P < .001 compared with the values obtained in the absence of b-SMase treatment.
ATP induces TF decryption and activates A-SMase in macrophages
Recent studies showed that ATP stimulation of P2X7 receptor results in TF decryption and release of TF-bearing MVs in bone derived-mouse macrophages.40,46 Bianco et al37 showed that ATP activation of the P2X7 receptor in glial cells resulted in rapid activation of A-SMase, which moves to the outer leaflet of the plasma membrane. Here, we investigated the possibility of ATP activation of P2X7 receptor decrypting cell surface TF and releasing TF+ MVs through the activation of A-SMase and hydrolysis of SM in the outer leaflet of the plasma membrane. To test this hypothesis, first, we investigated the effect of ATP stimulation of MDMs on cell surface TF activity and the release of TF+ MVs in our human macrophage model system. As shown in Figure 3A, ATP (200 µM) treatment increased cell surface TF activity of MDMs by approximately threefold to fivefold. The increase in cell surface-associated TF activity appeared to peak at 10 minutes of ATP treatment, and the increased cell surface TF activity was persistent for the entire duration of the experimental period (60 minutes). Immunofluorescence staining of TF and the subsequent quantification of TF levels by measuring fluorescence intensity signals on the cell surface showed no significant differences in TF protein levels in control and cells treated with ATP (200 µM for 15 minutes). The values are as follows: control, 71.6 ± 7.3; ATP stimulated, 78.7 ± 7.1 arbitrary units (n = 30 to 60 cells). Analysis of cell surface TF expression levels by FACS using nonpermeabilized MDMs further confirmed this finding (Figure 3B). ATP treatment also increased the release of TF+ MVs in a time-dependent fashion, progressively increasing TF activity from fivefold at 5 minutes to 20-fold at 60 minutes (Figure 3C). Measurement of cell surface prothrombinase activity showed that ATP stimulation also increased the prothrombinase activity, but the increase in prothrombinase activity is modest, 1.5-fold or less compared with control treatment (Figure 3D). It is surprising that prothrombinase activity did not increase more robustly over time with ATP treatment, particularly considering increasing PS externalization over time (see Figure 3E). This could reflect unstimulated MDMs expressing phospholipid surface cofactor activity suitable for prothrombinase, limited FVa or FXa binding sites on MDMs, or loss of FVa/FXa binding sites following ATP treatment. Immunofluorescence staining of MDMs with AF488–annexin V revealed that ATP treatment progressively increased PS exposure in a time-dependent manner (Figure 3E). It may be pertinent to note that, unlike the ATP-induced increase in cell surface TF activity that was peaked at 10 minutes, PS levels were increased progressively and continuously for the entire duration of the 60-minute treatment period. Annexin V treatment decreased the ATP-induced increased cell surface TF activity only partially (Figure 3F). In contrast, annexin V treatment completely attenuated the prothrombinase activity in both control and ATP-stimulated MDMs (Figure 3G).
Figure 3.

ATP stimulation enhances cell surface TF activity of macrophages and TF+MVs release. (A) MDMs were treated with benzoyl-ATP (BzATP) (200 µM) for varying time periods, supernatants were removed, and cells were washed once. Cell surface TF activity was measured by adding FVIIa (10 nM) and FX (175 nM) and determining the rate of FXa generation in a chromogenic assay. (B) MDMs were treated with control vehicle or BzATP (200 µM, 15 minutes). MDMs were labeled with control IgG (“C”) or rabbit anti-human TF IgG and subjected to FACS analysis. As a positive control, MDMs were stimulated with LPS for 4 hours. (C) MDMs were treated with BzATP as described in panel A, and the cell supernatants were collected. MVs were isolated, and TF activity in MVs was measured in FX activation as described in panel A. (D) MDMs were treated with BzATP as described in panel A, and prothrombinase activity on the cell surface was measured by adding FVa (10 nM), FXa (1 nM), and prothrombin (1.4 µM). (E) MDMs treated with BzATP (200 µM) for varying times were stained with AF488–annexin V and subjected to fluorescence microscopy. In control, MDMs were incubated with a control vehicle for 60 minutes, and this panel was labeled as “0” ATP. (F-G) MDMs were pretreated with annexin V (400 nM) for 30 minutes and then stimulated with a control vehicle or BzATP (200 µM) for 10 minutes. Cell surface TF (F) and prothrombinase (G) activities were measured as described previously. *P < .05; **P < .01; ***P < .001 compared with the values obtained in MDMs treated with a control vehicle or as specified by bars in the figures.
Next, we investigated whether ATP stimulation of macrophages results in the translocation of A-SMase from lysosomal compartments to the outer leaflet of the plasma membrane. MDMs treated with a control vehicle or ATP (200 µM) for 10 minutes were immunostained with A-SMase antibody. As shown in Figure 4A, A-SMase staining in MDMs treated with a control vehicle was limited primarily to the cytoplasm. Either very faint or no immunofluorescence of A-SMase staining was observed in the plasma membrane region in these cells. Following ATP stimulation, a clear signal for mobilization of A-SMase in the cytoplasm and the presence of A-SMase on the cell surface membrane were noted. These data illustrate that ATP signaling in MDMs induces the translocation of A-SMases to the cell surface from intracellular compartments (Figure 4A-B).
Figure 4.

ATP induces translocation of A-SMase to the plasma membrane in macrophages. (A) MDMs were treated with a control vehicle or BzATP (200 µM) for 10 minutes. The cells were fixed, permeabilized, and stained for A-SMase using A-SMase–specific antibodies followed by secondary antibodies conjugated with AF567 fluorescent dye. During the secondary antibody labeling, the cells were also labeled with a general cell membrane marker PHK67 green fluorescent cell linker. The immunofluorescence staining was analyzed by confocal microscopy. Arrowheads in the middle panel indicate immunostaining of A-SMase in the plasma membrane following ATP treatment. Yellow color in the right panel indicates colocalization of A-SMase with the membrane marker, illustrating mobilization of A-SMase from intracellular compartments to vesicular membranes for translocation to the other leaflet. (B) Quantification of A-SMase translocation to the plasma membrane. The A-SMase fluorescence intensity on the plasma membrane (30 cells or more) was measured using FIJI software (ImageJ2, Wayne Rasband, National Institute of Mental Health). Mann-Whitney U test was used to determine the significance between control and BzATP treatments. **P < .01.
ATP-induced A-SMase translocation increases SM hydrolysis and reduces SM content in the plasma membrane
Next, we investigated whether ATP-induced translocation of A-SMase results in increased hydrolysis of SM on the outer leaflet. To examine this, MDMs were metabolically labeled with [methyl-14C]-choline chloride, and then the labeled cells were treated with ATP for varying times. The hydrolysis of SM was evaluated by monitoring the release of [14C]-labeled phosphorylcholine head group into the supernatant medium. As shown in Figure 5A, ATP stimulation increased the hydrolysis of SM in a time-dependent manner. ATP treatment markedly reduced the binding of lysenin, a protein that specifically binds to SM, to intact macrophages (Figure 5B-C). These data indicate that ATP treatment reduces the SM content in the cell membrane. Inhibition of A-SMase by inhibitors desipramine and imipramine (Figure 5D) or silencing A-SMase expression by selective siRNA (Figure 5E) significantly reduced ATP-induced SM hydrolysis. These results indicate that ATP-induced SM hydrolysis is specific to A-SMase that was translocated to the outer leaflet following ATP stimulation.
Figure 5.

ATP-induced activation of A-SMase results in increased hydrolysis of SM on the cell surface. (A) MDMs were metabolically labeled with [methyl-14C]-choline chloride (0.2 μCi/mL) for 48 hours. The labeled cells were treated with a control vehicle (60 minutes) or BzATP (200 µM) for varying times and supernatants were collected. The cell supernatants were subjected to centrifugation at 400g for 5 minutes and then 21 000g for 60 minutes to remove cell debris and pellet MVs, respectively. The supernatants devoid of cell debris and MVs were counted for the radioactivity to determine the release of [14C]-phosphorylcholine. (B) MDMs treated with a control vehicle or BzATP (200 µM) for 10 minutes were analyzed for their ability to bind lysenin, a protein that specifically binds SM, as described in the legend to Figure 2. (C) The quantification of lysenin binding to cells. The fluorescence intensity of cells (30 cells or more) was measured using ImageJ2 software. (D) MDMs metabolically labeled with [methyl-14C]-choline chloride as described previously were treated with A-SMase inhibitors desipramine or imipramine for 1 hour and then stimulated with BzATP (200 µM) for 1 hour. The released radioactivity ([14C]-phosphorylcholine) was measured to monitor the hydrolysis of SM in the outer leaflet. (E) MDMs transfected with mock, scRNA, or siRNA specific for A-SMase were metabolically labeled with [methyl-14C]-choline chloride (0.2 μCi/mL) for 48 hours. The cells were then stimulated with a control vehicle or ATP for 60 minutes, and the release of radioactivity ([14C]-phosphorylcholine) was measured. Mann-Whitney U test was used to determine statistical significance between 2 groups. *P < .05; **P < .01; *** P < .001.
Involvement of A-SMase in ATP-induced TF decryption
To determine the role of A-SMase function in TF decryption and generation of TF+ MVs, MDMs were first treated with inhibitors of A-SMase, desipramine and imipramine, and then stimulated with ATP. As shown in Figure 6A, both desipramine and imipramine inhibited ATP-induced increased TF activity at the cell surface significantly. A-SMase inhibitors also inhibited ATP-induced TF+ MVs release (Figure 6B). To further strengthen the data on the involvement of A-SMase in ATP-induced TF decryption, A-SMase expression in MDMs was selectively inhibited by A-SMase (SMPD1) siRNA (Figure 6C) before their stimulation with ATP. No detectable increase in membrane translocation of A-SMase was found in MDMs transfected with A-SMase siRNA following ATP stimulation (Figure 6D-E). As shown in Figure 6F, the knockdown of A-SMase expression by siRNA significantly reduced the ATP-induced TF activity increase on the cell surface. A-SMase silencing also significantly impaired the ATP-induced TF+ MVs release (Figure 6G). These data provide strong evidence for the hypothesis that ATP-induced activation of A-SMase plays a critical role in TF decryption and the release of TF+ MVs. In additional studies, we investigated the effect of inhibition of A-SMase function on ATP-induced PS exposure. MDMs pretreated with specific inhibitors of A-SMase or transfected with A-SMase siRNA were stimulated with ATP, and PS exposure was evaluated by the binding of AF488–annexin V. As shown in Figure 6H, inhibition of A-SMase had no significant effect on ATP-induced PS exposure. Overall the data presented herein illustrate that ATP-induced A-SMase activation, which subsequently hydrolyzes SM and decreases the SM content in the outer leaflet, releases TF from its encryption controlled by SM. A-SMase–mediated TF activation is independent of ATP-induced PS externalization.
Figure 6.

Inhibition of A-SMase attenuates ATP-induced TF decryption and TF+MVs release in macrophages. (A-B) MDMs were treated with A-SMase inhibitors desipramine (Des) and imipramine (Imi) in SF-RPMI medium for 1 hour at 37°C. After that, MDMs were exposed to 200 μM BzATP in SF-RPMI at 37°C for 15 minutes, and cell surface TF activity (A), and MVs TF activity (B) were measured in FX activation assay using 10 nM FVIIa and 175 nM FX. (C) MDMs were subjected to mock transfection (transfection reagent alone) or transfected with scRNA or siRNA for A-SMase for 48 hours. Cell lysates were probed for A-SMase levels by immunoblot analysis. The same blot was also probed for β-actin as a loading control. (D) MDM cells transfected with a control scRNA or A-SMase siRNA were treated with a control vehicle or BzATP (200 µM) for 15 minutes. The cells were immunostained for A-SMase. (E) MDMs transfected with a mock transfection reagent, scRNA, and A-SMase siRNA were treated with a control vehicle or 200 μM BzATP for 15 minutes and immunostained for A-SMase. The fluorescence intensity of staining of A-SMase at the cell surface was quantified as described in the legend to Figure 4E. MDMs transfected with scRNA or siRNA for A-SMase were treated with a control vehicle or BzATP (200 µM) for 15 minutes, and TF activity at the cell surface (F) and MVs (G) were measured in FX activation assay. Mann-Whitney U test was used to determine the significance between control and experimental groups. ***P < .001. (H) MDMs pretreated with A-SMase inhibitors Des (10 µM) and Imi (5 µM) for 1 hour or transfected with scRNA or siRNA for A-SMase were treated with a control vehicle (−ATP) or BzATP (+ATP) for 15 minutes. The cells were stained with AF488–annexin V to evaluate PS exposure.
Role of PDI in b-SMase- and ATP-induced TF decryption and TF+ MVs release
Earlier studies suggest that PDI plays a role in ATP-induced TF decryption and TF+ MVs release in murine macrophages.40 To investigate the role of PDI in b-SMase–induced and ATP-induced TF decryption and TF+ MVs release, PDI activity was inhibited by inhibiting the expression of PDI by siRNA (Figure 7A) or using a specific inhibitor of PDI (PACMA 31). PDI silencing partly attenuated ATP-induced TF activity increase on the cell surface (Figure 7B) and TF+ MVs release (Figure 7C). In contrast to these results, PDI silencing had no significant effect on b-SMase–mediated TF decryption (Figure 7D) or TF+ MVs release (Figure 7E). Inhibition of PDI activity with PACMA 31 yielded similar results (Figure 7F-G). Analysis of SM hydrolysis showed that PDI silencing (Figure 7H) or PDI inhibitor PACMA31 (data not shown) had no effect on ATP-induced SM hydrolysis. Overall, our data indicate that ATP-induced activation of A-SMase and the resultant SM hydrolysis, TF decryption, and TF+ MVs release were independent of PDI. The observation that PDI inhibition had no effect on b-SMase–induced TF decryption and release of TF+ MVs supports the previous conclusion. However, our data also suggest that a PDI-mediated pathway may also play a role in ATP-induced TF decryption and TF+ MVs release, independent of A-SMase–mediated TF decryption and TF+ MVs release.
Figure 7.

Effect of PDI inhibition on ATP- and b-SMase-induced TF decryption, TF+MVs release, and SM hydrolysis. (A) Cell lysates of MDMs transfected with mock, scRNA, or siRNA specific for PDI were probed for PDI expression levels by western blot analysis. (B-C) MDMs transfected with mock, scRNA, or PDI siRNA were treated with a control vehicle or BzATP (200 µM) for 15 minutes. TF activity on the cell surface (B) or MVs (C) was measured in FX activation assay. (D-E) MDMs transfected with mock, scRNA, or PDI siRNA were treated with a control vehicle or b-SMase (1 U/mL) for 1 hour. Following the treatment, cell surface TF activity (D) or TF activity in MVs (E) was measured in FX activation assay. (F-G) MDMs were first treated with a control vehicle or PDI inhibitor PACMA31 (10 µM) for 1 hour, and then treated with control vehicle, BzATP (200 µM, 15 minutes), or b-SMase (1 U/mL, 1 hour). Cell surface TF activity (F) or MVs TF activity (G) was measured in FX activation assay. (H) MDMs transfected with mock, scRNA, or PDI siRNA were metabolically labeled with [methyl-14C]-choline chloride (0.2 μCi/mL) for 48 hours. Labeled MDMs were treated with a control vehicle, BzATP (200 µM, for 15 minutes), or b-SMase (1 U/mL, for 1 hour). Cell supernatants were collected and centrifuged to remove cell debris and MVs before they were counted for the radioactivity to determine the release of [14C]-phosphorylcholine as an index for SM hydrolysis.
Discussion
Encryption/decryption appears to be a universal mechanism that regulates TF activity on cell surfaces.47 At present, it is not entirely clear about the relative contribution of encryption and decryption in regulating TF activity and molecular mechanisms that regulate these processes, which may vary among cell types.9,11,48 A key question that has not been addressed so far is whether TF stays in a cryptic state on resting cells solely because of the limited availability of PS in the outer leaflet or specific membrane phospholipids that are present predominantly in the outer leaflet play an active role in maintaining TF in the cryptic state. Our present data show that SM, the predominant phospholipid present in the outer leaflet of the plasma membrane, is responsible for maintaining TF in the cryptic state on resting cells, and the hydrolysis of SM, without increasing PS exposure in the outer leaflet, enhances TF activity at the cell surface and releases procoagulant TF+ MVs. The present data connect ATP-induced activation of the P2X7 receptor that leads to TF activation and TF+ MVs release to SM metabolism triggered by ATP-induced translocation of A-SMase.
Studies performed with reconstituted TF established that the composition of lipid bilayer plays an important role in the activity of the TF-FVIIa complex, and maximal TF-FVIIa coagulant activity requires 30% or more of PS.20 However, other lipids not containing the bulky choline head group of PC or SM appear to “synergize” with PS and decrease % PS required for maximal TF-FVIIa coagulant activity.20 Our present data show that high concentrations of SM in the lipid bilayer suppress TF coagulant activity, which is consistent with an earlier preliminary observation.18 Because SM neither affects FVIIa binding to TF nor TF-FVIIa amidolytic activity, the SM effect appears to be specific for suppressing TF-FVIIa coagulant activity, which is akin to TF encryption. The choline head group of SM appears to be critical for the inhibitory effect because ceramide, which lacks the phosphorylcholine head group but contains the same backbone as SM, failed to inhibit TF activity. At present, it is unknown how SM inhibits TF activity. Apart from sharing the same hydrophilic phosphorylcholine head group, PC and SM are structurally quite different. SM has only 1 fatty acid in an amide linkage while the second hydrophobic chain is part of the sphingosine base structure.49 The interfacial part of SM contains a hydroxyl group at C-3 and a trans double bond between C-4 and C-5 (supplemental Figure 1). The interfacial hydroxyl and amide residues of the SM molecule are capable of donating and accepting hydrogen bonds.50 The strong hydrogen bonding properties characteristic of SM were shown to induce structural changes in the polar head group and interface regions.51 The sphingosine moiety and related hydrogen bonds along acyI composition of SM could influence phospholipid and protein packing and dynamic properties of the bilayer, which could affect protein function. Recent molecular modeling studies suggest that the TF ectodomain may directly interact with PS and thus modulate the TF-FVIIa conformation to be more favorable to FX activation.52 One could speculate that a direct interaction between TF and SM may keep the TF-FVIIa conformation unfavorable to FX activation.
In the mammalian plasma membrane, SM constitutes up to 50% of the total phospholipids present in the outer leaflet.22,23 The present study indicates that SM in the outer leaflet is responsible for maintaining TF in a noncoagulant/cryptic state because the hydrolysis of SM by exogenous SMase, which results in the removal of a choline group from SM, results in increased TF activity on the cell surface. The increased TF activity following SM hydrolysis appears to be unique as it is independent of PS. One of the vexing problems in elucidating the importance of various proposed mechanisms of TF decryption is that most of the activating stimuli that decrypt TF also increase PS externalization. Because PS exposure in itself leads to TF activation, it is difficult to distinguish the importance of other putative mechanisms of TF activation from that of the PS-dependent mechanism.11,53 The absence of any detectable increase in PS exposure in the outer leaflet of cells following b-SMase treatment in MDMs in the present study allows us to conclude convincingly that hydrolysis of SM in the outer leaflet leads to TF decryption, independent of PS. At present, it is unknown how SM hydrolysis or reducing SM content in the plasma membrane increases TF activity. In addition to potential mechanisms discussed previously in regard to SM’s inhibitory effect on TF coagulant activity in the reconstituted system, SM levels in the biological membrane could modulate TF activity by other modes. The plasma membrane not only has an asymmetric lipid distribution,54 but also exhibits heterogeneity in the lateral distribution of lipids. These lateral heterogeneities in the cell membranes envisaged as rafts are enriched with SM, other (glyco)sphingolipids, cholesterol, and specific membrane proteins.55-57 The tight interaction between SM and cholesterol through hydrogen bonding serves as the basis for raft formation.58 These membrane rafts are known to play a critical role in facilitating protein-protein and protein-lipid interactions and influence many cellular functions.59 Ultrastructural localization of TF in a variety of cell types showed that a fraction of TF in these cells is associated with lipid rafts/caveolae.60-63 It has been suggested that lipid raft/caveolae-associated TF represents the encrypted form, which can be activated rapidly by disruption of these structures.60,64,65 It is possible that TF decryption induced by SM hydrolysis may involve disruption of lipid rafts.
Recent studies showed that ATP stimulation of the P2X7 receptor induces TF decryption and shedding of procoagulant TF+ MVs in murine macrophages.40,66,67 TF activation and the release of MVs in murine macrophages appear to involve the production of reactive oxygen species and PDI-dependent extracellular thiol-disulfide exchange pathways.40 Bianco et al37 showed that ATP-induced MVs release from glial cells is associated with activation of A-SMase, which moves from intracellular compartments to the outer leaflet of the plasma membrane. Activation of A-SMase was found to be necessary and sufficient for MVs shedding in glial cells.37 Here, we provide convincing evidence that ATP-induced activation of TF and TF+ MVs release involve the activation of A-SMase. ATP stimulation is shown to translocate A-SMase to the outer leaflet, increase the hydrolysis of SM, and reduce SM content in the plasma membrane of macrophages. These data suggest that the A-SMase that is translocated to the outer leaflet upon ATP stimulation is capable of hydrolyzing SM in the plasma membrane. Inhibition of A-SMase expression by siRNA or inhibition of A-SMase activity with specific inhibitors attenuated not only ATP-induced SM hydrolysis but also inhibited ATP-induced TF activation and the release of TF+ MVs. These treatments had no effect on the known externalization of PS by ATP,40,68 indicating that ATP-induced A-SMase–mediated TF activation is independent of PS externalization. The present results are the first to demonstrate that SM metabolism in the plasma membrane controls TF encryption and decryption processes. Our studies identify a novel mechanism by which ATP activation of P2X7 leads to TF activation and release of TF+ MVs. However, our present study also suggests that ATP-induced PS externalization may partly contribute to TF decryption as annexin V partly inhibited ATP-induced TF activity increase on the cell surface.
Data from earlier studies, which used murine macrophages and mouse model systems, suggested that thioredoxin/thioredoxin reductase system and PDI-mediated thiol pathways play a role in ATP-induced P2X7 receptor–dependent TF activation and TF+ MVs release.40,66 Data reported in the present study support a role for PDI in ATP-induced TF decryption and TF+ MVs release. However, ATP-induced A-SMase activation, SM hydrolysis, and the subsequent TF activation and TF+ MVs release appear to be independent of PDI as PDI inhibition had no effect on ATP-induced SM hydrolysis and only partly inhibited TF activity increase on the cell surface and TF+ MVs release. The observation that PDI inhibition had no significant effect on b-SMase–induced TF decryption and TF+ MVs release is consistent with the previous concept. It is likely that ATP-induced signaling could promote TF decryption and TF+ MVs release by 2 independent mechanisms.
SM metabolism is altered in many thrombotic disease settings, including atherosclerosis, diabetes, sepsis, and cancer,26-34 raising a possibility that SM metabolism may play a key role in TF activation (see Visual Abstract). In the physiological setting, a high molar concentration of SM in the outer leaflet maintains TF in the cryptic state on resting cells by exerting its inhibitory effect. Pathophysiological stimuli, such as ATP secretion, cytokines induced by inflammation, or reactive oxygen/nitrogen species generated in oxidative stress, could activate secretory A-SMase that translocates to the outer leaflet.69 In bacterial infection, secretion or presence of SMase on the surface of certain bacteria70-72 may result in b-SMase coming in contact with the extracellular leaflet before inflammatory signals activate endogenous A-SMase in host cells. SMase contacting the outer leaflet of the plasma membrane hydrolyzes SM in the outer leaflet to ceramide and PC, releasing TF from SM’s inhibitory effect. It is also possible that SMase-produced ceramide could bind cholesterol and form ceramide-rich platforms that alter the membrane curvature and integrity.73,74 These changes in the membrane could also contribute to TF activation and MVs shedding. Clustering of ceramide into the inner leaflet following SM hydrolysis and the subsequent redistribution of ceramide within the lipid bilayer may facilitate the formation of plasma membrane protrusions75 that lead to MVs shedding. Our present observation that SMase is a key molecular effector in TF activation and prothrombotic TF+ MVs shedding constitutes as a novel finding and opens new strategies for the treatment of thrombotic diseases.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge that former fellows in the laboratory, Prosenjit Sen, Jagan Sundaram, and Hema Kothari, contributed to obtaining parts of the data shown in Figure 1 of the manuscript. The authors thank Sion Williams for proofreading the manuscript.
This work was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01-HL124055) (L.V.M.R.) and an American Heart Association Grant-in-Aid (15GRNT22620004) (U.R.P.).
Authorship
Contribution: J.W. performed experiments, summarized the data, and wrote the initial draft of the manuscript; U.R.P. contributed to designing experiments, reviewing data, and editing the manuscript; L.V.M.R. conceived and designed the study, analyzed the data, and wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, TX 75708; e-mail: vijay.rao@uthct.edu.
References
- 1.Rapaport SI, Rao LVM. The tissue factor pathway: how it has become a “prima ballerina”. Thromb Haemost. 1995;74(1):7-17. [PubMed] [Google Scholar]
- 2.Morrissey JH. Tissue factor: an enzyme cofactor and a true receptor. Thromb Haemost. 2001;86(1):66-74. [PubMed] [Google Scholar]
- 3.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(8):1687-1693. [DOI] [PubMed] [Google Scholar]
- 4.Taubman MB, Fallon JT, Schecter AD, et al. Tissue factor in the pathogenesis of atherosclerosis. Thromb Haemost. 1997;78(1):200-204. [PubMed] [Google Scholar]
- 5.Osterud B. Tissue factor: a complex biological role. Thromb Haemost. 1997;78(1):755-758. [PubMed] [Google Scholar]
- 6.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134(5):1087-1097. [PMC free article] [PubMed] [Google Scholar]
- 7.Fleck RA, Rao LVM, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59(2):421-437. [DOI] [PubMed] [Google Scholar]
- 8.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26(3):456-461. [DOI] [PubMed] [Google Scholar]
- 9.Rao LV, Pendurthi UR. Regulation of tissue factor coagulant activity on cell surfaces. J Thromb Haemost. 2012;10(11):2242-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Versteeg HH, Ruf W. Thiol pathways in the regulation of tissue factor prothrombotic activity. Curr Opin Hematol. 2011;18(5):343-348. [DOI] [PubMed] [Google Scholar]
- 11.Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost. 2014;111(4):590-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen VM, Hogg PJ. Encryption and decryption of tissue factor. J Thromb Haemost. 2013;11(suppl 1):277-284. [DOI] [PubMed] [Google Scholar]
- 13.Rao LV, Kothari H, Pendurthi UR. Tissue factor encryption and decryption: facts and controversies. Thromb Res. 2012;129(suppl 2):S13-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45(39):12020-12028. [DOI] [PubMed] [Google Scholar]
- 15.Ahamed J, Versteeg HH, Kerver M, et al. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103(38):13932-13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Wu Y, Li X, Ma X, Zhong L. Thioredoxin and thioredoxin reductase control tissue factor activity by thiol redox-dependent mechanism. J Biol Chem. 2013;288(5):3346-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116(6):993-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neuenschwander PF, Bianco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: enhancement of sensitivity to phosphatidylserine. Biochemistry. 1995;34(43):13988-13993. [DOI] [PubMed] [Google Scholar]
- 19.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007;282(9):6556-6563. [DOI] [PubMed] [Google Scholar]
- 20.Tavoosi N, Davis-Harrison RL, Pogorelov TV, et al. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286(26):23247-23253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89(4):1121-1132. [PubMed] [Google Scholar]
- 22.Quinn PJ. Plasma membrane phospholipid asymmetry. In: Quinn PJ, Kagan VE, eds. Phospholipid Metabolism in Apoptosis. Subcellular Biochemistry. Vol. 36. New York: Kluwer Academic; 2002:39-60. [DOI] [PubMed] [Google Scholar]
- 23.Adibhatla RM, Dempsy R, Hatcher JF. Integration of cytokine biology and lipid metabolism in stroke. Front Biosci. 2008;13:1250-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slotte JP. Biological functions of sphingomyelins. Prog Lipid Res. 2013;52(4):424-437. [DOI] [PubMed] [Google Scholar]
- 25.Taniguchi M, Okazaki T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration-from cell and animal models to human disorders. Biochim Biophys Acta. 2014;1841(5):692-703. [DOI] [PubMed] [Google Scholar]
- 26.Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19(11):2648-2658. [DOI] [PubMed] [Google Scholar]
- 27.Truman JP, Al Gadban MM, Smith KJ, Hammad SM. Acid sphingomyelinase in macrophage biology. Cell Mol Life Sci. 2011;68(20):3293-3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horres CR, Hannun YA. The roles of neutral sphingomyelinases in neurological pathologies. Neurochem Res. 2012;37(6):1137-1149. [DOI] [PubMed] [Google Scholar]
- 29.Milhas D, Clarke CJ, Hannun YA. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett. 2010;584(9):1887-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holthuis JC, Luberto C. Tales and mysteries of the enigmatic sphingomyelin synthase family. In: Chalfant C, Del Poeta M, eds. Sphingolipids as Signaling and Regulatory Molecules. Advances in Experimental Medicine and Biology. Vol. 688. New York: Springer; 2010;72-85. [DOI] [PubMed] [Google Scholar]
- 31.Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82(2):175-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong ML, Xie B, Beatini N, et al. Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. Proc Natl Acad Sci USA. 2000;97(15):8681-8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Górska M, Barańczuk E, Dobrzyń A. Secretory Zn2+-dependent sphingomyelinase activity in the serum of patients with type 2 diabetes is elevated. Horm Metab Res. 2003;35(8):506-507. [DOI] [PubMed] [Google Scholar]
- 34.Smith EL, Schuchman EH. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 2008;22(10):3419-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eilertsen KE, Østerud B. Tissue factor: (patho)physiology and cellular biology. Blood Coagul Fibrinolysis. 2004;15(7):521-538. [DOI] [PubMed] [Google Scholar]
- 36.Williams JC, Mackman N. Tissue factor in health and disease. Front Biosci (Elite Ed). 2012;E4:358-372. [DOI] [PubMed] [Google Scholar]
- 37.Bianco F, Perrotta C, Novellino L, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009;28(8):1043-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perrotta C, Clementi E. Biological roles of Acid and neutral sphingomyelinases and their regulation by nitric oxide. Physiology (Bethesda). 2010;25(2):64-71. [DOI] [PubMed] [Google Scholar]
- 39.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274(5294):1855-1859. [DOI] [PubMed] [Google Scholar]
- 40.Furlan-Freguia C, Marchese P, Gruber A, Ruggeri ZM, Ruf W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest. 2011;121(7):2932-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mimms LT, Zampighi G, Nozaki Y, Tanford C, Reynolds JA. Phospholipid vesicle formation and transmembrane protein incorporation using octyl glucoside. Biochemistry. 1981;20(4):833-840. [DOI] [PubMed] [Google Scholar]
- 42.Pendurthi UR, Williams JT, Rao LVM. Acidic and basic fibroblast growth factors suppress transcriptional activation of tissue factor and other inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17(5):940-946. [DOI] [PubMed] [Google Scholar]
- 43.Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110(12):3900-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kothari H, Rao LVM, Pendurthi UR. Cys186-Cys209 disulfide-mutated tissue factor does not equal cryptic tissue factor: no impairment in decryption of disulfide mutated tissue factor. Blood. 2010;116(3):502-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shogomori H, Kobayashi T. Lysenin: a sphingomyelin specific pore-forming toxin. Biochim Biophys Acta. 2008;1780(3):612-618. [DOI] [PubMed] [Google Scholar]
- 46.Baroni M, Pizzirani C, Pinotti M, et al. Stimulation of P2 (P2X7) receptors in human dendritic cells induces the release of tissue factor-bearing microparticles. FASEB J. 2007;21(8):1926-1933. [DOI] [PubMed] [Google Scholar]
- 47.Kothari H, Pendurthi UR, Rao LV. Analysis of tissue factor expression in various cell model systems: cryptic vs. active. J Thromb Haemost. 2013;11(7):1353-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang HPH, Hogg PJ. Critical importance of the cell system when studying tissue factor de-encryption. Blood. 2008;112(3):912-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barenholz Y, Thompson TE. Sphingomyelin: biophysical aspects. Chem Phys Lipids. 1999;102(1-2):29-34. [DOI] [PubMed] [Google Scholar]
- 50.Steinbauer B, Mehnert T, Beyer K. Hydration and lateral organization in phospholipid bilayers containing sphingomyelin: a 2H-NMR study. Biophys J. 2003;85(2):1013-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niemelä P, Hyvönen MT, Vattulainen I. Structure and dynamics of sphingomyelin bilayer: insight gained through systematic comparison to phosphatidylcholine. Biophys J. 2004;87(5):2976-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ke K, Yuan J, Morrissey JH. Tissue factor residues that putatively interact with membrane phospholipids. PLoS One. 2014;9(2):e88675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao LVM, Kothari H, Pendurthi UR. Tissue factor: mechanisms of decryption. Front Biosci (Elite Ed). 2012;E4:1513-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Meer G. Lipid traffic in animal cells. Annu Rev Cell Biol. 1989;5:247-275. [DOI] [PubMed] [Google Scholar]
- 55.Brown D. Structure and function of membrane rafts. Int J Med Microbiol. 2001;291(6-7):433-437. [DOI] [PubMed] [Google Scholar]
- 56.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569-572. [DOI] [PubMed] [Google Scholar]
- 57.Jacobson K, Sheets ED, Simson R. Revisiting the fluid mosaic model of membranes. Science. 1995;268(5216):1441-1442. [DOI] [PubMed] [Google Scholar]
- 58.Cremesti AE, Goni FM, Kolesnick R. Role of sphingomyelinase and ceramide in modulating rafts: do biophysical properties determine biologic outcome? FEBS Lett. 2002;531(1):47-53. [DOI] [PubMed] [Google Scholar]
- 59.Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7(6):456-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mulder AB, Smit JW, Bom VJJ, et al. Association of smooth muscle cell tissue factor with caveolae. Blood. 1996;88(4):1306-1313. [PubMed] [Google Scholar]
- 61.Mulder AB, Smit JW, Bom VJJ, Blom NR, Halie MR, van der Meer J. Association of endothelial tissue factor and thrombomodulin with caveolae. Blood. 1996;88(9):3667-3670. [PubMed] [Google Scholar]
- 62.Mandal SK, Pendurthi UR, Rao LVM. Cellular localization and trafficking of tissue factor. Blood. 2006;107(12):4746-4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Awasthi V, Mandal SK, Papanna V, Rao LV, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol. 2007;27(6):1447-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sevinsky JR, Rao LVM, Ruf W. Ligand-induced protease receptor translocation into caveolae: a mechanism for regulating cell surface proteolysis of the tissue factor-dependent coagulation pathway. J Cell Biol. 1996;133(2):293-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dietzen DJ, Page KL, Tetzloff TA. Lipid rafts are necessary for tonic inhibition of cellular tissue factor procoagulant activity. Blood. 2004;103(8):3038-3044. [DOI] [PubMed] [Google Scholar]
- 66.Rothmeier AS, Marchese P, Petrich BG, et al. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Invest. 2015;125(4):1471-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee R, Williams JC, Mackman N. P2X7 regulation of macrophage tissue factor activity and microparticle generation. J Thromb Haemost. 2012;10(9):1965-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moore SF, MacKenzie AB. Murine macrophage P2X7 receptors support rapid prothrombotic responses. Cell Signal. 2007;19(4):855-866. [DOI] [PubMed] [Google Scholar]
- 69.Kornhuber J, Rhein C, Müller CP, Mühle C. Secretory sphingomyelinase in health and disease. Biol Chem. 2015;396(6-7):707-736. [DOI] [PubMed] [Google Scholar]
- 70.Lin YL, Liu JS, Chen KT, Chen CT, Chan EC. Identification of neutral and acidic sphingomyelinases in Helicobacter pylori. FEBS Lett. 1998;423(2):249-253. [DOI] [PubMed] [Google Scholar]
- 71.Oda M, Hashimoto M, Takahashi M, et al. Role of sphingomyelinase in infectious diseases caused by Bacillus cereus. PLoS One. 2012;7(6):e38054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peñate Medina TA, Korhonen JT, Lahesmaa R, Puolakkainen M, Peñate Medina O, Kinnunen PKJ. Identification of sphingomyelinase on the surface of Chlamydia pneumoniae: possible role in the entry into its host cells. Interdiscip Perspect Infect Dis. 2014;2014:412827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goñi FM, Alonso A. Membrane fusion induced by phospholipase C and sphingomyelinases. Biosci Rep. 2000;20(6):443-463. [DOI] [PubMed] [Google Scholar]
- 74.López-Montero I, Monroy F, Vélez M, Devaux PF. Ceramide: from lateral segregation to mechanical stress. Biochim Biophys Acta. 2010;1798(7):1348-1356. [DOI] [PubMed] [Google Scholar]
- 75.Goñi FM, Alonso A. Biophysics of sphingolipids I. Membrane properties of sphingosine, ceramides and other simple sphingolipids. Biochim Biophys Acta. 2006;1758(12):1902-1921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.