

Abstract
Purpose
Children with high-risk neuroblastoma have a poor prognosis with chemotherapy alone and hematopoietic stem cell transplantation offers improved survival. As a dose escalation strategy, tandem transplants have been utilized, but are associated with persistent immune compromise. This study evaluated the provision of an autologous co-stimulated, activated T cell product to support immunologic function.
Experimental Design
Nineteen subjects with high risk neuroblastoma were enrolled in a pilot phase and twenty three subjects were entered in to the randomized study. Immunologic reconstitution was defined by flow cytometry and functional assays. Next generation sequencing was performed to identify changes to the T cell repertoire. Twenty-two patients were vaccinated to define effects on antibody responses.
Results
Subjects who received their autologous co-stimulated T cell product on Day 2 had significantly superior T cell counts and T cell proliferation compared to those who received T cells on Day 90. Early administration of autologous T cells suppressed oligoclonality and enhanced repertoire diversity. The subjects who received the Day 2 T cell product also had better responses to the pneumococcal vaccine.
Conclusions
The infusion of activated T cells can improve immunologic function especially when given early after transplant. This study demonstrated the benefit of providing cell therapies during periods of maximum lymphopenia.
Keywords: Neuroblastoma, T cell, antibody, opportunistic infections
Introduction
Neuroblastoma is the most common extracranial solid tumor in childhood (1). It is remarkable for its clinical range, encompassing spontaneous regression to progression and death. Approximately 12% of pediatric deaths from malignancy are due to neuroblastoma. The high-risk phenotype is generally characterized by disseminated disease and increased mortality (2, 3). Additional features associated with the high-risk phenotype include histopathological features and MYCN amplification (2, 4). In these high-risk patients, more than half of the patients relapse after multimodal therapy. Recently, anti-GD2 immunotherapy to treat minimal residual disease after achieving remission has led to additional improvements in survival, however, stem cell transplantation is considered standard-of-care currently for high-risk patients (5, 6).
Autologous stem cell transplantation for neuroblastoma has been under investigation for over 20 years and in 1997, a large retrospective analysis found improved survival in patients treated with stem cell transplant as opposed to conventional chemotherapy (7). The single largest randomized trial of autologous stem cell transplantation was the Children's Cancer Group 3891 study (8). In this study, all patients received a consolidation regimen and then were randomized to autologous transplantation or additional chemotherapy. The study established autologous transplantation and isotretinoin therapy as the standard-of-care for high-risk patients. In spite of this, more than half of the patients relapsed. In recent years, the autologous stem cell transplantation concept has been extended to sequential cycles. A tandem transplant allows for greater dose intensity and has shown promise as a strategy to improve outcomes with the largest study demonstrating a 3-year event-free survival of 55% (9-11).
One of the significant issues in patients who receive a tandem stem cell transplant is prolonged immune suppression. Post-transplant lymphoproliferative disease and other viral morbidities can occur (12)(13). This suggests that there is prolonged, medically significant immune suppression in this population. To mitigate immune suppression in other settings, autologous expanded T cells have been utilized to provide defense against viral infections (14-18). We examined immunologic reconstitution in a cohort of children with high risk neuroblastoma who received a tandem transplant and were then randomized to receive either an autologous expanded T cell product on Day 2 or Day 90. We carefully characterized immunologic parameters as well as their ability to respond to vaccines. The subjects that received the early autologous T cell product had superior T cell reconstitution and oligoclonality was suppressed during the vulnerable window post-transplant. Significant responses to vaccines were seen almost exclusively in subjects who received the early autologous T cell product.
Methods
Study Design
This study protocol was approved by the Institutional Review Boards at Children's Hospital of Philadelphia and Boston Children's and informed consent was obtained from all participants. The trial was preregistered at ClinicalTrials.gov as NCT00017368. The analysis plan was determined before study initiation and laboratory assays were performed in a blinded fashion. In an initial (pilot) safety and feasibility phase, patients received autologous expanded T cells on Day 2 (Early), Day 12 (Middle), or Day 90 (Late) after the second stem cell infusion. In a second phase of the study, patients were randomized after enrollment to either the Early (Day 2) or Late (Day 90) T cell infusion group (Figure 1). The seasonal inactivated influenza vaccine and the conjugated 7-valent pneumococcal vaccine were administered on Day 12 and Day 60 after the second stem cell infusion. This protocol did not include immunoglobulin administration. In the analyses, Baseline refers to samples obtained after diagnosis, but before chemotherapy. Days 30, 60, 90, 120, and 365 refer to the time after the second transplant.
Figure 1. Protocol description (randomized phase).
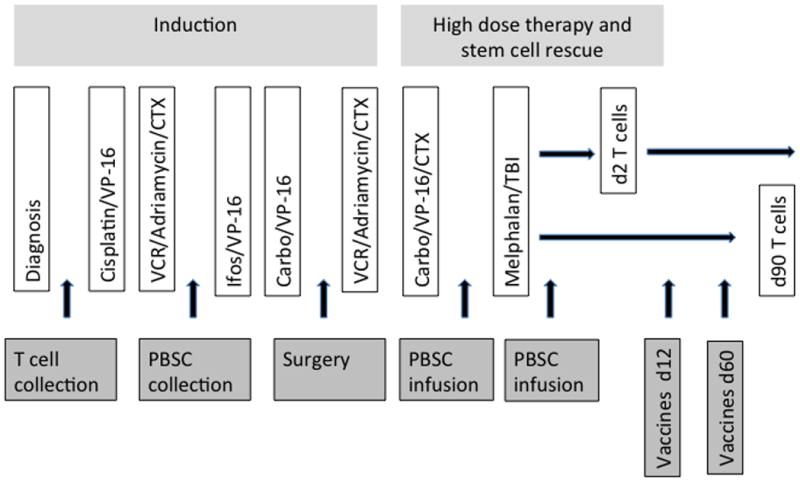
All patients received the induction regimen and high dose chemotherapy with stem cell rescue. Patients were randomized to either the Early group (autologous co-stimulated T cell product on Day 2 after the second stem cell rescue) or the Late Group (autologous co-stimulated T cell product on Day 90 after the second stem cell rescue). The conjugated 7-valent pneumococcal vaccine and the inactivated trivalent influenza vaccine were administered on days 12 and 60 after the second transplant. Evaluations of immunologic function were performed at the time of diagnosis and on days 30, 60, 90, 120 and 365. VCR=vincristine, CTX=cyclophosphamide, Ifos=ifosfamide, Carbo=carboplatin, TBI-total body irradiation.
In all, 22 patients received T cells at Day 2, 10 patients at Day 12, and12 patients at Day 90. Two patients were excluded for early relapse or treatment-related mortality, and the other 42 patients were included in the pilot study analysis of the impact of a T cell infusion, and the timing of the infusion, on T cell recovery. One Day 90 patient developed CMV disease after stem cell infusion and received T cells under a compassionate exception to the protocol at Day 60, and is included in the analysis of the Day 90 group, as assigned. In the analysis of the second, randomized phase, 22 patients were randomized and included in the immune response analysis, including vaccine response.
T cell Infusions
Patients underwent a single 2-6 blood volume leukapheresis at diagnosis (prior to receiving chemotherapy) to collect peripheral blood mononuclear cells (PBMC). The cells were depleted of monocytes and cryopreserved. The T cells underwent an established GMP co-stimulated activation and expansion protocol using CD3- and CD28-linked beads (19). All infused T cell products were required to meet FDA-specified safety and release criteria prior to infusion. The target number of T cells for infusion was 2-8 ×108 T cells/kg. As this was the first effort at expanding T cells from children, we compared the expanded product from children and adults (Supplemental Data 1)
Immunologic Assays
For the primary endpoint of T cell count post stem cell transplant in the pilot phase, samples were submitted to CLIA-approved clinical laboratories for a complete blood count with differential, as well as CD4 percentage and CD8 percentage according to standard clinical protocols. For the second randomized phase, detailed analyses of lymphocyte subsets were performed according to research protocols. T cell subset analyses were performed after fixation with 1% paraformaldehyde on an LSR II (BD Biosciences) using FlowJo software (TreeStar). CD4 Naïve cells were defined as CD45RA+CD31+, CD4 Central Memory T cells were defined as CD27+CD45RO+CCR7+, CD4 Effector Memory T cells were defined as CD45RO+/CD27+/CCR7-, CD4 Reverted Memory T cells were defined as CD45RA+/CD31-/CCR7+, and regulatory T cells (Treg) were defined as CD4+/CD25+/CTLA4+. CD8 Naïve cells were defined as CD45RA+CD31+, CD8 Central Memory T cells were defined as CD27+CD45RO+CCR7+, CD8 Effector Memory T cells were defined as CD45RO+/CD27+/CCR7-, and CD8 Reverted Memory T cells were defined as CD45RA+/CD31-/CCR7+.
For B cell phenotyping, fresh blood was stained with antibodies (all from BD Pharmingen, San Diego, CA), as described previously (20) and run on a FACSCalibur (Becton Dickinson) with CellQuest software (Version 5.2.1, Becton Dickenson, San Jose, CA). CD19+ lymphocytes were analyzed for CD27 and IgM. The absolute B cell count was obtained by multiplying the absolute lymphocyte count by the CD19+ fraction.
T cell proliferation, T cell repertoire, T cell ELISPOTs and B cell ELISPOTs were performed as previously described (18). The assessment of influenza and pneumococcal vaccine responses utilized a standard hemagglutination inhibition (HAI) assay optimized for the vaccine administered each year and a commercial laboratory for pneumococcal responses (21). Next generation sequencing was performed by Adaptive TCR technologies (www.immunoseq.com) using DNA from PBMC, TCRβ-specific primers, and a deep level of sequencing on the Illumina platform (22).
Statistical analysis
The linear regression model was utilized for analyzing repeated measures based on the Generalized Estimating Equation (GEE) method using SAS GENMOD. The regression models included Group effects, Time effects, and the interaction of Group*Time. Baseline differences were examined to exclude group differences related to randomization. To define the cumulative differences between the two groups, the area under the curve was calculated. The independent t-test, Kruskal-Wallis test, or the Mann-Whitney test were used for the comparisons of single time points. The small size of the patients and the severity of their illness occasionally prevented obtaining all study samples. Each analysis indicates the number of subjects included. The studies reported were pre-defined in the study design. Significance was set at p<0.05. Data analysis was performed by Drs. Sullivan, Grupp, Boyer, Luning Prak and Jawad. All authors had access to study data.
Results
Patient characteristics
All patients had high-risk neuroblastoma and were over one year of age. All but two patients had stage 4 disease. Consistent with the known epidemiology of the disease, the children ranged in age from 1-5 years of age. The subjects were roughly half female and the majority of patients were Caucasian. For the pilot study of T cell infusions, 8 patients received the infusion on Day 2 (8 were stage 4 and the median age was 2.4 years), 8 patients received the infusion on Day 12 (8 were stage 4 and the median age was 3.4 years), and 3 patients received the infusion on Day 90 (3 were stage 4 and the median age was 5.1 years). The randomized groups consisted of 12 subjects in the Early Group (Day 2 infusion, 11/12 stage 4, median age 1.9 years) and 10 subjects in the Late Group (Day 90 infusion, 9/10 stage 4, median age 2.7 years).
This study was not designed or powered to look at the impact of T cell infusion on survival. However, we performed a descriptive analysis of event-free survival (EFS) in the cohort and among patients assigned to day 12, day 2 and day 90 T cell infusion. Five year EFS in the cohort was 55%, which was similar to our prior report of 47% (12). No differences between T cell infusion subgroups were apparent, with 5 year EFS in the day 2, 12, and 90 subgroups being 55%, 50%, and 58%, respectively. One subject in each group experienced a post-transplant opportunistic infection. There was one case of CMV in the Late Group at Day 30 and one case of EBV lymphoproliferative disease in the Early Group on Day 21. Both of these infections were fatal.
Pilot study of T cell reconstitution, comparing three time points
We hypothesized that autologous, co-stimulated expanded T cells would alter the composition of the T cell compartment significantly. This CD3/CD28 bead-stimulated GMP cell therapy product has been previously demonstrated to produce CD4/CD8 ratios of approximately 1:1, and to maintain the T cell receptor repertoire of the input T cells (14, 15, 23). For analysis of the initial pilot phase, clinical measurements of ALC, CD3, CD4, and CD8 counts were used. These data are shown in Figure 2 and demonstrate a profound impact of T cell infusion on the T cell count. The group receiving T cells at Day 2 had improved CD4 and CD8 T cell counts compared to the Day 90 Group in the Day 2-90 interval. Interestingly, the Day 2 Group also had improved T cell recovery compared to the Day 12 Group, suggesting that very early T cell infusion might result in even more robust CD4 cell recovery. A similar effect across the groups was seen in terms of CD8+ cell recovery.
Figure 2. Schedule-dependent impact of co-stimulated, activated T cell transfer on T cell reconstitution after tandem stem cell transplant.
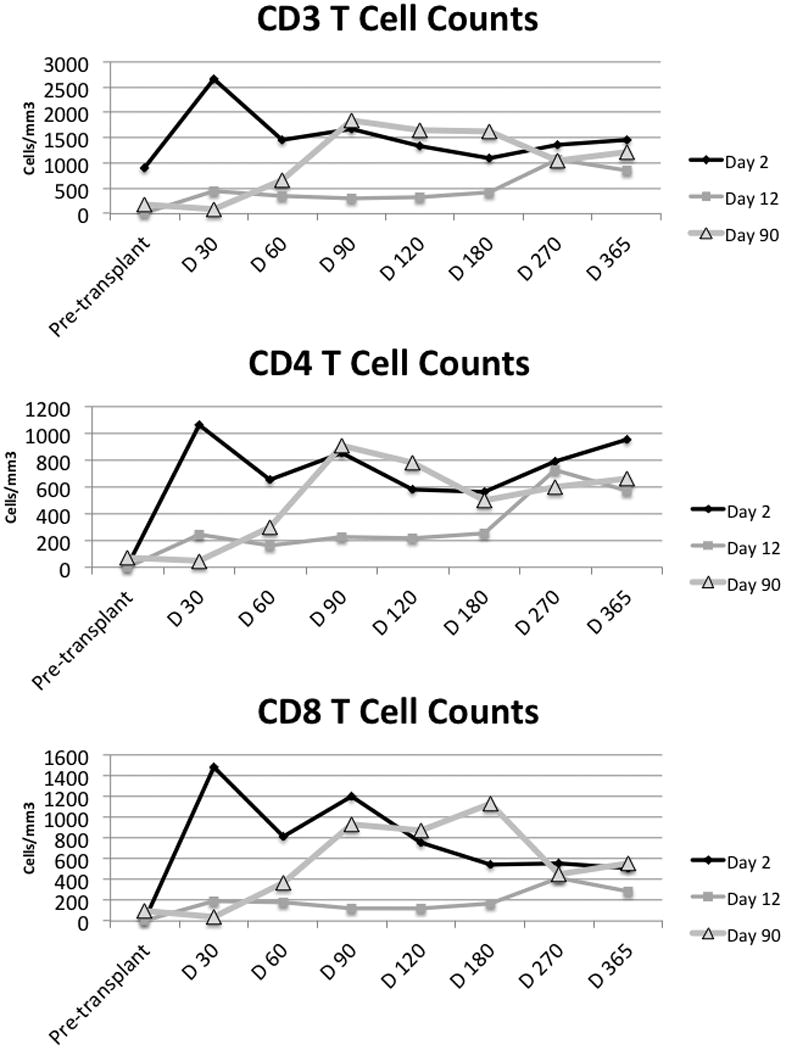
Patients received T cells at one of three points in this safety and feasibility phase: at Day 2, Day 12 and Day 90 post stem cell infusion. CD3+, CD3+CD4+ (CD4), CD3+CD8+ (CD8) T cells were enumerated on clinical samples at each time point. X-axis: Pre-transplant refers to blood drawn immediately prior to the first transplant; the other time points refer to days after the second transplant. The CD4 and CD8 T cell count differences between the three groups are significant at Day 30 (Kruskal Wallis p=0.0021, p=0.0033 respectively). At Day 60, CD3, CD4, and CD8 T cells are all significantly different with p=0.0005, 0.0003, p=0.0007 respectively.
T cell recovery after early or late T cell infusions
During the randomized phase of the study, we expanded the immunologic reconstitution assessments. We first characterized the T cells in the expanded autologous T cell product because there have been no previous studies of pediatric specimens. We identified the frequency of stem-like T cells (24), follicular helper T cells, regulatory T cells and other subsets of biological relevance. Surprisingly, there were almost no differences between the cell populations in the expanded products from children and adults, although naïve CD4 T cells were increased in the expanded product from the children compared to adults (Supplemental Data 1).
We characterized T cell subsets using flow cytometry and compared the Day 2 Early Group (n=12) and the Day 90 Late Group (n=7) (Figure 3). Absolute counts of both CD4 and CD8 T cells were higher in the Early Group compared to the Late Group in the early post-transplant interval (Figure 3). We measured the area under the curve to compare the advantage of the early T cell product over the late T cell product between days 0-90. The improvement in peripheral blood T cell counts was significant with p=0.0023 for CD4 T cells and a p=0.0010 for the CD8 T cells. To determine whether the excess cells in the Early Group were of a specific subset, we analyzed CD4 and CD8 T cell naïve and memory subsets (Figure 3). Examples of the naïve and memory flow cytometry gating are given in Supplemental Data 2. Naïve CD4 and CD8 T cells counts were higher in the Early Group compared to the Late Group at the 60 day time point (p=0.0380, p=0.0344 respectively). The CD4 central memory T cell count was higher in the Early Group compared to the Late Group with p=0.0311. Regulatory T cells were somewhat higher in the Early Group. After the T cell infusion in the Late Group at day 90, the advantage of the early infusion was lost. The T cell reconstitution was variable in both groups. We examined age, number of T cells infused, level of stem-like cells in the expanded product and regulatory T cells in the expanded product for associations with naïve CD4 T cell reconstitution. We did not identify any associations.
Figure 3. Accelerated and sustained reconstitution of T cells in the Early Group.
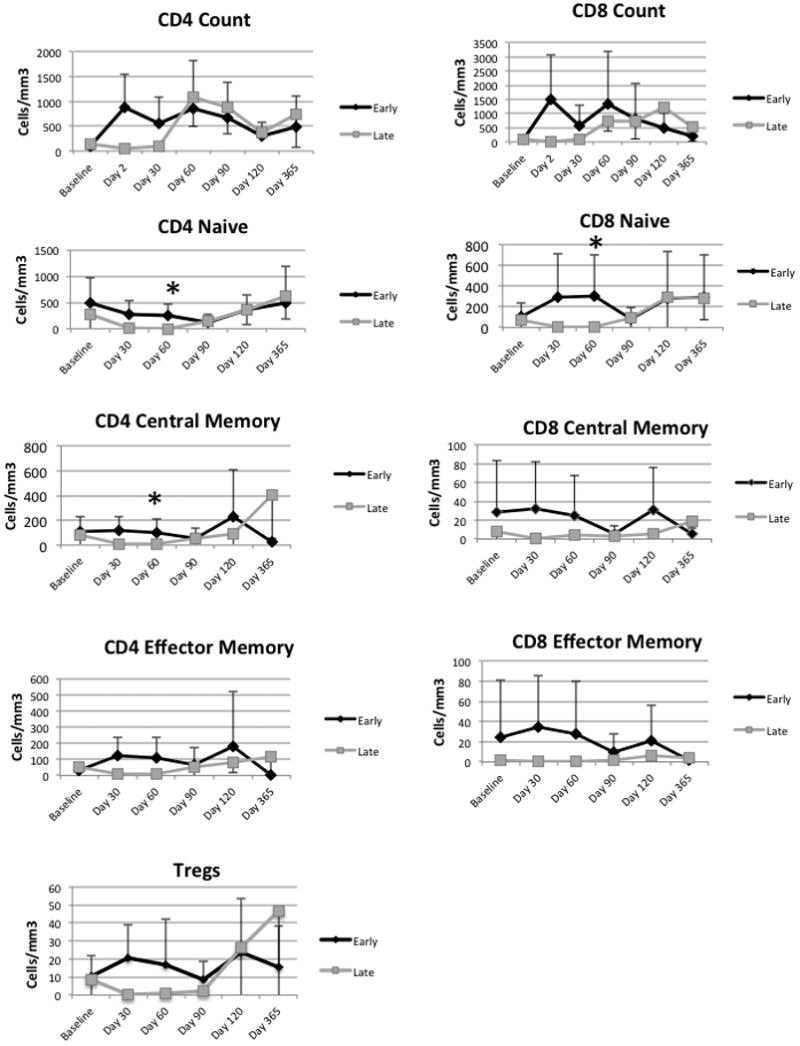
CD4 Central Memory, CD4 Naïve, and CD8 Naive cell counts were significantly higher at the 60 day time point in the Early Group compared to the late group (Kruskal Wallis p=0.03, 0.04, 0.006, respectively). The asterisks indicate significance.
Based on data from non-human primates, it is estimated that a typical 2-3 year old child has a total body content of approximately 2×1011 T cells (25). Therefore, the increase in T cell count in the peripheral blood was unlikely to be directly due to the addition of 2-8 ×108 co-stimulated, activated T cells/kg (approximately 3-12 × 109 cells total). The lymphocyte count was significantly higher in the Early Group at Day 30 compared to the Late Group (p=0.007). To determine whether T cells expanded in vivo, as has been seen in other settings (26, 27), we identified dividing CD3 positive T cells in the absence of stimulation by CFSE (Figure 4A). We reasoned that T cells undergoing homeostatic expansion in vivo would complete the response to that signal in vitro. Cells were incubated five days after staining with CFSE. There was enormous variability in the Early Group, whereas the Late Group consistently demonstrated low cell division in this assay. Nevertheless, the Early Group exhibited significantly more spontaneous proliferation than the Late Group, consistent with homeostatic expansion (Wilcoxon p value = 0.0010, Early Group n=12, Late Group n=7). To further examine the mechanism of expansion, serum IL-7 was measured in both groups. A slow rise in IL-7 was seen in the Late Group (Figure 4B). Chemotherapy driven release of cytokines may mediate early expansion while IL-7 was be responsible for the late expansion seen prior to the Day 90 infusion (28).
Figure 4. Preservation of peripheral blood T cell proliferation by early T cell transfer.
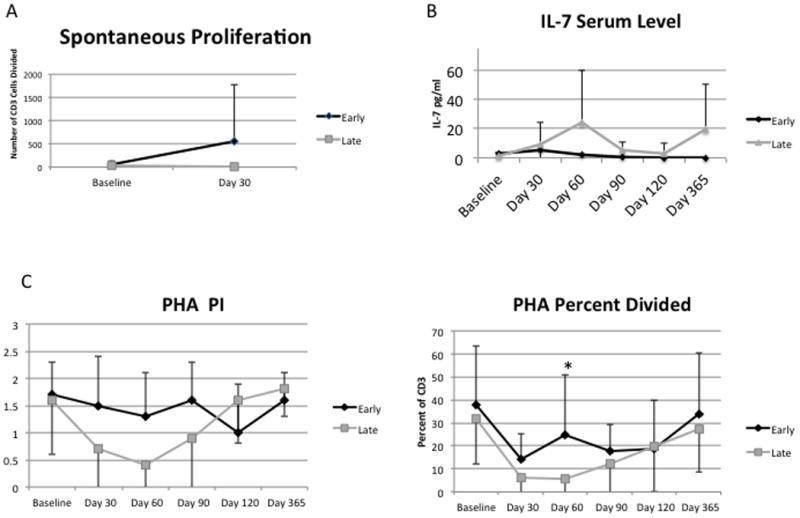
A) At Day 30, T cell proliferation occurs in the absence of stimulation in the Early Group, consistent with homeostatic proliferation. The difference between the Early and Late Groups was significant at day 30 (Wilcoxon p < 0.001). B) IL-7 serum levels, detected by ELISA, increase slowly in the Late Group. C) The Proliferation Index measures the competence to cycle after PHA stimulation and this was not different between the two groups. The Percent Divided metric measures the fraction of cells that entered into cell cycle after PHA stimulation. The Early Group had a higher fraction of responding cells compared to the Late Group at Day 30 (p=0.02).
To examine proliferative responses after stimulation, we incubated PBMC with PHA. CFSE dilution was defined using flow cytometry and the Proliferation Index (a measure of average number of cell divisions a responding cell has undergone) and the Percent Divided (a measure of the fraction of cells competent to divide) were calculated. The Proliferation Index was higher in the Early Group but this difference did not reach statistical significance (p=0.0522), however, the Percent Divided after PHA was significantly higher in the Early Group compared to the Late Group at day 60 with p=0.0232 (Figure 4C). Therefore, provision of an autologous T cell product at an early time point after the second transplantation provided superior quantitative and qualitative reconstitution of the T cell compartment early post-transplant.
Expansion of T cells could be associated with contraction of the T cell repertoire. We initially analyzed the diversity of the repertoire by spectratyping (29). Unexpectedly, the diversity was impressively restricted in the patients prior to any chemotherapy with over half of the Vβ families missing or having oligoclonal peaks (Figure 5). A previous study of adults, using this technology, found approximately 10% abnormal Vβ families (30). There was a modest increase in the frequency of abnormal Vβ families at days 30 and 60 in the Late Group, suggesting expansion of a small number of founder clones. To better explore the contribution of the infusion to the T cell repertoire, five patients in the Early Group and four patients in the Late Group were examined using next generation sequencing to better identify alterations to the repertoire. We hypothesized that baseline repertoire contraction could be due to expansion of a tumor-specific clone, however, at baseline, there were no T cell receptor (TCR) sequences shared by all patients. For all four patients who had baseline samples available, the number of unique reads was higher in the infusion sample compare to the baseline sample, supporting the previous finding that in vitro expansion using this method preserves polyclonality (15). There were relatively few clones from the infusion that appeared in later time points, < 5.5% in all cases. This is consistent with the small contribution expected from the infusion. Unexpectedly, provision of this small number of cells led to increased repertoire diversity (diminished oligoclonality) (Figure 5). For this analysis, we enumerated the number of clones comprising over 5% of the total reads as a marker of oligoclonality. There were no oligoclonal sequences at baseline, but at Day 30, the Late Group had an average of 4 oligoclonal sequences whereas the Early Group had an average of 0.25 oligoclonal TCR sequences (p=NS). Similarly, when the frequencies of the most common clones were analyzed, the Early Group had a mean frequency of 3.5% while the Late Group had a mean frequency of 13.5% (p=NS). As a final measure of diversity, we enumerated the number of distinct Vβ-J combinations at each time point. The Early Group had significantly more diversity at Day 30 compared to the Late Group (p=0.008, Figure 5).
Figure 5. T cell receptor repertoire analysis.
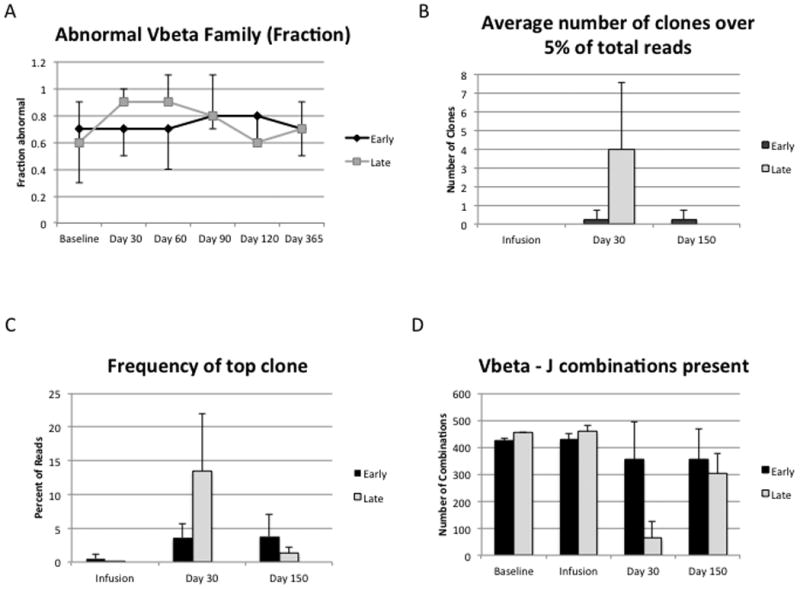
A) The fraction of abnormal (oligoclonal or absent) Vβ families is displayed on the y-axis. A total of 23 Vβ families were examined by PCR analysis (spectratyping). There were no differences between the Early Group and the Late Group, however, the baseline diversity was poor. B) To assess oligoclonality, next generation sequencing was used. As a measure of oligoclonality, the number of clones that represented at least 5% of the reads were enumerated. The infused product itself was polyclonal and oligoclonality in the Late Group seen at day 30 was corrected after infusion at day 90. C) The frequency of the most abundant clone was also assessed as a measure of oligoclonality. The Late Group has a higher mean frequency of the most abundant clone at Day 30 compared to the Early Group, with normalization by Day 150. D) The absolute number of Vβ-J combinations detected across the time points is plotted as a measure of diversity. There are 705 potential Vβ-J combinations. At Day 30, the Early Group has significantly more Vβ-J combinations than the Late Group (p=0.0082).
The suppression of oligoclonality could be due to greater “fitness” of the ex vivo expanded cells or could be due to enhancement of thymic output, such as has been previously suggested (31). Clones that were identified in both Baseline and Day 150 samples comprised an average of 1.1% of the total Day 150 clones and clones that were identified in both the Infusion and the Day 150 samples comprised an average of 0.65% of the total Day 150 clones, suggesting that the final composition of the repertoire was modestly derived from the infused cells. Thymic export of new T cells would be consistent with the naïve phenotype identified by flow cytometry (Figure 3). To explore this, we performed analyses of T cell receptor excision circles (TRECs). Four of nine subjects in the Early Group had increased TRECs at day 30 compared to their baseline, while 1/7 of the Late Group did. At day 150, 5/7 of the Late Group and 2/6 of the Early Group had increased TRECs compared to baseline.
Responses to vaccines
Improved T cell reconstitution could improve B cell function. To analyze this, we examined the response to the influenza vaccine for each of the three serotypes present in the inactivated vaccine. There was wide variability in the Early Group while the Late Group exhibited very little variability. There were no significant differences in the mean titer between the two groups and overall the responses were extremely low. We also calculated the seroconversion frequency at each time point by defining a four-fold increase from the baseline titer. Two patients in the Early Group (n=14) produced a four-fold titer increase after vaccination to all three serotypes. Three additional patients produced a four-fold increase in titer to at least one serotype in the Early Group. In contrast, only one patient in the Late Group (n=8) produced a four-fold response to a single serotype (p value not significant). The low titers at baseline suggest that this population was largely naïve to influenza, which may have compromised their ability to respond to the vaccine. We therefore examined responses to the conjugated pneumococcal vaccine, which the patients should have received as part of their child care prior to developing neuroblastoma.
Pneumococcal responses were evaluated as representative of an antigen previously encountered and a typical memory response (Figure 6). Fourteen serotypes were evaluated: seven contained in the conjugate vaccine and seven serotypes not in the vaccine. There were no responses to the serotypes that are not in the vaccine, as expected. For the serotypes in the vaccine, the baseline titers were higher than the non-vaccine serotypes, consistent with prior vaccination. There was a dichotomous response at the one year time point with the Early Group having significantly higher responses than the Late Group at one year. To determine whether this effect was due to a single serotype, the serotypes were individually evaluated. All serotypes individually were significantly higher at one year in the Early Group compared to the Late Group and all patients exhibited the same pattern. These data support the beneficial effect of the autologous T cell product on host humoral memory responses.
Figure 6. Early T cell reconstitution promotes B cell memory responses following pneumococcal vaccination.
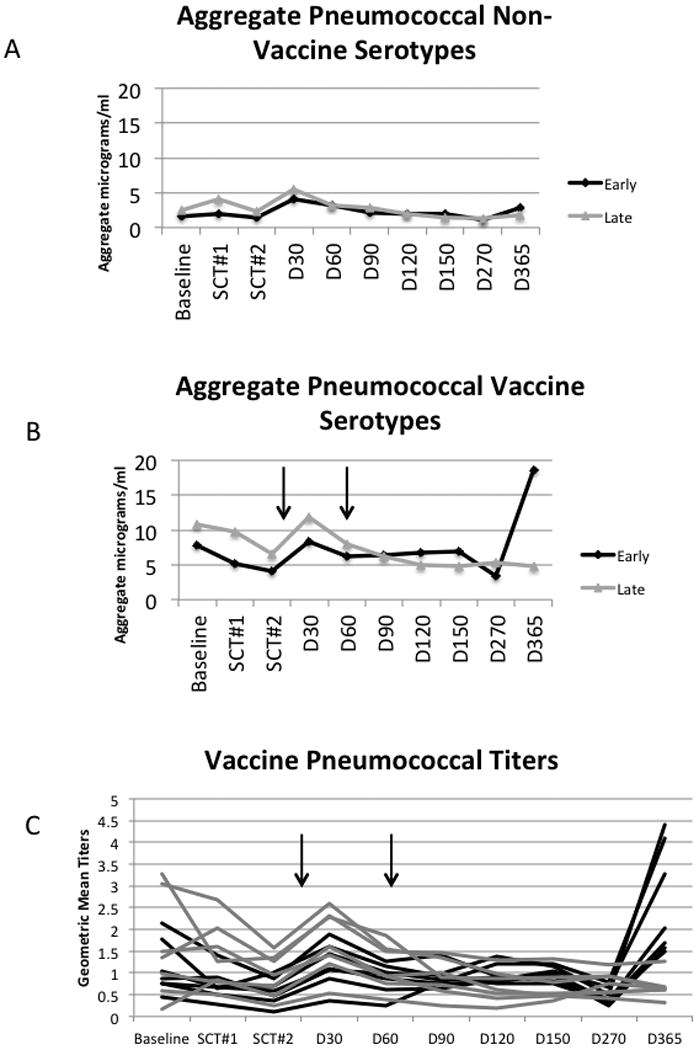
(A,B) Aggregate pneumococcal mean titers were calculated by summing the antibody responses to the seven serotypes not in the vaccine (A) and for the vaccine (B) serotypes (Early Group n=9, Late Group n=7). At baseline, the differences between the vaccine serotypes and non-vaccine serotypes were significant for both the Early Group (Mann-Whitney p=0.020) and the Late Group (p=0.002), confirming prior vaccination. At one year, the difference between the Early Group and the Late Group comparing the vaccine serotypes was significant (Kruskal Wallis p=0.002), while serologic response to non-vaccine serotypes did not differ. The vertical arrows denote the vaccine administration time points. (C) When each serotype was examined separately, the effect at Day 365 was consistent across all vaccine serotypes. The vertical arrows denote the vaccine administration time points. Each black line indicates a single vaccine serotype response and each gray line indicates a single non-vaccine serotype response.
B cell maturation
To better understand how the early autologous T cell infusion led to greater antibody production, we examined the B cell subsets in the peripheral blood at baseline and various time points after transplantation. Neither total B cells nor any of the subsets differed between the Early and Late Groups although B cell maturation appeared to be modestly accelerated in the Early Group (Supplemental Data 3). To examine B cell function, we utilized B cell ELISPOTs, measuring both the total IgG and the influenza-specific IgG produced. There were no differences between the Early Group and the Late Group but responses were universally low using this assay (data not shown).
Discussion
This study was designed to test the efficacy of an adoptive transfer of a co-stimulated, activated autologous T cell product as an approach to regenerate cellular immunity after high dose cytotoxic therapy. Patients who receive a tandem transplant for high-risk neuroblastoma are rendered severely immune deficient for a prolonged period of time (5, 9, 13, 32, 33). Consequences of the immune compromise are viral morbidity and mortality related to infection (13). The provision of an autologous T cell product can provide support during the vulnerable post-transplant period. In adults, autologous T cell products have been utilized with a significant improvement in immunologic function as measured by T cell counts and vaccine responses (18, 34-37). To our knowledge, no studies have tested the regenerative potential of adoptive T cell transfer in non-hematologic malignancies. Furthermore, no studies have assessed this in children where the kinetics of immune reconstitution may differ due to increased thymic potential (38).
This study clearly demonstrated a greater effect the earlier the T cell infusion was administered in the pilot phase although the improvement was greater than what we could account for by the infusion itself. Patients receiving infusions on Day 12 had the least marked T cell reconstitution and we hypothesize that this time point fell between inflammatory cytokine-induced expansion and IL-7-induced expansion. We found evidence of homeostatic expansion of the transferred cells at day 30, supporting a role for this process in expansion and reconstitution in the cytokine-rich, lymphopenic environment (28). There was a significant increase in naïve cells. Although strong homeostatic signals drive a naïve to memory conversion, not all homeostatic expansion is associated with conversion to a memory phenotype and we hypothesize that both thymic production and homeostatic expansion contributed to quantitative reconstitution (39-41). The TREC data were not uniform among subjects but the data were suggestive of the induction of thymopoiesis in some patients after the T cell infusion. Overall, the Early (Day 2) T cell infusions resulted in superior T cell reconstitution as assessed by quantitative and qualitative measures and we believe this will improve the defense against opportunistic infections.
To probe the breadth of improvement in immunologic function, we vaccinated children after their second transplant. We anticipated that most children would be naïve to influenza because of their young age and that nearly all children would have had the conjugated pneumococcal vaccine as part of routine childcare. We noted that the baseline aggregate titers to pneumococcal serotypes were significantly different between the vaccine serotypes and the non-vaccine serotypes, confirming that this cohort was previously vaccinated. There was a modest increase in titers at the Day 30 time point but the most remarkable result was the dramatic increase in vaccine-specific pneumococcal titers at the one-year time point, indicating that long term B cell immunity is dependent on antigen-specific T cell function.
The delayed kinetics were surprising, however, similar studies in adults demonstrated that priming prior to the T cell harvest markedly improves subsequent vaccine responses (18, 19). In those studies, the conditioning regimen did not deplete memory B cells and it could not be determined whether the priming of B cells was critical for the vaccine responses. In this study, the conditioning prior to transplant led to an absence of peripheral blood B cells on Day 2 suggesting that the infused primed T cells were largely responsible for the pneumococcal responses in the Early Group. The Early Group received the vaccines after infusion of the T cells while the Late Group received the vaccines prior to the cell infusion. The presence of primed T cells at the time of vaccination had a salutary if undefined effect on subsequent antibody responses to the vaccine. The delayed kinetics might reflect the recovery kinetics of the B cell compartment, which achieved normal numbers only at the one year time point. The T cells may have had a trophic effect on B cell development (42).
Harnessing the immune system for an anti-tumor effect is a long-term goal of immune reconstitution and infusion of genetically modified T cells expanded using CD3- and CD28-linked beads recently demonstrated dramatic anti-tumor responses (26, 27). The current data suggest that priming is important and a study of adults receiving a similar product also concluded that priming optimized vaccine responses (35). The most important lesson is that the autologous T cell product can provide important immunological support but it must be administered early. These results are also the first indication that the kinetics of immune reconstitution in the pediatric immune system has profound effects on long-term humoral immunity. These results may guide future studies to reconstitute immunity after high dose chemotherapy and perhaps for the induction of anti-tumor immunity.
Supplementary Material
Translational Relevance.
Neuroblastoma is one of the most common solid tumors of childhood. Over half of the high risk patents relapse and the mortality rate is high with multimodal therapy. Children can tolerate sequential or tandem hematopoietic stem cell transplants but morbidity and mortality are higher for tandem transplants than single transplants, largely due to prolonged immune suppression. This study examined the use of an autologous T cell infusion to support children who undergo tandem transplants for neuroblastoma. T cell infusions were demonstrated to increase T cell counts as expected, however, multiple measures of T cell function and B cell function were improved in a group that received their T cell infusions early after transplant. This study not only demonstrates the promise of autologous T cell infusion support after transplantation but also demonstrates there is critical time window to maximize the effect.
Acknowledgments
The authors would like to acknowledge the expert technical contributions of Kelly Maurer, the staff at the Human Immunology Core facility, the staff of the Clinical Cell and Vaccine Production Facility (Julio Cotte, Zhaohui Zheng, Andrea Brennan), Yang-Zhu Du, Noah Goodman, and Xiaoling Hou. We also wish to acknowledge the nurses and physicians caring for these patients as well as Dan Schullery for his organizational contributions.
Grant Support: This work was supported in part by NIH NO1-AI-50 024 and R01-CA105216.
Footnotes
Conflict of interest: C.H.J. and B. L. L. are inventors on patents related to the T-cell manufacturing process that was used for this protocol. This conflict has been disclosed and was managed in accordance with the policies of the University of Pennsylvania. No other authors have any conflict of interest to disclose.
References
- 1.Brodeur G, Maris J. Neuroblastoma. In: Pizzo P, Poplack D, editors. Principles and practice of pediatric oncology. 4th Edition ed. Philadelphia: Lippincott Williams and Wilkins; 2002. [Google Scholar]
- 2.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 3.Howlader N, Noone A, Krapcho M, Neyman N, Aminou R, Waldron W, et al. National Cancer Institute; 2011. SEER Cancer Statistics Review 1975-2008. http://seer.cancer.gov/csr/1975_2008/ [Google Scholar]
- 4.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–79. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 5.Fish JD, Grupp SA. Stem cell transplantation for neuroblastoma. Bone Marrow Transplant. 2008;41:159–65. doi: 10.1038/sj.bmt.1705929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu A, Gilman A, Ozkaynak M, London W, Kreissman S, Chen H, et al. Anti-GD2 antibody with GM-CSF, Interleukin-2 and isotretinoin for Neuroblastoma. The New England Journal of Medicine. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip T, Ladenstein R, Lasset C, Hartmann O, Zucker JM, Pinkerton R, et al. 1070 myeloablative megatherapy procedures followed by stem cell rescue for neuroblastoma: 17 years of European experience and conclusions. European Group for Blood and Marrow Transplant Registry Solid Tumour Working Party. Eur J Cancer. 1997;33:2130–5. doi: 10.1016/s0959-8049(97)00324-9. [DOI] [PubMed] [Google Scholar]
- 8.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med. 1999;341:1165–73. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 9.Grupp SA, Stern JW, Bunin N, Nancarrow C, Adams R, Gorlin JB, et al. Rapid-sequence tandem transplant for children with high-risk neuroblastoma. Med Pediatr Oncol. 2000;35:696–700. doi: 10.1002/1096-911x(20001201)35:6<696::aid-mpo46>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Grupp SA, Stern JW, Bunin N, Nancarrow C, Ross AA, Mogul M, et al. Tandem high-dose therapy in rapid sequence for children with high-risk neuroblastoma. J Clin Oncol. 2000;18:2567–75. doi: 10.1200/JCO.2000.18.13.2567. [DOI] [PubMed] [Google Scholar]
- 11.Kletzel M, Katzenstein HM, Haut PR, Yu AL, Morgan E, Reynolds M, et al. Treatment of high-risk neuroblastoma with triple-tandem high-dose therapy and stem-cell rescue: results of the Chicago Pilot II Study. J Clin Oncol. 2002;20:2284–92. doi: 10.1200/JCO.2002.06.060. [DOI] [PubMed] [Google Scholar]
- 12.George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, Shamberger RC, et al. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:2891–6. doi: 10.1200/JCO.2006.05.6986. [DOI] [PubMed] [Google Scholar]
- 13.Powell JL, Bunin NJ, Callahan C, Aplenc R, Griffin G, Grupp SA. An unexpectedly high incidence of Epstein-Barr virus lymphoproliferative disease after CD34+ selected autologous peripheral blood stem cell transplant in neuroblastoma. Bone Marrow Transplant. 2004;33:651–7. doi: 10.1038/sj.bmt.1704402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine BL, Cotte J, Small CC, Carroll RG, Riley JL, Bernstein WB, et al. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. Journal of Hematotherapy. 1998;7:437–48. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- 15.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–30. [PubMed] [Google Scholar]
- 16.Garlie NK, LeFever AV, Siebenlist RE, Levine BL, June CH, Lum LG. T cells coactivated with immobilized anti-CD3 and anti-CD-28 as potential immunotherapy for cancer. J Immunother. 1999;22:336–45. doi: 10.1097/00002371-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nature Biotechnology. 2002;20:143–8. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 18.Stadtmauer EA, Vogl DT, Luning Prak E, Boyer J, Aqui NA, Rapoport AP, et al. Transfer of influenza vaccine-primed costimulated autologous T cells after stem cell transplantation for multiple myeloma leads to reconstitution of influenza immunity: results of a randomized clinical trial. Blood. 2011;117:63–71. doi: 10.1182/blood-2010-07-296822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rapoport AP, Stadtmauer EA, Aqui N, Vogl D, Chew A, Fang HB, et al. Rapid immune recovery and graft-versus-host disease-like engraftment syndrome following adoptive transfer of Costimulated autologous T cells. Clin Cancer Res. 2009;15:4499–507. doi: 10.1158/1078-0432.CCR-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutter JA, Kwan-Morley J, Dunham J, Du YZ, Kamoun M, Albert D, et al. A longitudinal analysis of SLE patients treated with rituximab (anti-CD20): factors associated with B lymphocyte recovery. Clin Immunol. 2008;126:282–90. doi: 10.1016/j.clim.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Levin MJ, Song LY, Fenton T, Nachman S, Patterson J, Walker R, et al. Shedding of live vaccine virus, comparative safety, and influenza-specific antibody responses after administration of live attenuated and inactivated trivalent influenza vaccines to HIV-infected children. Vaccine. 2008;26:4210–7. doi: 10.1016/j.vaccine.2008.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weng N-P, Levine BL, June CH, Hodes RJ. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc Natl Acad Sci U S A. 1995;92:11091–4. doi: 10.1073/pnas.92.24.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nature Medicine. 2011;17:1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Mascio M, Paik CH, Carrasquillo JA, Maeng JS, Jang BS, Shin IS, et al. Noninvasive in vivo imaging of CD4 cells in simian-human immunodeficiency virus (SHIV)-infected nonhuman primates. Blood. 2009;114:328–37. doi: 10.1182/blood-2008-12-192203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Condomines M, Veyrune JL, Larroque M, Quittet P, Latry P, Lugagne C, et al. Increased plasma-immune cytokines throughout the high-dose melphalan-induced lymphodepletion in patients with multiple myeloma: a window for adoptive immunotherapy. J Immunol. 184:1079–84. doi: 10.4049/jimmunol.0804159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorski J, Yassai M, Zhu X, Kissela B, Kissella B, Keever C, et al. Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 size spectratyping. Correlation with immune status. J Immunol. 1994;152:5109–19. [PubMed] [Google Scholar]
- 30.Zemble R, Luning Prak ET, McDonald K, McDonald-McGinn DM, Zakcai E, Sullivan KE. Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/valocardiofacial syndrome) Clinical Immunology. 2010 doi: 10.1016/j.clim.2010.04.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochberg EP, Chillemi AC, Wu CJ, Neuberg D, Canning C, Hartman K, et al. Quantitation of T-cell neogenesis in vivo after allogeneic bone marrow transplantation in adults. Blood. 2001;98:1116–21. doi: 10.1182/blood.v98.4.1116. [DOI] [PubMed] [Google Scholar]
- 32.Hobbie WL, Moshang T, Carlson CA, Goldmuntz E, Sacks N, Goldfarb SB, et al. Late effects in survivors of tandem peripheral blood stem cell transplant for high-risk neuroblastoma. Pediatr Blood Cancer. 2008;51:679–83. doi: 10.1002/pbc.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, Shamberger RC, et al. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. J Clin Oncol. 2006;24:2891–6. doi: 10.1200/JCO.2006.05.6986. [DOI] [PubMed] [Google Scholar]
- 34.Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004–13. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 35.Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–7. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 36.Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119:188–94. doi: 10.1016/j.clim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Levine BL, Bernstein WB, Aronson NE, Schlienger K, Cotte J, Perfetto S, et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002;8:47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 38.Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. The New England journal of medicine. 1995;332:143–9. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 39.Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13306–11. doi: 10.1073/pnas.96.23.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. The Journal of experimental medicine. 2000;192:557–64. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Onoe T, Kalscheuer H, Chittenden M, Zhao G, Yang YG, Sykes M. Homeostatic expansion and phenotypic conversion of human T cells depend on peripheral interactions with APCs. Journal of immunology. 2010;184:6756–65. doi: 10.4049/jimmunol.0901711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choudhury A, Cohen PL, Eisenberg RA. B cells require “nurturing” by CD4 T cells during development in order to respond in chronic graft-versus-host model of systemic lupus erythematosus. Clin Immunol. 2010;136:105–15. doi: 10.1016/j.clim.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.