

Abstract
Novel N,N-dialkyl carboxy coumarins have been synthesized as potential anticancer agents via inhibition of monocarboxylate transporter 1 (MCT1). These coumarin carboxylic acids have been evaluated for their in vitro MCT1 inhibition, MTT cancer cell viability, bidirectional Caco-2 cell permeability, and stability in human and liver microsomes. These results indicate that one of the lead candidate compounds 4a has good absorption, metabolic stability, and a low drug efflux ratio. Systemic toxicity studies with lead compound 4a in healthy mice demonstrate that this inhibitor is well tolerated based on zero animal mortality and normal body weight gains compared to the control group. In vivo tumor growth inhibition studies in mice show that the candidate compound 4a exhibits significant single agent activity in MCT1 expressing GL261-luc2 syngraft model but doesn’t show significant activity in MCT4 expressing MDA-MB-231 xenograft model, indicating the selectivity of 4a for MCT1 expressing tumors.
Keywords: N,N-Dialkyl carboxy coumarin; Tumor glycolysis; Monocarboxylate transporter 1; MCT; Glioblastoma; GL261-luc2; Triple negative breast cancer; MDA-MB-231
Numerous cancer chemotherapeutics have been developed over the past 50 years to improve the quality of patient care and overall survival rate. However, cancer continues to be one of the major killers throughout the world. Suboptimal efficacy, unacceptable side effects, and development of chemo-resistance are some of the reasons for treatment failure and patient mortality. Hence, new candidate compounds with novel mechanisms of action, low side effects, and that work on all stages of tumors are urgently needed.
Recent studies indicate that alteration in cellular metabolism is a crucial hallmark of cancer development.1,2 Therefore, tumor metabolism is an attractive target for developing new cancer therapeutics. 3,4 The metabolic properties of cancer cells differ significantly from those of normal cells. In normal differentiated tissues, cellular energy is generated mainly via an efficient oxidative phosphorylation (OxPhos). In contrast, cancer cells pursue vigorous glycolysis even in the presence of sufficient amounts of oxygen (Warburg effect, WE).5–10 Glycolysis generates only two moles of ATP per one mole of glucose compared to OxPhos, which can produce up to 38 moles of ATP. To compensate this energy inefficiency, cancer cells upregulate glycolytic enzymes to keep up with the energy requirements of cell proliferation and tumor progression. Maintaining a high level of glycolytic activity is essential for survival, fueling anabolic pathways, tumor advancement, resistance to apoptosis, and invasion and metastasis of cancer cells.5–11
Because cancer cells are mainly dependent on glycolysis for their survival and propagation, they need to export the glycolysis end products pyruvic and lactic acids to avoid cytoplasmic acidification that may lead to apoptosis. To accomplish this task, cancer cells upregulate proton-coupled membrane proteins called monocarboxylate transporters (MCTs), which are responsible for transmembrane shuttling of small carboxylates such as lactate, pyruvate, and ketone bodies.12–18 There are 14 known isoforms of MCTs, but MCTs 1–4 are responsible for transporting these carboxylates. They are also implicated in influx and efflux of lactate by cancer-associated stromal fibroblasts and epithelial cancer cells for energy generation.19–22 Elevated expression of MCT1 has been identified in a large number of cancers and, therefore, this transporter is a major selective target for broad-spectrum cancer treatment.23–34 In this regard, we recently developed a series of MCT1 inhibitors based on the α-hydroxy-4-cyanocinnamic acid (CHC) template for potential anticancer applications.23 Although our earlier CHC based MCT1 inhibitors are potent, the lead compounds suffer from poor oral bioavailability and toxicity at high concentrations. In this regard, we envisioned to utilize a similar but bicyclic coumarin template to improve absorption, distribution, metabolism and elimination properties, decrease systemic toxicity and improve anticancer efficacy. Coumarin is a key structural unit with good pharmaceutical properties that is found in many important medicinal molecules.35–37 Structure-activity relationship studies using our first generation CHC template indicated that placing N,N-dialkyl/diaryl groups at the 4-position demonstrated the most optimized structural moiety for potent MCT1 inhibition. Therefore, we synthesized carboxy coumarins with N, N-dialkyl substitution at the 7-position as second generation MCT1 inhibitors (Fig. 1).25
Figure 1.

CHC and coumarin derivatives.
Here, we report on the pre-clinical evaluation of novel coumarin carboxylic acids. Included is the synthesis, in vitro MCT1 inhibition, cytotoxicity against two cancer cell lines, in vitro Caco-2 permeability and metabolic stability in mouse and human liver microsomes. Systemic toxicity studies in CD-1 mice, in vivo anticancer efficacy in glioblastoma GL261-luc2 syngraft and MDA-MB-231-luc xenograft mouse models are also reported.
We synthesized N,N-dialkyl carboxy coumarins starting from the alkylation of o-aminophenol 1 to obtain N,N-dialkylated-o-aminophenols 2. These alkylated aminophenols were formylated under Vilsmeier Haack conditions with POCl3 and DMF to obtain the corresponding aldehydes 3 in 60–70% yields. The aldehydes 3, upon Knoevenagel condensation with diethyl malonate, followed by the treatment with aqueous NaOH at 100 °C and adjusting pH to 7 at room temperature provided carboxy coumarins 4a–4e (Scheme 1).38
Scheme 1.

Synthesis of 7-(dialkylamino)-2-oxo-2H-chromene-3-carboxylic acid. (a) alkyl bromide, K2CO3, DMSO or EtOH, 80 °C, 12 h; (b) POCl3, DMF, 0 °C to 80 °C, 2–4 h; (c) (i) diethyl malonate, piperidine, CH3COOH, EtOH, 80 °C, 8–12 h (ii) 10% NaOH, 100 °C, 2 h (iii) 3 M HCl, pH 7.0; reported yields are from the reaction of 3 to 4.
We then carried out the MCT1 inhibition study of 4a–4e using L-[14C]-lactate uptake on rat brain endothelial-4 cells that predominantly express MCT1.23 This transport inhibition study revealed carboxy coumarins 4a–4e as potent inhibitors of MCT1 at nanomolar to low micromolar concentrations (Table 1).
Table 1.
MCT1 IC50 * (μM) values of amino carboxy coumarins
| Compounds | MCT1 IC50 in μM |
|---|---|
| 4a | 0.09 ± 0.01 |
| 4b | 0.38 ± 0.08 |
| 4c | 0.45 ± 0.05 |
| 4d | 0.17 ± 0.04 |
| 4e | 0.21 ± 0.02 |
Average ± SEM of minimum three separate experimental values.
Encouraged by excellent inhibition of MCT1, we then evaluated cytotoxicity of compounds 4a–4e in two cancer cell lines that are known to express either MCT1 or MCT4. For this purpose, a predominantly MCT1 expressing cell line, GL261-luc2, and an MCT4 expressing cell line, MDA-MB-231 were chosen (Fig. 2).39 GL261 is an important and a well-established mouse glioblastoma (GBM) cell line that closely mimics human gliomas.40,41 MDA-MB-231 is a triple negative breast cancer cell line derived from human tissue.
Figure 2.
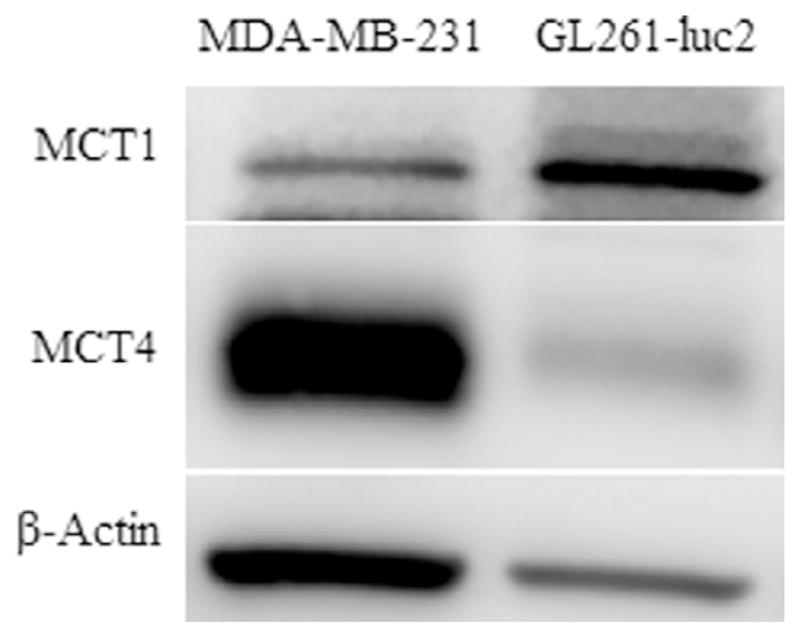
Western blot analysis of MCT1 and MCT4 expression in GL261-luc2 and MDA-MB-231 cell lines.
To determine in vitro toxicity, we evaluated cell viability using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.42,43 This colorimetric assay measures the reduction of MTT to formazan by cellular mitochondrial reductase as a measure of cell viability. Compounds 4a–4e were found to be generally non-cytotoxic at high concentrations as can be expected from MCT inhibitors (Table 2). These results suggest that under in vitro conditions, the cancer cells are able to adapt their metabolism to other pathways to maintain viability. Furthermore, it was reported that potent inhibition of MCT1 did not have any appreciable cytotoxic properties for several solid tumor cell lines.44 In contrast, in in vivo systems where tumor hypoxia is prevalent, cancer cells are dependent on ATP generation via vigorous glycolysis for essential metabolites, survival, and proliferation. Hence, chronic administration of an MCT1 inhibitor in vivo should hamper the glycolytic process, leading to severe energy crisis and tumor growth inhibition. Based on its excellent MCT1 inhibition, slightly enhanced cytotoxicity, and favorable solubility, compound 4a was selected for further in vitro and in vivo evaluation.
Table 2.
MTT assay IC50 * values of amino carboxy coumarins in MDA-MB-231, GL261-luc2 and MIA PaCa-2 cell lines
| Compounds | GL261-luc2 | MDA-MB-231 |
|---|---|---|
| 4a | >0.25 | 0.24 ± 0.01 |
| 4b | >0.25 | >0.25 |
| 4c | >0.25 | >0.25 |
| 4d | >0.25 | >0.25 |
| 4e | >0.25 | >0.25 |
IC50 values reported in mM, average ± SEM of minimum three separate experimental values.
We then determined its potential oral bioavailability using bidirectional Caco-2 cell monolayer permeability, and its metabolic stability in human and mouse liver microsomes (Tables 3–5).45,46 Caco-2 cell permeability assays for 4a in both apical to basolateral (A – B) and basolateral to apical (B – A) directions were carried out to predict its potential oral bioavailability. Caco-2 is a heterogeneous human epithelial colorectal adenocarcinoma cell line that mimics the human enterocytic intestinal layer.45 This assay estimates human intestinal permeability and drug efflux ratio of the compounds. 4a showed relatively low A – B and moderate B – A permeability (Table 3). Based on this study, 4a exhibits much better absorption (~30%) compared to poor absorption of first generation MCT1 inhibitors (3–6%, data not shown). Propranolol (highly permeable), labetalol (moderately permeable), ranitidine (poorly permeable), and colchicine (P-glycoprotein substrate) were used as controls. The efflux ratio was calculated using the formula Papp(B − A)/Papp(A − B) and efflux ratio >2 signifies that drug efflux is occurring. 4a has an efflux ratio of 0.2 indicating that the efflux rate is very low and 4a may not be a good substrate for drug efflux transporters, which is beneficial for providing anticancer efficacy.
Table 3.
In vitro Caco-2 monolayer permeability (pH 6.5/7.4, at 10 μM)
| A – B permeability (Caco-2, pH 6.5/7.4)
| ||
|---|---|---|
| Compounds | Permeability (10−6 cm/s) Mean* |
Percent recovery (%) Mean* |
| 4a | 32 | 34 |
| Propranolol | 40.9 | 67 |
| Labetalol | 8.5 | 66 |
| Ranitidine | ND** | ND** |
| Colchicine | 0.3 | 75 |
| B – A permeability (Caco-2, pH 6.5/7.4) | ||
| 4a | 6.4 | 55 |
| Propranolol | 41.5 | 80 |
| Labetalol | 36.5 | 72 |
| Ranitidine | 3.7 | 85 |
| Colchicine | 15.3 | 81 |
Mean of two experiments.
ND = not determined.
Table 5.
Intrinsic clearance in mice liver microsomes (0.1 μM)
| Intrinsic clearance (liver microsomes, mouse)
| ||||
|---|---|---|---|---|
| Compounds | Half-life*
|
Clint | ||
| Trial 1 | Trial 2 | Mean | ||
| 4a | 90.6 | 73.5 | >60 | <115.5 |
| Propranolol | 9.1 | 9.5 | 9 | 744.5 |
| Imipramine | 15.7 | 15.4 | 16 | 446.2 |
| Verapamil | 17.6 | 17.5 | 18 | 394.6 |
| Terfenadine | 7.5 | 6.1 | 7 | 1027.4 |
Half-life is reported in minutes, mean value of two experiments.
We then determined the metabolic stability (half-life) of 4a in human and mouse liver microsomes in order to assess its hepatic clearance rate because liver microsomes contain many enzymes that are responsible for drug metabolism. 4a exhibited good metabolic stability (half-life >60 min) in both human and mouse liver microsomes (Tables 4 and 5). Propranolol, imipramine, verapamil, and terfenadine were used as controls with high, high, medium, and low metabolic stabilities, respectively, in human liver microsomes.
Table 4.
Intrinsic clearance in human liver microsomes (0.1 μM)
| Intrinsic clearance (liver microsomes, human)
| ||||
|---|---|---|---|---|
| Compounds | Half-life*
|
Clint | ||
| Trial 1 | Trial 2 | Mean | ||
| 4a | 922.2 | >60 | >60 | <115.5 |
| Propranolol | 334.4 | 373 | >60 | <115.5 |
| Imipramine | 213.9 | 194.4 | >60 | <115.5 |
| Verapamil | 21.1 | 21.5 | 21 | 324.8 |
| Terfenadine | 9.8 | 9.1 | 9 | 736.8 |
Half-life is reported in minutes, mean value of two experiments.
We then evaluated the systemic toxicity profile of 4a in healthy CD-1 mice.47 Mice were treated once daily (qd) with 4a intraperitoneally (ip, 20 mg/kg) and via oral gavage (100 mg/kg). Control mice received the vehicle (10% DMSO in saline). At the end of the study, the treated groups did not show any visible toxic effects or mortality and had no significant difference in body weights compared to the control group (Fig. 3). This study indicates the nontoxic nature of the candidate compound 4a for in vivo anticancer applications.
Figure 3.

Body weight changes in systemic toxicity study of compound 4a in CD-1 mice.
We then evaluated anticancer efficacy of 4a in a flank-based GL261-luc2 tumor syngraft model.47–49 Group 1 was administered with 4a (20 mg/kg, ip, qd), group 2 was treated with the clinically used brain tumor drug temozolomide (20 mg/kg, ip, qd) and the control group was given vehicle. At the end of the study, the tumor growth inhibition of compound 4a and temozolomide was found to be 77% and 81%, respectively, compared to the control group (Fig. 4). This efficacy study demonstrated the therapeutic utility of 4a for potential anticancer applications.
Figure 4.

Tumor growth inhibition study with compound 4a in GL261-luc2 tumor syngraft model. N = 6; *P < 0.05.
To determine whether 4a is capable of exhibiting antitumor properties in high MCT4 and low MCT1 expressing tumors, we evaluated the tumor growth inhibition in an MDA-MB-231-luc flank-based xenograft model.47,49,50 Group 1 and group 2 mice were administered with compound 4a at two different dosages of 20 mg/kg, ip, qd and 100 mg/kg, oral gavage, qd. Group 3 was treated with the clinically used breast cancer drug doxorubicin (0.5 mg/kg, ip, 5 days/week). The control group was administered with vehicle (10% DMSO in saline). At the end of the study (day-19), the tumor growth inhibition in groups 1, 2, and 3 were found to be 10%, 13%, and 50% respectively, compared to the control group (Fig. 5). This study indicates that compound 4a doesn’t exhibit significant tumor growth reduction in a predominantly MCT4 expressing tumor model.
Figure 5.

Tumor growth inhibition study with compound 4a in MDA-MB-231-luc tumor xenograft model. N = 6; *P < 0.05.
In conclusion, we synthesized N,N-dialkyl carboxy coumarins as MCT1 inhibitors and carried out in vitro cytotoxicity studies in MCT1 and MCT4 expressing cell lines. Selected inhibitor 4a was further studied for its in vitro bidirectional Caco-2 cell permeability and metabolic stability in human and mouse liver microsomes. This lead compound 4a exhibited good absorption, metabolic stability, and a low drug efflux ratio. Systemic toxicity studies in healthy CD-1 mice indicated that compound 4a was well tolerated and treated animals gained normal body weights with no apparent side effects. The candidate compound 4a was evaluated for in vivo tumor growth inhibition in predominantly MCT1 expressing GL261-luc2 glioma syngraft, and MCT4 expressing MDA-MB-231 xenograft mouse models. These in vivo studies indicated that compound 4a significantly inhibited tumor growth in GL261-luc2 model, but did not exhibit any significant activity in MDA-MB-231-luc model. This emphasizes the importance of high MCT1 expression for the efficacy of these inhibitors. Owing to the importance of glycolysis in tumor progression and the elevated expression of MCT1 in several cancers, we believe that these inhibitors have good potential to be developed as broad-spectrum anticancer agents. 4a can be used as a single agent and can also be combined with other chemotherapeutic agents with different mechanisms of action to realize their synergistic potential in cancer treatment.
Acknowledgments
This work was supported by University of Minnesota Duluth; Department of Defense Breast Cancer Research Proposal, Break-through Award W81XWH-15-1-0047; Whiteside Clinical Research Institute, Duluth, Minnesota; Center for Pharmaceutical Development, University of Minnesota; Minnesota Partnership for Biotechnology & Medical Genomics; Masonic Cancer Center Brainstorm Mechanism, University of Minnesota; and KAM and Bighley Foundation, University of Minnesota.
References and notes
- 1.Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Erez A, DeBerardinis RJ. Nat Rev Cancer. 2015;15:440. doi: 10.1038/nrc3949. [DOI] [PubMed] [Google Scholar]
- 4.Cantor JR, Sabatini DM. Cancer Discovery. 2012;2:881. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peppicelli S, Bianchini F, Calorini L. Oncoscience. 2015;2:225. doi: 10.18632/oncoscience.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganapathy-Kanniappan S, Geschwind JF. Mol Cancer. 2013;12:152. doi: 10.1186/1476-4598-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganapathy V, Thangaraju M, Prasad PD. Pharmacol Ther. 2009;121:29. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Hsu PP, Sabatini DM. Cell. 2008;134:703. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. J Clin Invest. 2008;118:3930. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatenby RA, Gillies RJ. Nat Rev Cancer. 2004;4:891. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG. Nat Rev Drug Discovery. 2011;10:671. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 12.Halestrap AP. Mol Aspects Med. 2013;34:3379. doi: 10.1016/j.mam.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. J Bioenerg Biomembr. 2012;44:127. doi: 10.1007/s10863-012-9428-1. [DOI] [PubMed] [Google Scholar]
- 14.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Cell Cycle. 2009;8:3984. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 15.Spanier J, Drewes LR. In: Drug Transporters: Molecular Characterization and Role in Drug Disposition. You G, Morris ME, editors. Wiley Publishers; New York: 2007. p. 147. [Google Scholar]
- 16.Wang Q, Morris ME. Drug Metab Dispos. 2007;35:1393. doi: 10.1124/dmd.107.014852. [DOI] [PubMed] [Google Scholar]
- 17.Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, Bundick RV, Cook ID, Craggs RI, Edwards S, Evans LR, Harrison R, Holness E, Jackson AP, Jackson CG, Kingston LP, Perry MW, Ross AR, Rugman PA, Sidhu SS, Sullivan M, Taylor-Fishwick DA, Walker PC, Whitehead YM, Wilkinson DJ, Wright A, Donald DK. Nat Chem Biol. 2005;1:371. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP, Price NT. Biochem J. 1999;343:281. [PMC free article] [PubMed] [Google Scholar]
- 19.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Oncotarget. 2014;5:7982. doi: 10.18632/oncotarget.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Yang C, Doherty JR, Roush WR, Cleveland JL, Bannister TD. J Med Chem. 2014;57:7317. doi: 10.1021/jm500640x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Cell Cycle. 2012;11:1108. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, Witkiewicz A, Lin Z, Balliet R, Howell A, Sotgia F. Cancer Biol Ther. 2010;10:537. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurrapu S, Jonnalagadda SK, Alam MA, Nelson GL, Sneve MG, Drewes LR, Mereddy VR. ACS Med Chem Lett. 2015;6:558. doi: 10.1021/acsmedchemlett.5b00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Tortosa M, Genau HM, Rounbehler RJ, Lu Y, Dang CV, Kumar KG, Butler AA, Bannister TD, Hooper AT, Unsal-Kacmaz K, Roush WR, Cleveland JL. Cancer Res. 2014;74:908. doi: 10.1158/0008-5472.CAN-13-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.While this work was in progress, two publications have appeared on the utilization of aminocarboxy coumarins as MCT1 inhibitors. Draoui N, Schicke O, Seront E, Bouzin C, Sonveaux P, Riant O, Feron O. Mol Cancer Ther. 2014;13:1410. doi: 10.1158/1535-7163.MCT-13-0653.Draoui N, Schicke O, Fernandes A, Drozak X, Nahra F, Dumont A, Douxfils J, Hermans E, Dogne JM, Corbau R, Marchand A, Chaltin P, Sonveaux P, Feron O, Riant O. Bioorg Med Chem. 2013;21:7107. doi: 10.1016/j.bmc.2013.09.010.Draoui N, Feron O, Riant O, Sonveaux P, Schicke O, Fernandes A, Kilonda A, Vanherck J, Marchand A. WO 2014195507 A1 20141211. PCT Int Appl. 2014
- 26.Sanita P, Capulli M, Teti A, Galatioto GP, Vicentini C, Chiarugi P, Bologna M, Angelucci A. BMC Cancer. 2014;14:154. doi: 10.1186/1471-2407-14-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES, Witkiewicz AK. Cell Rep. 2014;9:2233. doi: 10.1016/j.celrep.2014.11.025. [DOI] [PubMed] [Google Scholar]
- 28.Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, Leiby B, Cognetti DM, Sotgia F, Lisanti MP, Martinez-Outschoorn UE. Cell Cycle. 2013;12:1371. doi: 10.4161/cc.24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mereddy VR, Drewes LR, Alam MA, Sravan KJ, Gurrapu S. WO2013109972. PCT Int Appl. 2013
- 30.Miranda-Goncalves V, Honavar M, Pinheiro C, Martinho O, Pires MM, Pinheiro C, Cordeiro M, Bebiano G, Costa P, Palmeirim I, Reis RM, Baltazar F. Neuro Oncol. 2013;15:172. doi: 10.1093/neuonc/nos298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pertega-Gomes N, Vizcaino JR, Miranda-Goncalves V, Pinheiro C, Silva J, Pereira H, Monteiro P, Henrique RM, Reis RM, Lopes C, Baltazar F. BMC Cancer. 2011;11:312. doi: 10.1186/1471-2407-11-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, Monterey MD, Galloway MP, Sloan AE, Mathupala SP. Neoplasia. 2011;13:620. doi: 10.1593/neo.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Draoui N, Feron O. Dis Model Mech. 2011;4:727. doi: 10.1242/dmm.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hao J, Chen H, Madigan MC, Cozzi PJ, Beretov J, Xiao W, Delprado WJ, Russell PJ, Li Y. Br J Cancer. 2010;103:1008. doi: 10.1038/sj.bjc.6605839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kale M, Patwardhan K. Curr Pharma Res. 2014;4:1150. [Google Scholar]
- 36.Pangal A, Gazge M, Mane V, Shaikh JA. Int J Pharm Res Biosci. 2013;2:168. [Google Scholar]
- 37.Jain PK, Joshi H. J Appl Pharm Sci. 2012;2:236. [Google Scholar]
- 38.Representative procedure for the synthesis of 7-(dibenzylamino)-2-oxo-2H-chromene-3-carboxylic acid 4a: To a solution of 3-aminophenol (10 mmol) in 10 mL DMSO (ethanol-H2O for other alkyl bromides), was added benzyl bromide (40 mmol), potassium carbonate (20 mmol) and refluxed at 80 °C for 12 h. Upon the completion of the reaction, the reaction mixture was extracted with ethyl acetate and water. The organic layer was dried with anhydrous Mg2SO4 and evaporated to obtain the 3-(dibenzylamino)phenol. The product was purified via column chromatography. To a solution of 3-(dibenzylamino) phenol (10 mmol) in DMF (60 mmol) was added phosphorous oxychloride dropwise at 0 °C and the reaction mixture was refluxed at 80 °C for 2–4 h. The reaction was quenched in a saturated solution of sodium carbonate and the solid was filtered and washed with hexanes to obtain 4-(dibenzylamino)-2-hydroxybenzaldehyde. To a solution of this aldehyde (10 mmol) in 20 ml ethanol, was added diethyl malonate (20 mmol), acetic acid (5 drops) and piperidine (13 mmol) and refluxed for 8–12 h at 80 °C. Upon the completion of the reaction, the above solution was evaporated and extracted with ethyl acetate. The organic layers were dried with anhydrous MgSO4 and evaporated. The product obtained was further refluxed in 20 ml of 10% NaOH solution. The reaction was quenched with 3 M HCl (pH 7.0) and extracted with ethyl acetate. The compound was purified by column chromatography (eluted with 100% ethyl acetate) and recrystallized. 1H NMR (500 MHz, CDCl3): δ 12.31 (s, 1H), 8.69 (s, 1H), 7.48–7.23 (m, 11H), 6.86 (dd, J = 8.5, 1.5 Hz, 1H), 6.67 (s, 1H), 4.84 (s, 4H); 13C NMR (125 MHz, CDCl3): 165.53, 164.24, 157.89, 155.60, 150.75, 135.84, 132.23, 129.46, 128.16, 126.52, 112.02, 109.79, 107.24, 98.63, 55.03; Anal. Calcd for C24H19NO4·HCl (421.88): C, 68.33; H, 4.78; N, 3.32. Found: C, 69.22; H, 4.88; N, 3.43.
- 39.Western blot analysis of MCT1 and MCT4 expression: Cultured cells were scraped from culture flasks and immediately frozen. Cells were thawed on ice and solubilized in 200 μL SDS boiling buffer, then centrifuged for 2 min at high speed. The supernatant was collected and diluted 1:5 with deionized H2O and assayed for protein using the BCA protocol. A volume containing 10 μg was loaded on SDS PAGE gels (Novex) for standard electrophoresis (40 min at 200 V). Proteins were transferred from the gel to nitrocellulose membrane under denaturing conditions (200 mA for 1.75 h). MCT1 and MCT4 were detected using specific antibodies (rabbit polyclonal IgG antibody for MCT1, sc-50324; rabbit polyclonal IgG antibody for MCT4, sc50329; Santa Cruz, Inc.) and visualized using chemiluminescence. For relative quantitation, actin was detected and measured as a control protein.
- 40.Newcomb EW, Lymberis SC, Lukyanov Y, Shao Y, Schnee T, Devitt M, Rosenstein BS, Zagzag D, Formenti SC. Cell Cycle. 2006;5:93. doi: 10.4161/cc.5.1.2271. [DOI] [PubMed] [Google Scholar]
- 41.Winkler F, Kienast Y, Fuhrmann M, Von Baumgarten L, Burgold S, Mitteregger G, Kretzschmar H, Herms J. Glia. 2009;57:1306. doi: 10.1002/glia.20850. [DOI] [PubMed] [Google Scholar]
- 42.Niks M, Otto M. J Immunol Methods. 1990;130:149. doi: 10.1016/0022-1759(90)90309-j. [DOI] [PubMed] [Google Scholar]
- 43.Cell culture conditions and MTT cytotoxicity: MDA-MB-231 cells (ATCC) were grown in DMEM supplemented with 10% FBS and penicillin-streptomycin (50 U/ml, 50 μg/ml). GL261-luc2 cells (Perkin Elmer) were cultured in DMEM, 10% FBS, 50 μg/mL geneticin and penicillin-streptomycin (50 U/ml, 50 μg/ml). Cells (5 × 103 cells/well) were seeded in 96-well plate and incubated at 37 °C and 5% CO2 for 24 h. Test compounds were added to the wells in replicate and incubated for 72 h. 10 μL MTT (12 mM in 1X PBS) was added in each well and incubated for 4 h before adding 100 μL SDS (1 g/10 mL 0.01 N HCl) to dissolve formazan precipitate and incubated for further 4 h. Absorbance was recorded at 570 nm. Percent survival was calculated using the formula %survival = (abs test compound/abs of DMSO control) × 100. IC50 was obtained using GraphPad by plotting log[concentration] on x-axis and% survival on y-axis.
- 44.Critchlow SE, Tate L. WO 2010089580A1 20100812. PCT Int Appl. 2010
- 45.Hidalgo IJ, Raub TJ, Borchardt RT. Gastroenterology. 1989;96:736. [PubMed] [Google Scholar]
- 46.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. J Pharmacol Exp Ther. 1997;283:46. [PubMed] [Google Scholar]
- 47.Ethical considerations: The experimental procedure involving animals that were conducted at the University of Minnesota Duluth was in compliance with the U.S. National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC). Studies with protocols 1311-31063A (systemic toxicity, Fig. 3) and 1312-31108A (GL261-luc2 syngraft, Fig. 4) were conducted at University of Minnesota. MDA-MB-231-luc xenograft study (Fig. 5) was conducted by GenScript Corporation (Piscataway, NJ) according to their approved IACUC protocol GS-PAMD1401SN052.
- 48.Tumor growth inhibition studies in GL261-luc2 syngraft model: Tumor cells suspended in 1:1 matrigel-PBS were injected onto the right flank of C57BL/6 mice (5 × 106 GL261-luc2 cells). The mice were randomly assigned into groups (n = 6, male and female mice). Treatment was started when tumor volume reached 200 mm3. Tumors were measured by caliper every two or three days and tumor volumes calculated using the formula V = (ab2)/2 where ‘a’ is the long diameter of the tumor and ‘b’ is the short diameter of the tumor. Mice were euthanized at the end of the study and tumors were isolated and weighed. The inhibition amount was determined using the formula % inhibition = [(C − T)/C] × 100 where C is average tumor weight of the control group and T is the average tumor weight of the test group.
- 49.Statistical analysis: Statistics were computed using GraphPad Prism version 6.0. Mann-Whitney test was used to compare the treated and untreated groups for all in vivo tumor syngraft/xenograft studies. A P-value of <0.05 was considered significant.
- 50.Tumor growth inhibition studies in MDA-MB-231 xenograft model: Tumor cells suspended in 1:1 matrigel-PBS were injected on right flank of female SCID mice (10 × 106 MDA-MB-231 cells). The mice were randomly assigned into groups (n = 6). Treatment was initiated when the average tumor volume was ~100 mm3. Mice were euthanized at the end of the study and tumors were isolated and weighed. Tumor growth inhibition was calculated as above in Ref. 49.