

Abstract
Two new bispecific T cell engaging (BiTE) molecules with specificity for NKG2D ligands were developed and functionally characterized. One, huNKG2D-OKT3, was derived from the extracellular portion of the human NKG2D receptor fused to a CD3ε binding single-chain variable fragment (scFv), known as OKT3. NKG2D has multiple ligands, including MICA, which are expressed by a variety of malignant cells. A second molecule, B2-OKT3, was created in the tandem scFv BiTE format that targets MICA on tumor cells and CD3ε on human T cells. Both BiTEs specifically activated T cells to kill human tumor cell lines. Cytotoxicity by B2-OKT3 but not huNKG2D-OKT3 is blocked by soluble rMICA. The huNKG2D-OKT3 induced greater T cell cytokine production in comparison to B2-OKT3. No T cell pre-treatment was required for IFNγ production upon co-culture of B2-OKT3 or huNKG2D-OKT3 with T cells and target cells. The effector memory T cell compartment was the primary source of IFNγ, and culture of T cells and these BiTEs with plate-bound rMICA showed ligand density dependent production of IFNγ from both CD4+ and CD8+ T cells. There was two-fold more IFNγ produced per CD8+ T cell and five-fold greater percentage of CD8+ T cells producing IFNγ compared to CD4+ T cells. In addition, both BiTEs elicited significant anti-tumor responses against human metastatic melanoma tumor samples using autologous or healthy donor T cells. These data demonstrate the robust anti-tumor activity of these NKG2D ligand binding bispecific proteins and support their further development for clinical use.
Keywords: BiTE, MICA, interferon-gamma, immunotherapy, cytotoxicity
Introduction
Immunotherapies modulating T cell activity have proven to be effective in cancer treatment (1). T cells contribute to tumor immune-surveillance, but as tumors progress they develop immune escape mechanisms that limit T cell efficacy (2). Immunotherapies that alleviate T cell exhaustion, such as checkpoint blockade, or redirect T cells against tumors can shift the balance back in favor of the immune system (3). One type of bispecific protein that has met with success for cancer treatment is the bispecific T cell engager (BiTE) (4, 5). A BiTE is traditionally composed of the heavy and light chains of two single chain variable fragments (scFv) joined together through flexible serine-glycine linkers in a format known as a tandem scFv (6). Typically, one arm engages T cells through binding to CD3ε while the other scFv binds a ligand on a tumor cell. This dual binding acts as a bridge resulting in the formation of a synapse between the T cell and tumor cell. The binding of both sites activates the T cell resulting in robust cytokine production and perforin/granzyme mediated killing of the tumor cell. This mechanism of action gives BiTEs several properties including efficacy at very low doses, no requirement for T cell co-stimulation, and T cell activation only upon engagement of both receptors (7, 8).
While BiTEs have great promise for the treatment of cancer, the selection of tumor-specific targets can be challenging. Tumor antigens can be specific to a cell lineage, such as CD19 on B cells, or found on the surface of many different types of cancer. The first BiTE approved by the Food and Drug Administration (FDA) was blinatumomab, an anti-CD19 × anti-CD3 BiTE, for use in patients with Philadelphia chromosome-negative relapsed or refractory precursor B-cell acute lymphoblastic leukemia (ALL) (9). There are a number of other BiTEs in development that target carcinoembryonic antigen (CEA), CD33, and epithelial cell adhesion molecule (EpCAM), which are found on several types of tumors (10-12).
In this study, two BiTEs that target NKG2D ligands were functionally characterized and evaluated. NKG2D ligands are often expressed on human tumor cells, and it has been estimated that more than 90% of human tumors may express at least one NKG2D ligand (13). There are two families of NKG2D ligands in humans, MHC class I chain-related proteins A (MICA) and B (MICB) as well as UL16 binding proteins (ULBP1 - 6) (13). The reported affinity of human NKG2D to its ligands is in the range of 1μM (13-15). The expression of NKG2D ligands is controlled at the transcriptional, translational and post-translational levels. Under conditions of cell stress, such as infection or transformation, these ligands can become expressed on the cell surface (16, 17). In healthy tissues, mRNA and intracellular protein expression has been observed, however, miRNA and proteasome degradation prevents cell surface expression of these ligands (13, 18-20). The wide expression of NKG2D ligands on many different tumor types, with limited expression on normal tissues, makes them attractive targets for immunotherapy. To redirect T cells to MICA+ tumors, the B2-OKT3 BiTE was created using a tandem scFv BiTE format – targeting MICA on tumor cells and CD3ε on human T cells. The huNKG2D-OKT3 BiTE is composed of the extracellular portion of the human NKG2D receptor linked to a CD3ε binding scFv. Both human NKG2D and MICA-specific BiTEs are able to target a wide variety of tumors because MICA has broad expression on multiple tumor types, including cervical, colon, breast, lung, ovarian, prostate, renal cell, and pancreatic carcinomas (16, 17, 21). MICA is also expressed on 75% of cutaneous melanoma samples and 50% of metastatic melanoma (13, 22). In this study, two structurally different BiTEs were evaluated for anti-tumor activity, antigen dependency, and ability to recognize human melanoma tumors.
Materials and Methods
Cell culture and Cell lines
K652 (human myeloid leukemia cell line) was obtained from the American Type Culture Collection (Rockville, MD). Breast cancer cell lines MCF-7 and T47D were provided by Dr. James Direnzo (Dartmouth Medical School, Lebanon, NH). Pancreatic cancer cell line Panc-1 was provided by Dr. Murray Korc (Dartmouth Medical School). Prostate cancer cell line PC-3 was provided by Dr. Marc Ernstoff (Dartmouth Medical School). The B16F10 cell line was from Richard Barth (Dartmouth Medical School). These cell lines were obtained between 2002 and 2006. All human cell lines were authenticated by American Type Culture Collection between August – November 2016 by short tandem repeat (STR) analysis. Cell lines are tested routinely for mycoplasma by PCR around the time of use in experiments and retested periodically. The most recent mycoplasma testing was conducted July 2015 (MCF7 and PC3), November 2015 (T47D), September 2016 (Panc1) and March 2017 (B16F10 and K562). After thawing, cells were passaged 2-5 times before use in experiments. K562, K562-Luc and PC3 were cultured in complete RPMI - RPMI 1640 (GE Hyclone Laboratories, SH30027.01) supplemented with 10% heat-inactivated FBS (GE Hyclone Laboratories, SH30910.03), 100 U/ml penicillin, 100 μg/ml streptomycin (GE Hyclone Laboratories, SV30010), 1 mM sodium pyruvate (Corning Cellgro, 25-000-Cl), 10 mM HEPES (Corning Cellgro, 25-060-Cl), 0.1 mM MEM non-essential amino acids (Corning Cellgro, 25-025-Cl) and 50 μM 2-mercaptoethanol (Sigma, M6250-100ML). B16F10, B16F10-MICA, Panc1, Panc1-Luc, T47D, and MCF7 cell lines were cultured in complete Dulbecco's Modified Eagle Medium with a high glucose concentration (4.5 g/L) (DMEM, GE Hyclone Laboratories, SH30022.01) containing the same supplements. K562-Luc and Panc1-Luc were generated by retrovirus transduction of their parental cell line K562 or Panc1 cells, respectively, using retroviral vectors containing a firefly luciferase gene. B16F10-MICA was generated by retrovirus transduction of the parental cell line B16F10 using a retroviral vector containing the MICA gene, as previously described (23).
Healthy donor peripheral blood mononuclear cells (PBMCs) were isolated from cell cones, obtained from plateletpheresis and provided anonymously by the Dartmouth-Hitchcock Medical Center Blood Donor Center. Cells were separated using Lymphocyte Separation Media (Corning, 25-072-CV) density gradient centrifugation, then stored frozen in 90% FBS, 10% dimethyl sulfoxide (DMSO, Fisher Scientific, D128-500) in liquid nitrogen. For experiments, PBMCs were thawed, washed with HBSS (GE Hyclone Laboratories, SH30031.02) then cultured in complete RPMI. Activated T cells were obtained by culturing PBMCs at 106 cells/ml for three days in the presence of Ultra-LEAF purified anti-human CD3 antibody clone OKT3 at 40 ng/ml (Biolegend, 317304) and human IL-2 at 50 U/ml (NCI Biological Resource Division). After three days, cells were washed twice with HBSS then cultured at 106 cells/mL in complete RPMI with fresh human IL-2 at 50 U/ml for an additional five days before use.
Generation of BiTE Constructs
Anti-human CD3ε scFv was constructed by fusing VH [aa 20–138] and VL [aa 23–128] region of an anti-human CD3ε hybridoma OKT3 with a (G4S)3 linker. HuNKG2D-OKT3 was created by joining the anti-CD3ε scFv to the extracellular domain of human NKG2D [aa 78-216] with a second (G4S)3 linker. B2-OKT3 was generated by fusing an scFv that recognized MICA with the OKT3 scFv via a (G4S)3 linker. The scFv recognizing MICA was selected through a yeast scFv library screen (24). Purified, recombinant MICA allele *001, mutated at C273S to remove an unpaired cysteine, was used for the yeast library screen after biotinylation with EZ-Link™ Sulfo-NHS-LC-Biotin (Pierce, 21435). The scFv selected in the screen was amplified from the library vector. Each construct contains a histidine tag (6×His) in order to facilitate protein purification. The fusion genes were then cloned into a CMV promoter based expression vector (Clontech). All PCR reactions were performed using a high-fidelity DNA polymerase Phusion™ (New England Biolabs, M0530S). All oligonucleotides were synthesized by either Integrated DNA Technologies (Coralville, IA) or Sigma-Genosys (Woodsland, TX).
Purification of BiTEs
BiTEs were purified either using the Expi293™ expression system or from transiently transfected suspension-adapted Freestyle ™ 293-F cells (ThermoFisher Scientific, A14635, R79007). Manufacturer's instructions were followed for BiTEs purified using the Expi293™ expression system. For BiTEs purified from 293-F cells the following protocol was used: 293-F cells cultured in Gibco FreeStyle 293™ Expression Medium (ThermoFisher, 12338018) were transfected with BiTE specific DNA constructs by 40 kD PEI (Polysciences, Inc, 24765). Transfection was done by gently mixing 293-F cells with DNA and PEI at a final concentration of 106 cells/ml, 1.4 μg/ml DNA, and 25 μg/ml PEI. DNA and PEI were mixed separately in Opti-MEM ™ (ThermoFisher Scientific, 31985070), and incubated at room temperature for 10 minutes. The DNA-PEI solution was then added dropwise to cell cultures. The culture was maintained in 37°C, 8% CO2 with shaking at 100 rpm for five days, and cell-free culture supernatant was harvested and filtered using 0.45 μm filter units and two glass 1.0 micron fiber prefilters (EMD Millipore Corporation, SCHVU05RE, AP1507500). The supernatant was mixed with 4× nickel column binding buffer (1.2 M NaCl, 200 mM NaH2PO4, 80 mM, imidazole, pH=7.4) and loaded onto a 1 ml HisTrap HP column (GE Healthcare, 29-0510-21) using an AKTA Start purification system (GE Healthcare, 29-0220-94). The column was washed with 10 column volumes of nickel column binding buffer (300 mM NaCl, 50 mM NaH2PO4, 20 mM imidazole, pH=7.4). HuNKG2D-OKT3 elution was performed with 20 column volumes of elution buffer (300 mM NaCl, 50 mM NaH2PO4, 480 mM imidazole pH= 7.4) with a linear gradient of imidazole from 20 to 480 mM. B2-OKT3 and the negative control BiTE (TZ47-OKT3) were eluted using a stepwise gradient, 186 mM of imidazole for three column volumes and 388mM of imidazole for 17 column volumes using the elution buffer (300 mM NaCl, 50 mM NaH2PO4, 480 mM imidazole pH= 7.4). Eluted fractions were collected and examined by SDS-PAGE gel stained with SYPRO Orange (ThermoFisher Scientific, S6650). Individual fractions containing BiTE protein were combined and buffer exchanged into 1× PBS (Corning Cellgro, 21-040-CV) using two 5 ml HiTrap desalting columns (GE Healthcare, 29-0486-84) connected in tandem using the AKTA Start purification system. Concentration of BiTE protein was quantified absorbance using a Nanodrop at 280 nm. Extinction coefficients (0.1% Abs) used to calculate concentrations were: 2.293 for huNKG2D-OKT3 and 2.137 for B2-OKT3. Average BiTE yield was 1,020 μg/L for B2-OKT3 and 1,005 μg/L for huNKG2D-OKT3 using the Expi293™ expression system. To produce soluble rMICA, the extracellular domain of MICA*001 allele was cloned into a pCMV based protein expression vector containing a 6×His Tag. The QuickChange II XL kit (Agilent Technologies, 200521) was used to convert the free cysteine to a serine (C273S) by mutation at nucleotide site 818 (G818C). 293-F cells were transfected with the rMICA construct and rMICA was eluted stepwise with 180mM, followed by 400mM imidazole.
ELISA
Effector cells (105 cells/well) and tumor cells were plated in 96-well flat bottom plates at an effector to target cell ratio (E:T) ratio of 4:1. When human melanoma tumor cells were used as target cells, the tumor cells were plated and allowed to culture overnight (37°C), then washed twice with HBSS before effector cells or BiTEs were added. For assays where rMICA was used as a target, rMICA was diluted in 1× PBS, plated at 100 μl/well, incubated overnight at 4°C, and washed three times before effector cells or BiTEs were added. After all cells and reagents were combined, plates were cultured (37°C) and cell-free medium was collected after 24 - 48 hours, as indicated. IFNγ was quantified by ELISA, according to the manufacturer's instructions (Biolegend, 430106).
Luciferase Cytotoxicity Assay
Cultured human T cells were plated in 96-well plates with luciferase expressing tumor cells at specified E:T ratios in triplicate in Costar round-bottom non-tissue culture treated white polystyrene plates (Corning Incorporated, 3789) and BiTE was added. After overnight incubation, 50 μl of 200 μg/ml luciferin (Gold Biotechnology, 115144-35-9), diluted in complete RPMI was added to each well. The plates were incubated for 30 min at 37°C. Each well was read for 2s on a Centro LB 960 luminometer (Berthold Technologies, Oak Ridge, TN). Controls included T cells alone, luciferase expressing cell lines alone, and T cells co-cultured with tumor cell lines without BiTE. For the rMICA blocking experiments, rMICA was added in five-fold dilutions at a maximum of 5000ng/ml immediately after the addition of the BiTEs.
Human melanoma samples
Human melanoma tumor cell samples were derived from patient metastatic tumor specimens at the time of surgical resection. Tumor cells were isolated by mechanical and enzymatic digestion and cryopreserved in 90% heat inactivated Human AB serum (Gemini Bioproducts, 100-512) with 10% DMSO freeze media at -140°C. Samples were thawed and allowed to recover overnight before use in the described assays. All patients signed informed consent that was approved by the Committee for the Protection of Human Subjects at Dartmouth Hitchcock Medical Center, to allow use of their melanoma tumor tissue for research purposes.
Flow Cytometry
Cell surface stains were performed by washing cells twice with FACS buffer, 1% FBS in 1× PBS, followed by incubation with human Cohn's fraction IgG (Sigma-Aldrich, G4386) for 10 minutes to reduce non-specific binding. Samples were then washed with FACS buffer twice before incubation in the appropriate mixture of antibodies for 30 min at 4°C. After incubation, the cells were washed three times and resuspended in FACS buffer before samples were acquired on a C6 Accuri flow cytometer (BD Biosciences, Ann Arbor, MI). Expression of MICA/B on cell lines was quantified using PE-conjugated anti-MICA/MICB antibody clone 6D4 (Biolegend, 320906). T cell subsets were identified using anti-CD3 clone OKT3 conjugated with PerCP 710 (eBiosciences, 46-0037-42), PE conjugated anti-CD45RO clone UCHL1 (Biolegend, 304206), and FITC-conjugated anti-CCR7 clone 150503 (R&D Systems, FAB197F). When intracellular staining was performed, cell surface stains were completed before cells were fixed and permeabilized using a Fix/Perm staining kit (eBioscience, 00-5523-00) followed by intracellular staining for IFNγ with APC anti-human IFNγ clone B27, (Biolegend, 506510) or isotype control APC msIgG1 clone MOPC-21 (Biolegend, 400120) per the manufacturer's instructions. All data acquisition and analysis was done using the Accuri flow cytometry software (BD Biosciences, Ann Arbor, MI).
Bio-Layer Interferometry
The huNKG2D-OKT3 and B2-OKT3 binding to rMICA was measured by Bio-Layer Interferometry using an Octet RED96 System (ForteBio, Menlo Park, CA). For each experiment, two sets of eight streptavidin (SA) biosensor tips (ForteBio, 18-5019) were used, one set for measurement and one set as a reference to subtract background binding. The biosensors were equilibrated with 1× PBS containing 0.5% BSA and 0.1% Tween®20 (Sigma Aldrich, P1379 and Fisher Scientific BP1600-100) for 100s. The measurement biosensors were loaded with 0.1mg/ml biotin-labeled rMICA protein for 200s, dipped into buffer to reach baseline for 100s, followed by association of the first analyte for 600s, immediately followed by association in either the same BiTE, an anti-MICA/B antibody clone 6D4 (Biolegend, 320906), or the test BiTE for 600s. Fortebio Data Analysis 7.0 software (ForteBio, Menlo Park, CA) was used to analyze the data and generate response curves.
Statistics
A paired t-test was used to evaluate significance for cytotoxicity assays (Figure 1A,1B, 2C, 2D), changes in IFNγ Mean Fluorescence Intensity (MFI) following BiTE treatment (Figure 4C), and with human melanoma samples (Figure 5A-C). For comparison in T cell subset distribution, significance was determined by one-way ANOVA with Newman-Keuls multiple comparisons analysis (Figure 3B). Two-way ANOVA was used to determine significance of IFNγ dose response and between treatment groups (Figure 4A). A p value < 0.05 was considered statistically significant.
Figure 1. BiTEs specifically activate T cells and induce killing of MICA+ cell lines.
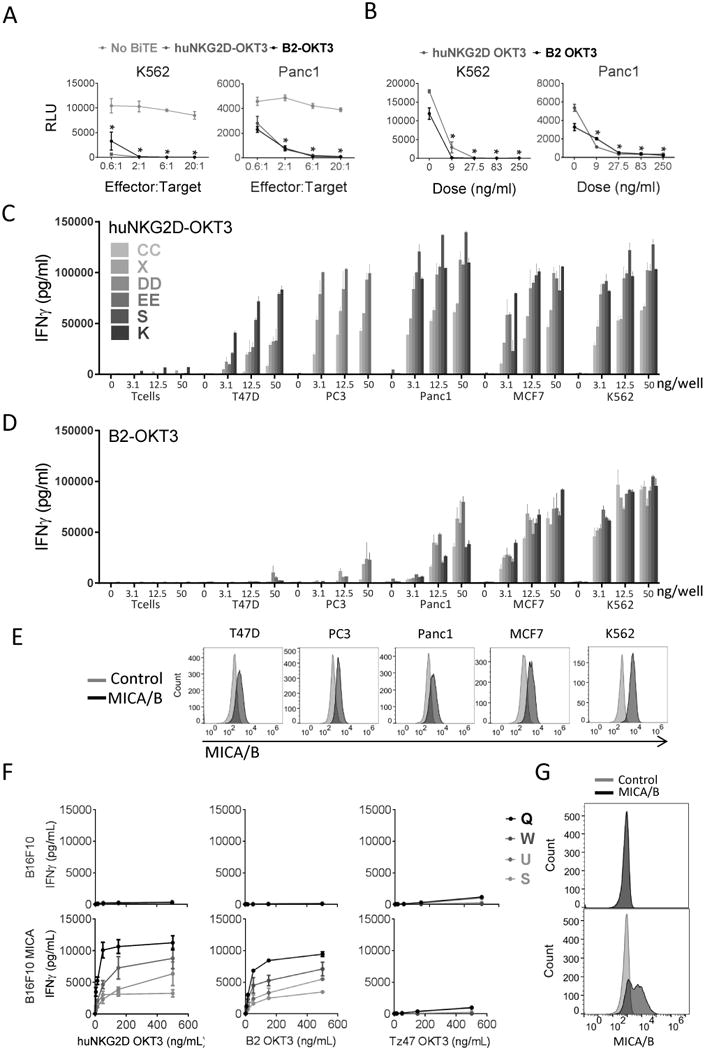
(A) Cytotoxicity of human T cells induced by BiTEs against MICA+ human cell lines over a range of effector: target (E:T) cell ratios. Cultured T cells from four healthy donors were incubated with K562-Luc (left) or Panc1-Luc (right) tumor cells with 250 ng/mL B2-OKT3 or huNKG2D-OKT3, or no BiTE in triplicate wells. Relative light units (RLU) were measured after overnight culture. Representative plots with averaged RLU are shown. Error bars reflect the standard deviation. *p≤0.05 compared to no BiTE treatment. (B) Dose response of cytotoxicity induced by BiTE. Cultured T cells from two healthy donors were incubated with K562-Luc (left) or Panc1-Luc (right) cells at an E:T of 10:1 with the indicated concentrations of B2-OKT3 or huNKG2D-OKT3 in triplicate wells. Representative plots with averaged RLU are shown. Error bars reflect the standard deviation. *p≤0.05 compared to 0ng/ml BiTE. T cell IFNγ production when cultured with huNKG2D-OKT3 (C) or B2-OKT3 (D) against different tumor cell lines. Cultured T cells from four to six healthy donors (K, S, X, CC, DD, or EE) were incubated with human tumor cell lines, K562, PC3, Panc1, MCF7, or T47D. IFNγ was measured in cell-free medium after 24 hours. Error bars reflect the standard deviation. (E) Representative flow plots showing expression of MICA/B (black) compared to unstained controls (grey) for the cell lines used in C and D. (F) T cell IFNγ production (from healthy donors Q, S, W, U) when cultured with huNKG2D-OKT3 (left) B2-OKT3 (middle) or a TZ47-OKT3 (control BiTE; right) against either MICA- cell line B16F10 or B16F10 cells transduced to express MICA (B16F10-MICA). (G) Flow plots showing expression of MICA/B (black) compared to unstained controls (grey) for B16F10 and B16F10-MICA.
Figure 4. Antigen density directly impacts T cell activation by B2-OKT3 and huNKG2D-OKT3 BiTEs.
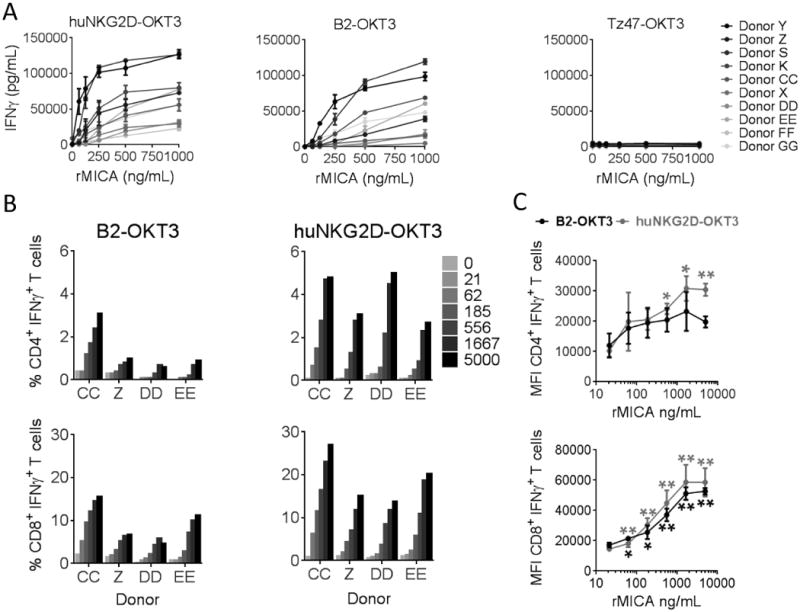
A) Activated T cells from ten donors were incubated with immobilized rMICA at the indicated concentrations and either huNKG2D-OKT3 (left), B2-OKT3 (center) or negative control BiTE (right) at 250 ng/mL for 24 hours. IFNγ from cell free supernatant was measured by ELISA. (B) Cells from 2-4 donors were treated as in A, but after a six hour incubation in the presence of brefeldin A, cells were stained for expression of CD4, CD8 and intracellular IFNγ, and data were acquired by flow cytometry. Percentage of activated cells as indicated by IFNγ+ staining are shown in B, while MFI for each T cell subset over the range of antigen densities tested is shown in C. MFI were compared to 0 ng/ml group by paired t test (* p ≤ 0.05, **p ≤ 0.01). Error bars represent SD of triplicates in (A) and between donors in (C). Blood donors are designated by letters (K, S, P, X, Y, Z, CC, DD, EE, FF, GG).
Figure 5. huNKG2D-OKT3 and B2-OKT3 BiTEs are able to activate healthy donor T cells, PBMCs and donor matched T cells against human melanoma tumor cell samples.
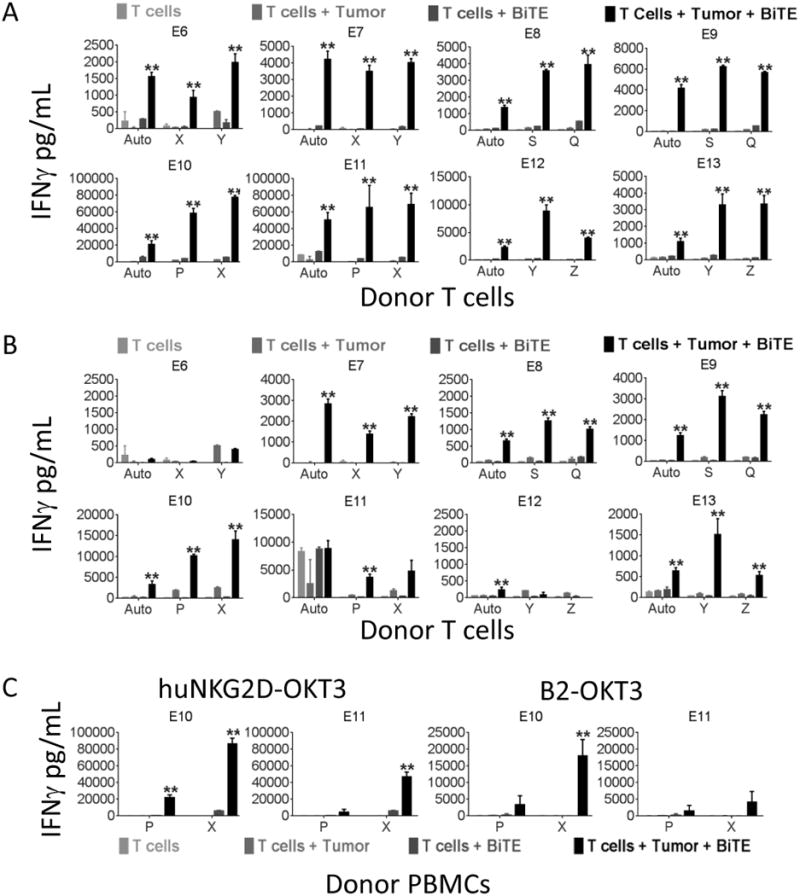
T cells from two different healthy donors and autologous T cells were plated with each melanoma tumor cell sample at an E:T ratio of 4:1 and A) huNKG2D-OKT3 or B) B2-OKT3 BiTE at 250 ng/ml and incubated for 24 hours. IFNγ production was measured by ELISA. C) Unstimulated PBMCs from two healthy donors were incubated with each primary melanoma sample as in A and B, but IFNγ production in the supernatants was measured after a 48 hour incubation. Treatment with huNKG2D-OKT3 is shown on the left, and B2-OKT3 on the right. Each sample was run in triplicate and data are shown + SD of triplicates. A * indicates p ≤ 0.05 or ** indicates p ≤ 0.01 compared to all control groups. Auto = autologous; tumors are designated E8-13, blood donors are designated by letters (S, Q, P, X, Y, Z).
Figure 3. After BiTE treatment, IFNγ producing T cells are enriched for effector memory T cells.
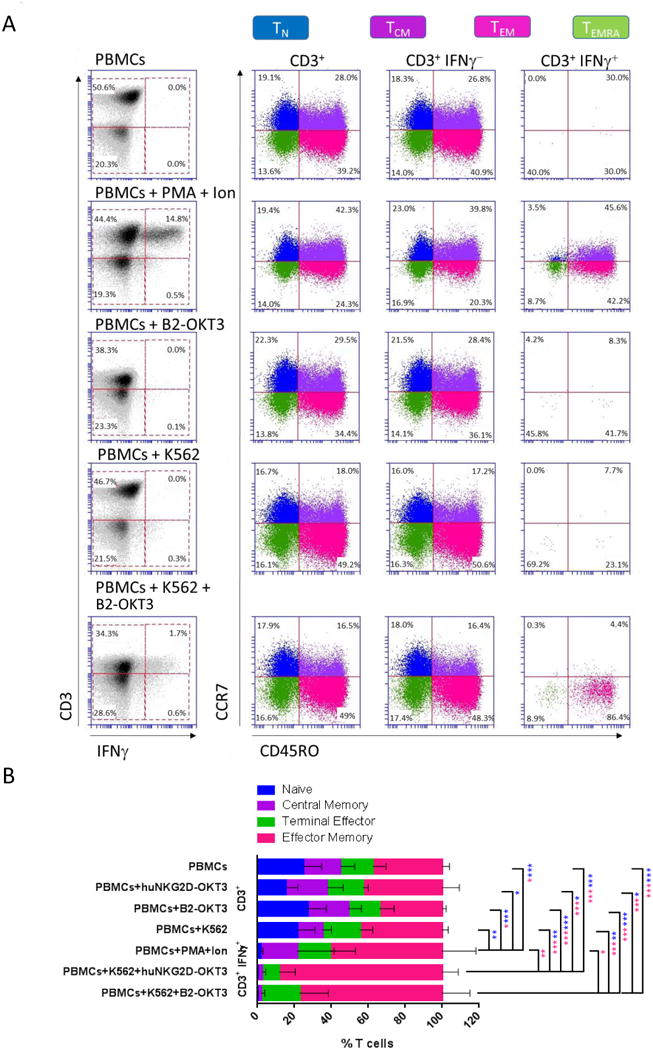
A) Gating strategy used to identify functionally active T cell subsets based on IFNγ+ intracellular staining. In the indicated treatment groups, PBMCs and target K562 cells were cultured at an E:T ratio of 4:1, and BiTE was used at a dose of 250 ng/ml. Treatment with PMA and Ionomycin (Ion) was used as a positive control. Samples were stained with CD3, CD45RO, CCR7 and IFNγ. T cell subsets were designated as follows: Naïve (TN) CD3+CD45RO-CCR7+, Central memory (TCM) CD3+CD45RO+CCR7+, Effector memory (TEM) CD3+CD45RO+CCR7-, and Terminal Effector T cells (TEMRA) CD3+CD45RO-CCR7-. Data are representative of experiments performed with four different PBMC Donors. B) Distribution of cells in each T cell subset in the treatment groups described in (A). Data are shown for mean value of the four donors + standard deviation. Statistical significance determined by one-way ANOVA with Newman-Keuls multiple comparisons analysis. The color of the asterisk indicates the population that is statistically different. * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Results
HuNKG2D-OKT3 and B2-OKT3 BiTEs specifically activate T cells and induce killing of MICA ligand-expressing tumor cells
The extracellular portion of the human NKG2D receptor was fused to a human CD3ε binding scFv derived from the antibody clone OKT3, in order to develop a BiTE that binds all NKG2D ligands. Thus, this huNKG2D-OKT3 protein is able to bind CD3ε on human T cells with one arm and the multiple tumor antigens recognized through NKG2D (MICA/B and ULBP1 - 6) with the other arm. A second BiTE in the traditional tandem format was also developed. This BiTE, B2-OKT3, is specific for the NKG2D ligand MICA. Out of four MICA binding scFvs isolated by screening a human scFv library against soluble rMICA, B2 was selected for further analysis due to its robust activity.
To determine whether huNKG2D-OKT3 and B2-OKT3 redirected primary T cells to specifically recognize and kill ligand expressing tumor cells, effectors (T cells) and tumor cells were cultured together at different ratios of effectors to target cells (E:T) with 250 ng/ml of either BiTE (Figure 1A). Cytotoxicity was measured using a luciferase based assay, where reduction of luciferase signal corresponded to a decrease in viable target cells. At all E:T ratios tested, significant decreases in tumor survival were observed in cultures with K562-Luc (chronic myelogenous leukemia) or Panc1-Luc (pancreatic cancer) tumor cells with BiTE compared to no BiTE treatment. B2-OKT3 and huNKG2D-OKT3 were almost equivalent in their ability to elicit cytotoxicity from T cells under these conditions. To determine the dose dependency of the cytotoxic activity, each BiTE was co-cultured with T cells and tumor cells at BiTE concentrations from 0 to 250 ng/ml (Figure 1B). Cytotoxicity was observed at all doses of each BiTE. B2-OKT3 and huNKG2D-OKT3 had equivalent cytotoxicity at doses above 28 ng/ml. These results demonstrate that both BiTEs are effective at low concentrations against MICA/B expressing cell lines derived from liquid or solid tumors.
To determine the ability of BiTEs to elicit T cell responses against a panel of human cell lines from different tissues, T cells from four to six donors were cultured with the BiTEs and tumor cell lines and cytokine production was measured (Figure 1C, 1D). Interferon gamma (IFNγ) production was observed with all tumor target cells, at even the lowest dose (3.1 ng/well) of the huNKG2D-OKT3 BiTE (Figure 1C). The B2-OKT3 BiTE resulted in T cell IFNγ production in a dose-dependent manner when co-cultured with K562, Panc1, or MCF7 tumor cells, and triggered IFNγ production to a lesser extent with the PC3 tumor cell line. In contrast to huNKG2D-OKT3, minimal IFNγ was produced by T cells co-cultured with the cell line T47D and B2-OKT3 (Figure 1D). The expression of MICA/B are shown for these cell lines, and overall those with the highest expression induced greater IFNγ production (Figure 1E). Because NKG2D can recognize both MICA and MICB, along with other ligands, it is not surprising that the NKG2D BiTE would lead to greater IFNγ production against these cell lines than the B2-OKT3 BiTE. Importantly, both BiTEs caused T cells to produce little IFNγ when cultured with T cells alone. As an additional control for MICA specificity, we tested the induction of IFNγ by human T cells cultured with increasing amounts of these BiTEs or an irrelevant BiTE (B7H6-specific TZ47-OKT3) against murine tumor cells that do (B16F10-MICA) or do not (B16F10) express MICA. Specificity for MICA was demonstrated by showing a dose-dependent increase in IFNγ only when MICA was expressed on the target cells (Figure 1F & 1G). Results of these analyses show that both huNKG2D-OKT3 and B2-OKT3 induce cytotoxicity and robust proinflammatory cytokine production from human T cells against a diversity of tumor cells.
Cytotoxicity by B2-OKT3 but not huNKG2D-OKT3 is blocked by soluble rMICA
We tested competition of huNKG2D-OKT3 and B2-OKT3 for binding to rMICA to determine if they bind at overlapping or distinct epitopes by Biolayer Interferometry (BLI). (Figure 2A, 2B). Briefly, SA biosensors were activated with biotinylated rMICA, then allowed to saturate with one BiTE before testing for association with the second BiTE or a control analyte. The results show that blocking with either BiTE did not prevent association of the second BiTE, whereas each BiTE prevented the binding of additional BiTE of the same type. These data are consistent with distinct epitopes on MICA for the NKG2D and B2 BiTEs.
Figure 2. B2-OKT3 and huNKG2D-OKT3 BiTEs bind different epitopes on MICA and B2-OKT3 cytotoxic activity is blocked at supraphysiological concentrations of rMICA.
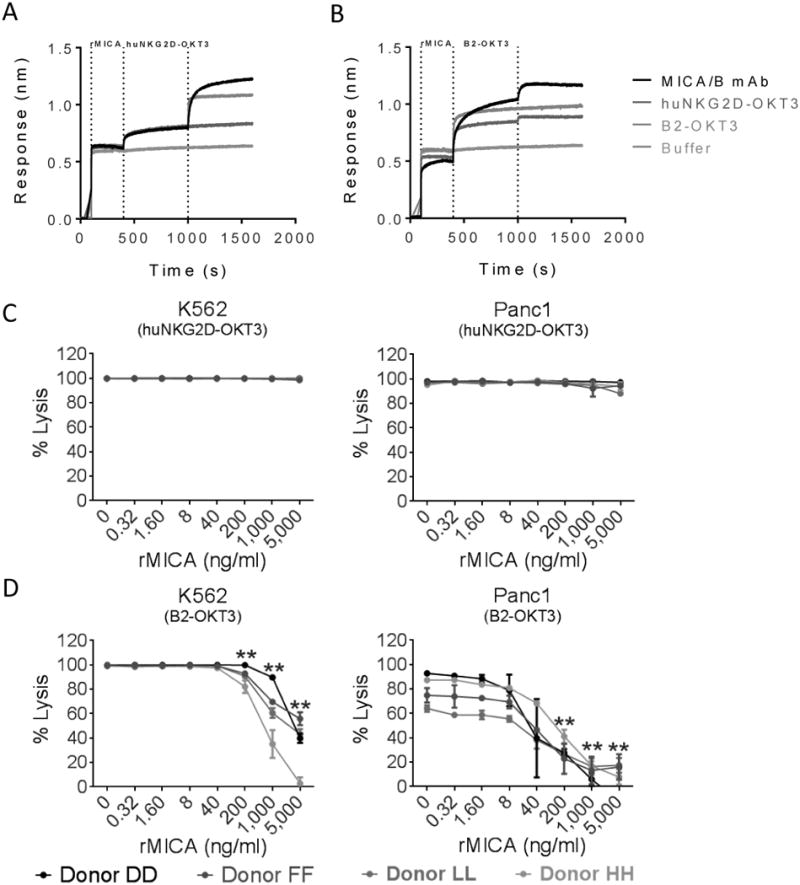
B2-OKT3 and huNKG2D-OKT3 binding to rMICA was measured by biolayer interferometry. Streptavidin biosensors were loaded with biotinylated rMICA, saturated with the blocking BiTE (A) huNKG2D-OKT3 or (B) B2-OKT3 and then dipped into the test analytes: anti-MICA/B, huNKG2D-OKT3, B2-OKT3, and buffer only. Data are representative of at least two independent experiments. Cultured human T cells from four different donors were plated with either K562-Luc or Panc1-Luc at a 4:1 ratio. (C) huNKG2D-OKT3 or (D) B2-OKT3 was added at 50ng/ml. rMICA was at the indicated concentrations. ** Indicates concentrations where the cytotoxicity was significantly lower than no rMICA controls, p < 0.01.
In some patients, tumors are known to shed MICA protein into the bloodstream with the potential to inhibit NKG2D function (25). We tested whether soluble rMICA inhibited BiTE triggered cytotoxicity of K562 or Panc1 tumor cells (Figure 2C & 2D). When low amounts of BiTE were used (50ng/ml) a decrease in lysis triggered by the B2-OKT3 BiTE was observed when a 5-fold or greater amount of rMICA was present. This is due to the specificity of this BiTE for MICA, reinforced by the need for higher amounts of rMICA to cause a similar level of inhibition for the K562 target cell line which expresses more surface MICA than the Panc1 targets (Figure 2D). In contrast, the huNKG2D-OKT3 was not inhibited by rMICA under these conditions (Figure 2C). This may be due to the weaker binding of NKG2D for MICA and/or its ability to recognize multiple ligands on tumor cells. Although soluble MICA may be present in patient sera, it is typically less than 10ng/ml and often under 1ng/ml (25). At this concentration, it may not alter the ability of BiTEs to trigger T cell cytotoxicity until local amounts become very high, which has not been reported.
Both cultured human T cells and resting PBMCs respond to ligand-positive cells in the presence of B2-OKT3 and huNKG2D-OKT3
It has been shown that BiTEs are able to activate T cells in the absence of any T cell pre-conditioning or co-stimulation (7). To determine the ability of B2-OKT3 and huNKG2D-OKT3 to activate both cultured and resting T cells against ligand-expressing tumor cells, resting (PBMCs) or cultured (anti-CD3 stimulated) T cells from five different human donors were treated with a range of BiTE concentrations in the presence of MICA+ K562 tumor cells. T cell activation was assessed by IFNγ production. T cells from all five donors showed robust IFNγ production in response to BiTE treatment. Maximum IFNγ production was observed after 24 hours for the activated T cells, while T cells among resting PBMCs reached maximum IFNγ production after 48 hours (our unpublished observations). EC50 values were calculated from the dose response curves for each donor at the time of maximum IFNγ production (Table 1). The EC50 values show that both PBMCs and cultured T cells were able to be activated at low ng/ml doses with either BiTE. Additionally, comparison of the EC50 values between the two constructs showed a trend of lower EC50 values for huNKG2D-OKT3 compared to B2-OKT3 demonstrating an ability of huNKG2D to elicit T cell activity at very low concentrations.
Table 1. HuNKG2D-OKT3 and B2-OKT3 elicit IFNγ production from both cultured T cells and PBMCs in the presence of MICA+ tumor cells.
| Cultured T cells | Resting PBMCs | |||
|---|---|---|---|---|
|
| ||||
| Donor | huNKG2D-OKT3 | B2-OKT3 | huNKG2D-OKT3 | B2-OKT3 |
| Q | 22a | 103 | 7 | 146 |
| S | 10 | 15 | 9 | 187 |
| W | 46 | 66 | 6 | 5 |
| X | 2 | 21 | 15 | 2246 |
| FF | 1 | 9 | 3 | 92 |
|
| ||||
| Mean | 15 | 43 | 8 | 535 |
| SD | 21 | 40 | 5 | 959 |
Numbers represent EC50 (ng/ml BiTE) required for half maximal IFNγ response measured by ELISA after 24 hours (T cells) or 48 hours (PBMCs) of culture at an E:T ratio of 4:1 and BiTE concentrations of 0, 5, 15, 50, 150, and 500 ng/ml
After BiTE treatment, effector memory T cells are the primary producers of IFNγ
To identify the T cell subsets activated by the B2-OKT3 and huNKG2D-OKT3 BiTEs, resting PBMCs and K562 cells were treated with BiTE, and specific subsets of activated T cells were identified by intracellular staining of IFNγ. CD3+ T cells were subcategorized into naïve (TN), central memory (TCM), effector memory (TEM) or terminal effector (TEMRA) T cells based on surface staining of CD45RO/CCR7. An example of the gating strategy used to identify the different T cell subsets is shown in Figure 3A. Quantification of the percent of cells in each T cell subset from four donors, under control conditions including PBMCs alone, PBMCs with either BiTE, and PBMCs with K562 cells had no statistical difference in distribution of T cell subsets. As a control for T cell activation, the T cell subset distribution of PBMCs stimulated with PMA and Ionomycin (Ion) was also examined. CD3+ IFNγ + PMA/Ion-treated PBMCs had a significant reduction in TN cells compared to all control conditions and some enrichment of the TEM population. However, IFNγ was produced nearly exclusively by TEM cells upon co-culture of either BiTE with K562 cells and PBMCs. This was a significant change in T cell distribution compared to the initial distribution in any of the control conditions as well as compared to PMA/Ion-treated PBMCs (Figure 3B). The TEM population is preferentially activated by these two BiTEs because the same change in cell distribution was not induced by non-specific T cell activation with PMA/Ion.
Antigen density affects T cell activation by B2-OKT3 and huNKG2D-OKT3 BiTEs
HuNKG2D-OKT3 can recognize multiple NKG2D ligands, which may be one reason for the higher activity of this BiTE against tumor cells. To determine the contribution that antigen density plays in the activation of T cells by the two BiTEs, plate bound rMICA was used as a ligand in the absence of tumor cells. The use of rMICA eliminated the possibility that huNKG2D-OKT3 recognized another ligand in the assay. The rMICA was immobilized at multiple concentrations, and T cells were cultured in the presence of huNKG2D-OKT3 or B2-OKT3. T cell activation was subsequently measured by IFNγ production and by intracellular IFNγ staining. Analysis of total IFNγ production for ten donors demonstrated an antigen concentration dependent increase for T cells treated with either huNKG2D-OKT3 or B2-OKT3. TZ47-OKT3 (B7H6-specific) served as a negative BiTE control protein (Figure 4A). Comparison of the dose response curves revealed that 8 of the 10 donors produced less IFNγ when treated with B2-OKT3 compared to treatment with huNKG2D-OKT3 (p <0.01), this indicates that less MICA was needed to trigger T cell activation with the huNKG2D-OKT3 BiTE.
Analysis of intracellular IFNγ + cells from T cells cultured in the presence of either BiTE showed that the percentage of activated CD4+ and CD8+ T cells increased in concert with increasing rMICA antigen concentration (Figure 4B). However, treatment with huNKG2D-OKT3 resulted on average in a 4-fold greater percentage of CD4+ and 2.2-fold greater percentage of CD8+ T cells producing IFNγ as compared to treatment with B2-OKT3 (Figure 4B). Treatment with the huNKG2D-OKT3 BiTE also elicited an antigen dependent increase in IFNγ production on a per cell basis, as indicated by an increased mean fluorescence intensity (MFI) for both CD4+ and CD8+ T cells. While treatment with B2-OKT3 only showed this increase in IFNγ production on a per cell basis for the CD8+ T cells but not in the CD4+ compartment (Figure 4C). Overall these results show that T cell activation by both BiTEs was dependent on antigen density, that huNKG2D-OKT3 induced greater T cell activation compared with B2-OKT3, and this increase in T cell activation occurred when MICA was the target ligand.
HuNKG2D-OKT3 and B2-OKT3 BiTEs are able to activate T cells against human melanoma tumor samples
To determine the ability of these two NKG2D ligand binding BiTEs to trigger T cell responses to human tumor cells, cultured T cells and resting PBMCs from healthy donors, and autologous T cells from metastatic melanoma patients were tested against a panel of eight matched human melanoma tumor samples. Each tumor sample was co-cultured with effector cells at an E:T ratio of 4:1, in the presence or absence of the BiTEs, and T cell IFNγ production was measured. Upon treatment with huNKG2D-OKT3 significant IFNγ was produced by all cultured T cells tested against each of the eight tumor samples (100% response) (Figure 5A). Treatment with B2-OKT3 resulted in significant IFNγ production from T cells for five out of the eight tested donors (62.5% response) (Figure 5B). It is interesting to note that autologous T cell responses were often equally as robust as those from healthy donors, indicating that melanoma patient T cells can be activated with these therapeutic proteins.
The BiTEs were also able to activate resting healthy donor PBMCs against melanoma tumor cell samples. Two of the melanoma samples, E10 and E11, were co-cultured with resting PBMCs from two healthy donors at an E:T ratio of 4:1 with and without BiTEs, and IFNγ production was measured (Figure 5C). Significant IFNγ was produced upon treatment with huNKG2D-OKT3 from both PBMC donors co-cultured with E10 tumor cells and with one donor when co-cultured with E11 tumor cells. Treatment with B2-OKT3 elicited significant IFNγ production from one PBMC donor against tumor E10. Notably, there was greater IFNγ production with both donors against each of the melanoma tumor samples compared to T cells with BiTE or T cells with tumor samples alone. These data show that prior T cell stimulation was not required for these BiTEs to elicit T cell cytokine production against human melanoma tumor cells.
Discussion
In this study, we have characterized two new bispecific proteins with ligands recognized by the human NKG2D receptor. NKG2D functions in the recognition of carcinogenic transformation of cells (18). NKG2D ligands can be polymorphic, heterogeneous, and vary in expression during tumor progression (13, 26-28). There are more than 100 alleles of MICA that code for 82 different proteins and 40 alleles of MICB that encode 28 different proteins (29). The MICA targeting BiTE, B2-OKT3, was screened against the MICA*001 allele. The ability of this BiTE to bind other alleles of MICA is not known, but broad activity against tumor cell lines and human melanoma tumor cells suggests that the B2-OKT3 protein may have therapeutic application for a broad range of tumors. The huNKG2D-OKT3 BiTE has the potential to be effective for a high percentage of patients, based on the 100% response rate of T cells with this protein against each of the human melanoma tumor samples. It is possible that chemotherapy or infection may transiently upregulate NKG2D ligands on tissues, and this represents a safety concern for on-target off-tumor effects. As clinical data are published on targeting NKG2D ligands, the nature of these potential concerns will be better understood and mitigated.
Many T cell redirecting bispecifics are being clinically and preclinically evaluated, and BiTEs have emerged as a successful format. This is partially due to the ability of this format to activate T cells in the absence of any pre-activation or costimulation (7, 30). This costimulation independent activation of T cells is thought to be the reason for the preferential expansion of the effector memory T cell population upon BiTE treatment. It has been shown that the TEM cell population expanded during treatment of ALL patients with blinatumomab, and TEM expansion was also shown in long term T cell cultures expanded with BiTE ESK-1 (31, 32). Treatment with 6PHU3, a BiTE that targets claudin 6, was also found to increase the population of TEM in the tumors of xenografted mice (33). The TEM cell population has also been shown to contribute to BiTE redirected target cell lysis. The importance of the TEM population in cytotoxicity has been shown with MT110, an EpCAM targeting BiTE. In an in vitro study of MT110, isolated TEM cells made the largest contribution to T cell redirected lysis as compared to naïve or terminal effectors (34). TEM are long-lived and retain their effector function, unlike terminal effector cells. It is possible that the expansion of TEM may be necessary for BiTE efficacy in patients where there is reduced T cell function, which has been shown in CLL (35). In this study, TEM cells were the major contributor to IFNγ production after BiTE activation, which is important because IFNγ is an important mediator of a proinflammatory microenvironment and is known to be essential for BiTE efficacy in vivo (36).
Another important modulator of BiTE efficacy is ligand density. Differences in ligand expression on cells can affect T cell cytotoxicity induced by BiTEs (10). Ligand density dependent IFNγ production by T cells was observed with plate bound rMICA in the presence of either BiTE, which shows that plate bound antigen alone is sufficient to trigger T cell activity. Additionally, these experiments showed that the percentage of both CD4+ and CD8+ T cells producing IFNγ, and the amount of IFNγ produced per CD8+ T cell, was dependent on ligand density. CD4+ T cells have been shown to contribute to BiTE activity, but have a delayed activation as compared to CD8+ T cells (7, 37), so it may be that at time points beyond those tested, B2-OKT3 could induce greater CD4+ T cell activity.
The huNKG2D-OKT3 BiTE has several advantages when compared to the B2-OKT3 BiTE, including recognition of multiple ligands that allows for application to a wider range of tumors. While T cell responses were able to be induced against 100% of the tested melanoma tumor samples with huNKG2D-OKT3 treatment, more than half of the samples produced IFNγ when B2-OKT3 was present (62.5%). This is congruent with the observation that 50-75% of melanoma tumors are expected to express MICA, while at least one NKG2D ligand may be present on up to 90% of human tumors (13). HuNKG2D-based targeting may reduce tumor escape through heterogeneous ligand expression or down regulation of ligands as compared to B2-OKT3 where one ligand is targeted, as has been seen after treatment with blinatumomab (38). Furthermore, the NKG2D-OKT3 was not readily inhibited by soluble rMICA compared to the B2-OKT3 and thus may avoid this tumor escape mechanism. This may be due to the lower affinity of NKG2D for MICA or how the NKG2D complex interacts with MICA. However, the B2-OKT3 construct has other properties that should not be overlooked. The tandem scFv format typically has a higher protein yield during production (our unpublished observations), which may make the B2-OKT3 BiTE a better candidate for GMP manufacturing. In addition, the safety concern for on-target off-tumor effects arising from chemotherapeutic or infection-induced upregulation of NKG2D ligands, makes the use of the MICA-targeting BiTE desirable if it leads to a more favorable safety profile. These results support the conclusion that both BiTEs are excellent candidates for clinical application and further testing will elucidate the best choice based on efficacy, safety and protein production.
Acknowledgments
We would like to thank the NCI Biological Resource Division (Frederick, MD) for IL2. We would also like to thank Dr. Margaret Ackerman for the use of the ForteBio Octet and Dr. Charles Wira for the use of the Centro LB 960 luminometer.
Financial Support: This work was supported in part by grants from the National Institutes of Health to C.L. Sentman CA164178 and A.M. Huehls was supported by T32 AI007363.
Abbreviations
- BiTE
Bispecific T cell Engager
- scFv
single-chain variable fragment
- huNKG2D-OKT3
human NKG2D receptor fused to anti-human CD3
- NKG2D
Natural Killer Group 2D
- B2-OKT3
anti-MICA × anti-human CD3
- MICA
major histocompatibility complex (MHC) class I chain-related A
- rMICA
recombinant MICA
- TZ47-OKT3
anti-B7H6 × anti-human CD3
References
- 1.Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. 2016 Jan;13(1):25–40. doi: 10.1038/nrclinonc.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. 2017 Jan 24;8(4):7175–80. doi: 10.18632/oncotarget.12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016 Jun;13(6):394. doi: 10.1038/nrclinonc.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N, et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016 Mar 17;127(11):1410–6. doi: 10.1182/blood-2015-06-651380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantarjian HM, Stein AS, Bargou RC, Grande Garcia C, Larson RA, Stelljes M, et al. Blinatumomab treatment of older adults with relapsed/refractory B-precursor acute lymphoblastic leukemia: Results from 2 phase 2 studies. Cancer. 2016 Jul 15;122(14):2178–85. doi: 10.1002/cncr.30031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015 Oct;67(2 Pt A):95–106. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005 Sep 15;10(18):1237–44. doi: 10.1016/S1359-6446(05)03554-3. [DOI] [PubMed] [Google Scholar]
- 8.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015 Mar;93(3):290–6. doi: 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Przepiorka D, Ko CW, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu HJ, et al. FDA Approval: Blinatumomab. Clin Cancer Res. 2015 Sep 15;21(18):4035–9. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 10.Ferrari F, Bellone S, Black J, Schwab CL, Lopez S, Cocco E, et al. Solitomab, an EpCAM/CD3 bispecific antibody construct (BiTE(R)), is highly active against primary uterine and ovarian carcinosarcoma cell lines in vitro. J Exp Clin Cancer Res. 2015 Oct 17;34:123. doi: 10.1186/s13046-015-0241-7. 015-0241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laszlo GS, Gudgeon CJ, Harrington KH, Dell'Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014 Jan 23;123(4):554–61. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberst MD, Fuhrmann S, Mulgrew K, Amann M, Cheng L, Lutterbuese P, et al. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs. 2014;6(6):1571–84. doi: 10.4161/19420862.2014.975660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immun. 2013 May 1;13:8. [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol. 2001 May;2(5):443–51. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- 15.McFarland BJ, Strong RK. Thermodynamic analysis of degenerate recognition by the NKG2D immunoreceptor: not induced fit but rigid adaptation. Immunity. 2003 Dec;19(6):803–12. doi: 10.1016/s1074-7613(03)00320-0. [DOI] [PubMed] [Google Scholar]
- 16.Del Toro-Arreola S, Arreygue-Garcia N, Aguilar-Lemarroy A, Cid-Arregui A, Jimenez-Perez M, Haramati J, et al. MHC class I-related chain A and B ligands are differentially expressed in human cervical cancer cell lines. Cancer Cell Int. 2011 Jun 1;11:15. doi: 10.1186/1475-2867-11-15. 2867-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A. 1999 Jun 8;96(12):6879–84. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanier LL. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol Res. 2015 Jun;3(6):575–82. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001 Feb;14(2):123–33. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 20.Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004 Sep;21(3):367–77. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Shi P, Yin T, Zhou F, Cui P, Gou S, Wang C. Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer. 2014 May 25;14:370. doi: 10.1186/1471-2407-14-370. 2407-14-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vetter CS, Groh V, thor Straten P, Spies T, Brocker EB, Becker JC. Expression of stress-induced MHC class I related chain molecules on human melanoma. J Invest Dermatol. 2002 Apr;118(4):600–5. doi: 10.1046/j.1523-1747.2002.01700.x. [DOI] [PubMed] [Google Scholar]
- 23.Wu MR, Zhang T, DeMars LR, Sentman CL. B7H6-specific chimeric antigen receptors lead to tumor elimination and host antitumor immunity. Gene Ther. 2015 Aug;22(8):675–84. doi: 10.1038/gt.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldhaus M, Siegel R. Flow cytometric screening of yeast surface display libraries. Methods Mol Biol. 2004;263:311–32. doi: 10.1385/1-59259-773-4:311. [DOI] [PubMed] [Google Scholar]
- 25.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer. 2006 Feb 1;118(3):684–7. doi: 10.1002/ijc.21382. [DOI] [PubMed] [Google Scholar]
- 26.Stephens HA. MICA and MICB genes: can the enigma of their polymorphism be resolved? Trends Immunol. 2001 Jul;22(7):378–85. doi: 10.1016/s1471-4906(01)01960-3. [DOI] [PubMed] [Google Scholar]
- 27.Carlsten M, Bjorkstrom NK, Norell H, Bryceson Y, van Hall T, Baumann BC, et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007 Feb 1;67(3):1317–25. doi: 10.1158/0008-5472.CAN-06-2264. [DOI] [PubMed] [Google Scholar]
- 28.McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res. 2009 Nov 15;15(22):6993–7002. doi: 10.1158/1078-0432.CCR-09-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015 Jan;43(Database issue):D423–31. doi: 10.1093/nar/gku1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kufer P, Zettl F, Borschert K, Lutterbuse R, Kischel R, Riethmuller G. Minimal costimulatory requirements for T cell priming and TH1 differentiation: activation of naive human T lymphocytes by tumor cells armed with bifunctional antibody constructs. Cancer Immun. 2001 Nov 12;1:10. [PubMed] [Google Scholar]
- 31.Zugmaier G, Gokbuget N, Klinger M, Viardot A, Stelljes M, Neumann S, et al. Long-term survival and T-cell kinetics in relapsed/refractory ALL patients who achieved MRD response after blinatumomab treatment. Blood. 2015 Dec 10;126(24):2578–84. doi: 10.1182/blood-2015-06-649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015 Oct;33(10):1079–86. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stadler CR, Bahr-Mahmud H, Plum LM, Schmoldt K, Kolsch AC, Tureci O, et al. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology. 2015 Oct 29;5(3):e1091555. doi: 10.1080/2162402X.2015.1091555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kischel R, Hausmann S, Baeuerle P, Kufer P. Abstract #3252: Effector memory T cells make a major contribution to redirected target cell lysis by T cell-engaging BiTE antibody MT110. Cancer Res. 2014;69(9 Supplement):3252. [Google Scholar]
- 35.Wong R, Pepper C, Brennan P, Nagorsen D, Man S, Fegan C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica. 2013 Dec;98(12):1930–8. doi: 10.3324/haematol.2012.082248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu MR, Zhang T, Gacerez AT, Coupet TA, DeMars LR, Sentman CL. B7H6-Specific Bispecific T Cell Engagers Lead to Tumor Elimination and Host Antitumor Immunity. J Immunol. 2015 Jun 1;194(11):5305–11. doi: 10.4049/jimmunol.1402517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mack M, Gruber R, Schmidt S, Riethmuller G, Kufer P. Biologic properties of a bispecific single-chain antibody directed against 17-1A (EpCAM) and CD3: tumor cell-dependent T cell stimulation and cytotoxic activity. J Immunol. 1997 Apr 15;158(8):3965–70. [PubMed] [Google Scholar]
- 38.Yannakou CK, Came N, Bajel AR, Juneja S. CD19 Negative Relapse in B-ALL Treated with Blinatumomab Therapy: Avoiding the Trap. Blood. 2015;126(23):4983. [Google Scholar]