

Abstract
Using a commercially available Umemoto's reagent, the metal-free trifluoromethylation of nitroalkanes is now possible. This method provides a general, high yielding synthesis of α-trifluoromethylnitroalkanes. The quaternary α-trifluoromethylnitroalkanes obtained from this transformation can be elaborated to a variety of complex nitrogen-containing molecules, including α-trifluoromethylamines.
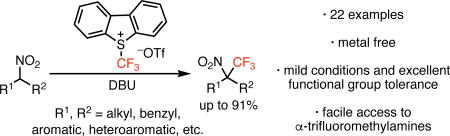
Organofluorine compounds play a vital role in the chemical enterprise, including in pharmaceuticals, agrochemicals, liquid crystals, dyes, and polymers.1 Trifluoromethyl groups in particular have been shown to impart unique physiological properties, including modulation of binding affinity, metabolic stability, lipophilicity, and bioavailability when introduced into small molecules.2 For example, the introduction of a trifluoromethyl group alpha to nitrogen has been shown to modulate the biological properties of numerous small molecules compared to their nonfluorinated analogs.3
A potentially efficient entry into such α-trifluoromethylamino compounds would involve the trifluoromethylation of a nitroalkane.4 In 2007, Togni reported that α-nitroesters can be trifluoromethylated in reasonable yields under copper catalysis (Figure 1, top).5 Unfortunately, this method is not applicable to nitroalkanes lacking the adjacent activating ester group. A general protocol for the trifluoromethylation of nitroalkanes has not yet been described.6
Figure 1.

Trifluoromethylation of Nitroalkanes
Recent studies from our group have demonstrated a variety of reactions for the alkylation of nitroalkanes using copper catalysis and radical-stabilizing alkyl halide electrophiles.7 Given the variety of recent examples of transformations involving trifluoromethyl radicals,8 and the broad utility of nitroalkanes,9 we were inspired to investigate the trifluoromethylation of nitroalkanes as a potential entry into α-trifluoromethylamino compounds. Herein we report simple, transition-metal free conditions for trifluoromethylation of secondary nitroalkanes. These conditions provide high-yielding access to fully substituted α-trifluoromethylnitroalkanes, which can be readily converted into the corresponding α-trifluoromethylamines.
In analogy to our prior studies, our initial efforts focused on the use of copper catalysts in combination with a variety of reagents known to generate trifluoromethyl radicals.10 Using nitroalkane 1 as a model substrate, we were initially pleased to find that the combination of catalytic CuBr and a diketiminate ligand with Umemoto’s reagent (2) and base led to detectable levels of the desired product 3 (Table 1, entry 1).11 Control experiments, however, quickly revealed that the reaction did not require the catalytic additives (entry 2). Switching the base from sodium trimethylsilanolate to DBU increased the yield to 52% (entry 3). The reaction proved most efficient when conducted in methylene chloride.10 Finally, lowering the temperature from 40 °C to −25 °C afforded optimal amounts of the desired product 3 (entries 3–5).
Table 1.
Optimization of Reaction Conditions
| ||||
|---|---|---|---|---|
|
| ||||
| entry | base | additive | temp (°C) | yield 3 (%)a |
| 1 | NaOSiMe3 | 20 mol % Cu/Lb | 40 | 22 |
| 2 | NaOSiMe3 | none | 40 | 24 |
| 3 | DBU | none | 40 | 52 |
| 4 | DBU | none | rt | 58 |
| 5 | DBU | none | −25 | 90 |
1.3 equiv 2; yields determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
20 mol % CuBr, 20 mol % bis-N,N′-(2,6-dimethylphenyl)-2,4-diiminopentane added to reaction.
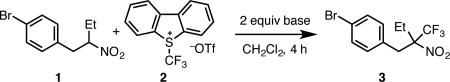
With optimized conditions in hand, the scope of the transformation was investigated (Scheme 1). The reaction is general for a broad range of secondary nitroalkanes. The model substrate was isolated in 83% yield (3).7a Other homobenzylic nitroalkanes (4) led to similar results. Both benzylic substrates (5 and 6)12 and Michael reaction adducts (e.g., 9, 10, and 11) were also well tolerated. Sterically demanding substrates could also be used. For example, even neopentylic substrates led to appreciable yield of products (14) containing vicinal fully substituted centers. In contrast to secondary substrates, primary nitroalkanes provide very little reactivity. For example, only traces of 13 were observed. Further studies will be directed at expanding the scope of the reaction to primary nitroalkanes.
Scheme 1.

Scope of the trifluoromethylation of secondary nitroalkanes.
a Isolated yields unless otherwise noted. Diastereomeric ratios (dr) determined by 1H NMR analysis of crude reaction. b 1.5 equiv 2 used. c 18 h. d Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. e 48 h. f 24 h.
Significantly, nitroalkanes bearing a tertiary stereocenter beta to the nitro group proved to be excellent substrates.7b In these cases, good to excellent levels of diastereoselection were observed. For example, amide 15 was formed with greater that >95:5 selectivity favoring the diastereomer shown.13 Similar selectivity was observed for the Weinreb amides (16). Related ester products could also be prepared (17, 18, and 19), albeit with slightly lower levels of diastereoselection. These results mirror the selectivities previously observed in Michael additions of β-nitrocarbonyls.14 Henry reaction products (20),9a as well as those from conjugate addition of nitroalkenes (21),15 could both be trifluoromethylated with good to excellent levels of diastereoselectivity. In the latter case, stereoselectivity is consistent with addition of the CF3 group away from the large aromatic ring.
The functional group tolerance of the reaction is very high. In addition to those already mentioned, tolerated functional groups include aryl halides (3 and 15), heterocycles (4, 7, 8, and 20), alkenes (10), aryl ethers (5), nitriles (9), ketones (6), protected and free alcohols (7, 17, 18, and 20), sulfones (11), and protic nitrogen function groups (14 and 15).
The method does show some limitations with respect to nitroalkanes bearing acidic and sterically accessible β-protons. In such cases, elimination of an equivalent of nitrous acid from the trifluoromethylated product can be observed. For example, under standard conditions using DBU as base, reaction of 22 did not lead to the trifluoromethyl nitroalkane 23 (Scheme 2, top). Instead, the trifluormethyl alkene 24 was observed in moderate yield. In some cases, the use of the bulkier base, tetramethylguanidine (TMG), enabled access to the desired product without significant elimination - albeit with less than ideal conversion and yield. In other cases, such as with ester 25, elimination could not be avoided regardless of the base used (Scheme 2, bottom).
 |
(1) |
Scheme 2.

Competitive alkene formation and role of base.
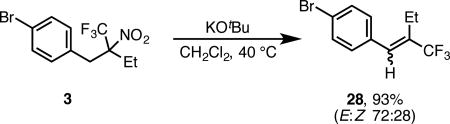
Interestingly, the trifluoromethylalkenes described in Scheme 2 all formed with significant selectivity for the E-isomer (as determined by 1H-19F HOSEY NMR).10 We attribute this selectivity to the larger steric size of the CF3 group compared to an n-alkyl group.2f Recognizing the possible utility of this process for preparing trifluoromethyl alkenes,16 we investigated if this base promoted process can be triggered in less acidic products. Using substrate 3 as a model system, we found that exposure to KOtBu at 40 °C led to nearly quantitative yield of corresponding vinyl trifluoromethylalkene 28 with modest E:Z selectivity (eq 1). This method potentially provides a mild, high yielding, two step synthesis of vinyl trifluoromethylalkenes from a variety of complex nitroalkanes.
Trifluoromethylnitroalkanes are readily reduced to α-trifluoromethylamines. As shown in Scheme 3 (top and middle), both Zn/AcOH reduction and hydrogenolysis can be effective. However, we note that with α-aryl nitroalkanes, which are prone to denitration,17 hydrogenolysis is the preferred method for reduction (Scheme 3, bottom).
 |
(2) |
Scheme 3.

Preparation of α-trifluoromethylamines.
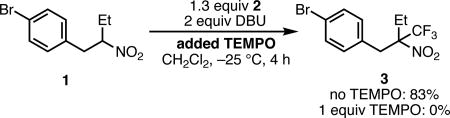
Consistent with our earlier results,7 preliminary mechanistic studies suggest that the trifluoromethylation reaction proceeds via a radical mechanism. When the radical inhibitor TEMPO is introduced into the reaction, no desired trifluoromethylated product was observed (eq 2). Further, in situ 1H NMR studies in CD2Cl2 have revealed many of the details of the reaction mechanism. First, combining DBU and nitroalkane 1 at low temperature reveals that a significant equilibrium concentration of nitronate anion 32 is produced, and that the deprotonation is relatively slow (it takes about 10 min for a 2:1 mixture of DBU and 1 to reach equilibrium at −25 °C). Second, when DBU and 2 are combined at −25 °C, 2 is instantly consumed and a new complex bearing related aromatic signals is produced. Prior studies have shown that 2 forms electron-donor-acceptor (EDA) complexes with basic amines,18 and we have tentatively assigned this as the EDA complex 2·DBU. Third, monitoring the trifluoromethylation reaction by 1H NMR of 1 under slightly modified conditions (−25 °C, half optimal concentration) reveals an initially fast rate of production of 3 that slows considerably as the reaction progresses. Under these conditions, two reactive intermediates are observed. The first, which is maximally present at the first observation point (ca. 2 min) and then decays as the reaction proceeds, has signals that match 2·DBU. The second builds in early in the reaction and then decays as the reaction progresses. This complex bears 1H NMR signals that are related both to 1 and 2. We tentatively assign this as the associated ion pair 33.19
Based upon these observations we propose the following reaction mechanism (Figure 2). Early in the reaction, DBU and 2 form the EDA complex 2·DBU. As the nitronate anion 32 is formed, 2·DBU is consumed and the ion pair 33 is formed. The salt complex 33 then undergoes slow decomposition to a nitronate radical 34, CF3-radical, and dibenzothiophene via electron transfer. Rapid recombination of the two radicals results in the formation of the observed product 3.20,21
Figure 2.

Proposed mechanism for nitroalkane trifluoromethylation.
In conclusion, we have developed mild reaction conditions for the trifluoromethylation of secondary nitroalkanes using a commercially available trifluoromethylating reagent. This procedurally simple protocol allows rapid access to highly complex quaternary α–trifluoromethylnitroalkanes in good yields and diastereoselectivity. The wide functional group tolerance highlights the power of this transformation as a method for late-stage installation of a trifluoromethyl group. In addition, we have demonstrated that these products can be reduced to medicinally interesting α-trifluoromethylamines. Finally, we have also shown that, in at least some cases, base-induced elimination of HNO2 allows the products to be converted to highly substituted trifluoromethylalkenes with good levels of stereocontrol. Further studies will be aimed at expanding the scope of these transformations.
Supplementary Material
Acknowledgments
The University of Delaware (UD), the University of Delaware Research Foundation, the Research Corp. Cottrell Scholars Program, and the NIH NIGMS (R01 GM102358) are gratefully acknowledged for support. Dr. Peter Gildner (UD) is acknowledged for initial experiments in this area, as are Dr. Glenn Yap (UD) and Dr. Shi Bai (UD) for assistant with X-ray crystallography and NMR analysis. Data was acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF CHE0421224, CHE0840401, CHE1229234, and CHE1048367; NIH S10 OD016267-01, S10 RR026962-01, P20GM104316, P30GM110758)
Footnotes
ASSOCIATED CONTENT
Experimental procedures, crystallographic and spectral data. The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
References
- 1.Kirsch DP. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications. Wiley-VCH; Weinheim: 2013. [Google Scholar]
- 2.a Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; b Kirk KL. Org. Proc. Res. Dev. 2008;12:305–321. [Google Scholar]; c Hagmann WK. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; d Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; e Schlosser M. Angew. Chem. Int. Ed. 2006;45:5432–5446. doi: 10.1002/anie.200600449. [DOI] [PubMed] [Google Scholar]; f Smart BE. J. Fluorine Chem. 2001;109:3–11. [Google Scholar]
- 3.a Lim J, Taoka B, Otte RD, Spencer K, Dinsmore CJ, Altman MD, Chan G, Rosenstein C, Sharma S, Su H-P, Szewczak AA, Xu L, Yin H, Zugay-Murphy J, Marshall CG, Young JR. J. Med. Chem. 2011;54:7334–7349. doi: 10.1021/jm200909u. [DOI] [PubMed] [Google Scholar]; b Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong LT, Falgueyret J-P, Kimmel DB, Lamontagne S, Léger S, LeRiche T, Li CS, Massé F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Thérien M, Truong V-L, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC. Bioorg. Med. Chem. Lett. 2008;18:923–928. doi: 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]; c Black WC, Bayly CI, Davis DE, Desmarais S, Falgueyret J-P, Léger S, Li CS, Massé F, McKay DJ, Palmer JT, Percival MD, Robichaud J, Tsou N, Zamboni R. Bioorg. Med. Chem. Lett. 2005;15:4741–4744. doi: 10.1016/j.bmcl.2005.07.071. [DOI] [PubMed] [Google Scholar]; d Dal Pozzo A, Ni M, Muzi L, de Castiglione R, Mondelli R, Mazzini S, Penco S, Pisano C, Castorina M, Giannini G. J. Med. Chem. 2006;49:1808–1817. doi: 10.1021/jm0511334. [DOI] [PubMed] [Google Scholar]; e Ojima I, Slater JC, Pera P, Veith JM, Abouabdellah A, Bégué J-P, Bernacki RJ. Bioorg. Med. Chem. Lett. 1997;7:133–138. [Google Scholar]
- 4.Electrophiliic fluoronitration of difluoroalkenes has previously been shown to allow limited access to these compounds, see: Knunyants IL, German LS, Rozhkov IN. Russ. Chem. Bull. 1963;12:1794–1797.
- 5.a Kieltsch I, Eisenberger P, Togni A. Angew. Chem. Int. Ed. 2007;46:754–757. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; b Charpentier J, Früh N, Togni A. Chem. Rev. 2015;115:650–682. doi: 10.1021/cr500223h. [DOI] [PubMed] [Google Scholar]
- 6.Also see: Umemoto T, Kuriu Y. Tetrahedron Lett. 1981;22:5197–5200.Feiring AE. J. Org. Chem. 1983;48:347–354.
- 7.a Gildner PG, Gietter AAS, Cui D, Watson DA. J. Am. Chem. Soc. 2012;134:9942–9945. doi: 10.1021/ja304561c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gietter AAS, Gildner PG, Cinderella AP, Watson DA. Org. Lett. 2014;16:3166–3169. doi: 10.1021/ol5014153. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shimkin KW, Gildner PG, Watson DA. Org. Lett. 2016;18:988–991. doi: 10.1021/acs.orglett.6b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Studer A. Angew. Chem. Int. Ed. 2012;51:8950–8958. doi: 10.1002/anie.201202624. [DOI] [PubMed] [Google Scholar]; b Barata-Vallejo S, Lantaño B, Postigo A. Chem. - Eur. J. 2014;20:16806–16829. doi: 10.1002/chem.201404005. [DOI] [PubMed] [Google Scholar]; c Alonso C, Martínez de Marigorta E, Rubiales G, Palacios F. Chem. Rev. 2015;115:1847–1935. doi: 10.1021/cr500368h. [DOI] [PubMed] [Google Scholar]; d Wang S-M, Han J-B, Zhang C-P, Qin H-L, Xiao J-C. Tetrahedron. 2015;71:7949–7976. [Google Scholar]
- 9.a Ono N. The Nitro Group In Organic Synthesis. John Wiley And Sons; New York: 2001. [Google Scholar]; b Ooi T, Takada S, Doda K, Maruoka K. Angew. Chem. Int. Ed. 2006;45:7606–7608. doi: 10.1002/anie.200602787. [DOI] [PubMed] [Google Scholar]; c Dobish MC, Johnston JN. Org. Lett. 2010;12:5744–5747. doi: 10.1021/ol1025712. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Grenning AJ, Tunge JA. Org. Lett. 2010;12:740–742. doi: 10.1021/ol902828p. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Noole A, Lippur K, Metsala A, Lopp M, Kanger T. J. Org. Chem. 2010;75:1313–1316. doi: 10.1021/jo902664v. [DOI] [PubMed] [Google Scholar]; f Noble A, Anderson JC. Chem. Rev. 2013;113:2887. doi: 10.1021/cr300272t. [DOI] [PubMed] [Google Scholar]; g Qian H, Yu X, Zhang J, Sun J. J. Am. Chem. Soc. 2013;135:18020–18023. doi: 10.1021/ja409080v. [DOI] [PubMed] [Google Scholar]; h Li J, Lear MJ, Kawamoto Y, Umemiya S, Wong AR, Kwon E, Sato I, Hayashi Y. Angew. Chem. Int. Ed. 2015;54:12986–12990. doi: 10.1002/anie.201505192. [DOI] [PubMed] [Google Scholar]; i Manna MS, Mukherjee S. J. Am. Chem. Soc. 2015;137:130–133. doi: 10.1021/ja5117556. [DOI] [PubMed] [Google Scholar]; j Schwieter KE, Johnston JN. Chem. Commun. 2016;52:152–155. doi: 10.1039/c5cc08415f. [DOI] [PubMed] [Google Scholar]; k Vara BA, Johnston JN. J. Am. Chem. Soc. 2016;138:13794–13797. doi: 10.1021/jacs.6b07731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.See Supporting Information.
- 11.a Umemoto T, Ishihara S. J. Am. Chem. Soc. 1993;115:2156–2164. [Google Scholar]; b Zhang C. Org. Biomol. Chem. 2014;12:6580–6589. doi: 10.1039/c4ob00671b. [DOI] [PubMed] [Google Scholar]
- 12.a Vogl EM, Buchwald SL. J. Org. Chem. 2001;67:106–111. doi: 10.1021/jo010953v. [DOI] [PubMed] [Google Scholar]; b Walvoord RR, Kozlowski MC. J. Org. Chem. 2013;78:8859–8864. doi: 10.1021/jo401249y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The relative configuration of 15 was established by single crystal X-ray analysis.
- 14.Gietter-Burch AAS, Mitrut RE, Watson DA. Org. Lett. 2015;17:5468–5471. doi: 10.1021/acs.orglett.5b02832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi T, Senda T, Ogasawara M. J. Am. Chem. Soc. 2000;122:10716–10717. [Google Scholar]
- 16.For recent examples of their preparation see: Kathiravan S, Nicholls IA. Org. Lett. 2015;17:1874–1877. doi: 10.1021/acs.orglett.5b00551.Ramachandran PV, Mitsuhashi W. Org. Lett. 2015;17:1252–1255. doi: 10.1021/acs.orglett.5b00235.
- 17.Fessard TC, Motoyoshi H, Carreira EM. Angew. Chem. Int. Ed. 2007;46:2078–2081. doi: 10.1002/anie.200604263. [DOI] [PubMed] [Google Scholar]
- 18.a Cheng Y, Yuan X, Ma J, Yu S. Chem. - Eur. J. 2015;21:8355–8359. doi: 10.1002/chem.201500896. [DOI] [PubMed] [Google Scholar]; b Cheng Y, Yu S. Org. Lett. 2016;18:2962–2965. doi: 10.1021/acs.orglett.6b01301. [DOI] [PubMed] [Google Scholar]
- 19.A similar reaction profile is observed at ∓25 °C under optimized reaction conditions however the reaction is too fast to adequately observed by NMR over the first half-life and is complicated by initial heterogeneity during the first mins of the reactions. See Supporting Information.
- 20.Radical substitution mechansims are well precedented for nitro compounds. For an early example see: Kornblum N, Swiger RT, Earl GW, Pinnick HW, Stuchal FW. J. Am. Chem. Soc. 1970;92:5513–5514.
- 21.We cannot exclude the possibilty of an alternative radical chain mechanism.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.