

Abstract
Intraventricular hemorrhage causes spatial memory loss, but the mechanism remains unknown. Our recent studies demonstrated that traumatic brain injury activates Src family kinases, which cause spatial memory loss. To test whether the spatial memory loss was due to blood in the ventricles, which activated Src family kinases, we infused autologous whole blood or thrombin into the lateral ventricles of adult rats to model non-traumatic intraventricular hemorrhage. Hippocampal neuron loss was examined 1 day to 5 weeks later. Spatial memory function was assessed 29 to 33 days later using the Morris water maze. Five weeks after the ventricular injections of blood or thrombin, there was death of most hippocampal neurons and significant memory deficits compared with sham operated controls. These data show that intraventricular thrombin is sufficient to kill hippocampal neurons and produce spatial memory loss. In addition, systemic administration of the non-specific Src family kinase inhibitor PP2 or intraventricular injection of siRNA-Fyn, a Src family kinase family member, prevented hippocampal neuronal loss and spatial memory deficits following intraventricular hemorrhage. The data support the conclusions that thrombin mediates the hippocampal neuronal cell death and spatial memory deficits produced by intraventricular blood and that these can be blocked by non-specific inhibition of Src family kinases or by inhibiting Fyn.
Keywords: Intraventricular hemorrhage, Src family kinases, Fyn, spatial memory deficits, PP2
Introduction
Intraventricular hemorrhage (IVH) is defined by the presence of blood in the cerebral ventricles. IVH in humans usually has an acute onset, has several different causes, and is associated with a high morbidity and mortality.1–3 Causes of IVH in humans include hypertension, intracerebral hemorrhage, subarachnoid hemorrhage, arteriovenous malformations, traumatic brain injury (TBI), coagulopathies, and others. The major complications after IVH include hematoma induced brain injury, brain edema, and a variety of neurological deficits.4 Increasing evidence also shows that many patients suffer from significant cognitive impairment, which markedly decreases quality of life of patients over the long-term.5,6 However, the mechanism of cognitive impairment following IVH is not understood and thus there is no treatment.
Once IVH occurs, the blood causes release of several toxic molecules into the cerebrospinal fluid (CSF), including thrombin, oxyhemoglobin (oxyHb), iron, cytokines, reactive oxygen species (ROS), and others. Since many molecules and signaling pathways are implicated in the brain injury and cognitive deficits post-IVH, blocking a single mediator or single pathway may not be clinically effective.7 This led us to consider an approach that would block multiple pathways.
Src family kinases (SFKs), a class of non-receptor tyrosine kinases, consist of eight family members including c-Src, c-Yes, Fyn, Lck, Lyn, Hck, Blk, and c-Fgr.8 SFKs can be activated by many trans-membrane receptors, such as adhesion receptors, tyrosine kinase receptors, G protein-coupled receptors, cytokine receptors, and others.9 This feature of the SFKs makes them a point of convergence for many toxic molecules (e.g. thrombin, oxyHb, ROS, cytokines, and others) that are released and mediate secondary injury after IVH. Indeed, thrombin activation of Protease Activated Receptors (PARs) leads to SFK activation.10 The activated SFKs cause death of hippocampal and other neurons resulting in cognitive deficits through several neurotoxic down-stream signaling pathways including RhoA-Rho-kinase1 (ROCK1), Jun N-terminal kinase (JNK), P38 and Erk mitogen-activated protein kinases (MAPKs), and phosphorylation of NMDA receptors.11–20
To follow up our recent studies that have demonstrated that cognitive decline occurs following TBI,11 we hypothesized that the TBI-induced cognitive decline is related to the toxic effects of blood released into the cerebral ventricles after TBI, which activates thrombin which in turn activates SFKs and results in the cell death of hippocampal neurons. To address this hypothesis, we infused autologous whole blood or thrombin into the lateral ventricles to determine whether intraventricular autologous whole blood or thrombin alone causes death of hippocampal neurons and cognitive deficits via SFK pathways. Spatial memory function was assessed using the Morris Water Maze 1 month later. Cell survival in hippocampus was also assessed. To evaluate the role of SFKs in animals injected with fresh whole blood or thrombin into the ventricles, rats were injected with a systemic non-specific SFK inhibitor, PP2 or vehicle; or were injected intraventricularly with siRNA-Fyn (one SFK family member) or scrambled siRNA. The data show that intraventricular thrombin or fresh blood produces cell death of hippocampal neurons and loss of spatial memory via activation of SFKs following IVH. Moreover, we show that inhibition of a specific SFK kinase Fyn (one SFK subtype) prevents the spatial memory deficits produced by IVH.
Materials and methods
Male Sprague-Dawley rats, weighing 300–320 g, were purchased from Harlan Laboratories. Animals were assigned to experimental and control groups randomly. All experiments were performed blindly and all analyses were done by individuals blinded to the groups. Group sizes of 8 were used for behavioral studies of animals injected with whole blood or thrombin.
Injections of autologous whole blood into lateral cerebral ventricle
The animals (n = 58) were anesthetized with isoflurane (Minrad, New York, NY) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). A heating blanket maintained body temperature at 37℃. Autologous whole blood (200 µl) drawn from the tail of each individual rat was infused into brain over 10 min into the left lateral ventricle (coordinates: −0.9 mm anterior–posterior; −1.4 mm medial–lateral; −4.6 mm dorsal–ventral, with respect to bregma).21 The sham controls received a burr hole and needle placement without injection. The rats who received intraventricular (i.c.v.) autologous whole blood injections were immediately given one intraperitoneal injection of the non-specific SFKs inhibitor PP2 (1.0 mg/kg, Biomol International LP, Plymouth Meeting, PA) or vehicle control (5% DMSO in Saline). The PP2 dose was based upon our previous studies showing it to be the most effective dose for thrombin or TBI-induced brain injury.11,21,22
A separate group of rats had intracerebroventricular injections of siRNA-Fyn (Sense, GCUUGUACAGCAUUACUCATT; Antisense, UGAGUAAUGCUGUACAAGCTG) (100 µg/animal, Ambion, Austin, TX) or scrambled-siRNA 1 day (24 h) prior to intraventricular injections of autologous whole blood (n = 16). The siRNA-Fyn and scrambled-siRNA were wrapped using nanoparticles to improve cell penetration and prolong half-life (Altogen Biosystems, Las Vegas, NV) and incubated at room temperature for 30 min prior to being injected into the ventricles of the rats.
After closure of the operative sites, rats were allowed to recover in an incubator maintained at 37℃, and then returned to their home cages with free access to food and water. All experimental procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee, University of California at Davis. The manuscript was written up in compliance with the ARRIVE guidelines.
Assessment of knocking down efficiency of nanoparticle-siRNA using RT-PCR and Western blots for Fyn
Sprague-Dawley rats of both sexes (n = 12), weighing 300–350 g, were divided into four groups (three rats/group). Apart from naïve rats used for blank control, animals had i.c.v. injections of either 10 µl nanoparticle coupled 100 µg scrambled siRNA controls, or 10 µl nanoparticle-alone as vehicle controls, or 10 µl nanoparticle siRNA to Fyn (100 µg/rat, i.c.v.). One day after i.c.v. injections, rats were euthanized, and the bilateral rat hippocampi and parietal cortex were dissected, frozen in liquid nitrogen, and stored at −80℃.
Total RNA was extracted from the brain tissue using mirVana PARIS kit (Lifetech, Carlsbbad, CA). Reagents and primers specific for Fyn (Fyn 270 FWD: CTGACGGAGGAGAGGGACG; Fyn 399 REV: GGCTGCGTGGAAGTTGTTGTA) were purchased from Lifetech (Carlsbbad, CA) and standard SYBR Green RT-PCR protocol followed. 18 s rRNA was used as internal controls for RT-PCR. The Fyn values were normalized with internal controls and calculated as a percentage of mRNA expression for the siRNA-Fyn treated sample relative to the naïve sample. Statistical differences were determined using ANOVA followed by Dunnett’s post hoc test.
Injections of thrombin into the lateral ventricle
The experimental procedures described above to infuse autologous whole blood were duplicated for these studies. However, instead of autologous whole blood, male Sprague-Dawley rats (n = 32) had intraventricular injections (i.c.v) of thrombin (20 U in 5 µl saline/animal, Sigma, St. Louis, MO). Rats that received i.c.v. thrombin were concurrently given either i.c.v. injections of the thrombin inhibitor hirudin (20 U/animal), or immediately given one intraperitoneal injection of the SFK inhibitor PP2 (1 mg/kg) or injected with vehicle.
Morris water maze
Acquisition of spatial memory learning was assessed on days 29–33 after IVH in the Morris water maze (MWM) as previously described.11 The test apparatus consisted of a large white circular tank (183 cm diameter by 60 cm high) filled with water to a depth of 22 cm. Water temperature was maintained at 24–28℃. A transparent circular escape platform (12.8 cm diameter, 20 cm high) was placed in the tank 2 cm below the water surface. Four consistent visual cues were located in the test room outside of the maze. Rats were released from one of four starting points (selected randomly on each day for each rat) and allowed 120 s to find and mount the escape platform. If the rat did not find the platform within 120 s, the experimenter placed the rat on the platform. The rat remained on the platform for 30 s before being removed from the maze. The rat received a 4-min inter-trial interval in a warmed holding cage before being returned to the maze for subsequent trials. Rats received a total of four trials per day, one from each starting point, over five consecutive days. Mean latency to find the platform was calculated for each day to assess learning. Data from all trials were recorded using a video tracking system (Poly-Track Video Tracking System version 2.1, San Diego Instruments Inc., San Diego, CA). Statistical differences were determined using repeated measures ANOVA with assessment days as the repeated variable within subjects followed by Dunnett’s post hoc test.
Hippocampal histology/NeuN immunohistochemistry
A separate group of adult Sprague-Dawley rats (n = 24) were divided into six groups for histology (four rats/group). The animals either had a sham operation or had thrombin injected into the lateral ventricle (20 U) followed immediately by intraperitoneal injection of vehicle (saline) or PP2 (1 mg/kg, i.p.). Animals were allowed to survive 1 day, 7 days, 14 days, or 5 weeks. They were then anesthetized and perfused via the aorta with 4% paraformaldehyde in phosphate buffer. Brains were removed, sectioned, and immunostained for NeuN.
The avidin-biotin-peroxidase complex (Vectastain Elite ABC Kit, Vector Laboratories, Inc., Burlingame, CA) method was used to perform NeuN (a marker of mature neurons) immunohistochemistry. Brain sections were incubated at room temperature in 0.3% H2O2 in methanol for 30 min to quench endogenous peroxidase. After two 5 min rinses in PBS, sections were incubated with 3% horse blocking serum for 20 min, and then incubated for 0.5 h in primary antibody (mouse anti-NeuN, 1:150, Millipore MAB377, Billerica, MA) diluted in PBS containing 0.1% Triton X-100 and 3% horse serum. After being washed in PBS, sections were incubated in biotinylated secondary antibody (Goat anti-mouse 1:1000, Vector BA 9200, Burlingame, CA) for 0.5 h. After three 5 min rinses in PBS, sections were placed in Vectastain ABC reagent for 0.5 h. After two more 5 min washes, sections were incubated in peroxidase substrate 3,3′-diaminobenzidine (DAB) solution for 10 min. DAB staining was examined using a Nikon E600 microscope.
Results
The effects of PP2 on cognitive function after i.c.v. autologous whole blood
The group of animals that had intraventricular injections of autologous blood had significantly longer latencies to find the hidden platform over the 5 days of testing compared with the sham group (P < 0.05) (Blood/Vehicle vs Sham/Vehicle) (Figure 1). Intraperitoneal injections of PP2 (1 mg/kg, i.p.) significantly improved performance compared with intraperitoneal vehicle injections (P < 0.05) in animals with intraventricular blood injections (Blood/PP2 vs Blood/Vehicle) (Figure 1). Administration of PP2 did not affect cognitive function of naïve rats without intraventricular injections of blood (Naïve/PP2 vs Naïve/Vehicle) (Figure 1). There were no significant statistical differences between the naïve/vehicle, blood/PP2, and naïve/PP2 groups (Figure 1).
Figure 1.

PP2 attenuated autologous whole blood-induced spatial memory loss in rats. Cognitive function was examined at 29 through 33 days after autologous fresh blood injections using the Morris Water Maze. The y-axis shows latency to finding platform (s). Each point represents the mean ± standard error (n = 8). Blue line: naïve control (naïve/vehicle); red line: sham control (sham/vehicle); orange line: vehicle-treated IVH (blood/vehicle); light blue line: PP2-treated IVH (blood/PP2); purple line: PP2-treated naïve (naïve/vehicle). #P < 0.05 (blood/vehicle) vs (sham/vehicle); *P < 0.05 (blood/PP2) vs (blood/vehicle). (Repeated measures ANOVA followed by Dunnett’s post hoc test).
The effects of siRNA-Fyn on cognitive function after i.c.v. autologous whole blood
The nanoparticle-wrapped siRNAs (100 µg/rat, i.c.v.) to Fyn significantly decreased Fyn mRNA > 70% compared with naïve, nanoparticle transfection reagent alone (negative control, TR-alone), and scrambled si-RNA (si-NC, P < 0.01) (Figure 2). The scrambled siRNA had no effect on hippocampal Fyn mRNA (Figure 2). We also observed that siRNA-Fyn produced a modest knock down of Fyn protein expression at 24 hrs, though the decrease of Fyn protein was not as remarkable as Fyn mRNA knockdown (Supplementary Figure 1).
Figure 2.

The nanoparticle-wrapped siRNA-Fyn knocks down Fyn mRNA. Adult Sprague-Dawley rats had either si-RNA to Fyn or scrambled-siRNA (siNC) or nanoparticle transfection reagent alone (vehicle control, TR-alone) injected into the left lateral ventricle. Naïve animals had no cerebral injections. One day later, the hippocampus was removed and mRNA extracted from the left hippocampus. RT-PCR for Fyn mRNA was then performed. Note that siRNA-Fyn decreased Fyn mRNA compared with the naïve, vehicle control (TR-alone), and scrambled si-RNA (si-NC). **P < 0.01, si-Fyn vs si-NC (ANOVA followed by Dunnett’s post hoc test).
The group of animals that had intraventricular injections of blood and scrambled-siRNA had significantly longer latencies to find the hidden platform over the 5 days of testing compared with the sham-operated group (P < 0.05) (Blood/siRNA scramble vs Sham/Vehicle) (Figure 3). Of the animals that received intraventricular injections of blood, treatment with siRNA-Fyn (100 µg, i.c.v.) significantly improved performance compared with the scrambled-siRNA (P < 0.05) (Blood/siRNA scramble vs Blood/siRNA-Fyn) (Figure 3). There were no significant differences between the sham/vehicle group and the blood/siRNA-Fyn group (Figure 3).
Figure 3.

Nanoparticle wrapped siRNA-Fyn attenuated autologous whole blood-induced spatial memory loss in rats. Cognitive function was examined at 29 through 33 days after autologous fresh blood injections using the Morris Water Maze. The y-axis shows latency to finding platform (s). Each point represents the mean ± standard error (n = 8). Blue line: sham control (sham/vehicle); red line: scrambled-siRNA-treated IVH (blood/scrambled-siRNA); purple line: siRNA-Fyn-treated IVH (blood/siRNA-Fyn). #P < 0.05 (blood/scrambled-siRNA) vs (sham/vehicle); *P < 0.05 (blood/siRNA-Fyn) vs (blood/scrambled-siRNA). (Repeated measures ANOVA followed by Dunnett’s post hoc test).
The effects of PP2 on cognitive function after intraventricular thrombin
The group of animals with intraventricular injections of thrombin and intraperitoneal vehicle had significantly longer latencies to find the hidden platform over the 5 days of testing compared with the sham group (P < 0.05) (Thrombin/Vehicle vs Sham/Vehicle) (Figure 4). Treatment of animals with intraventricular thrombin with intraperitoneal PP2 (1 mg/kg, i.p.) or intraventricular hirudin (20 U, i.c.v.), a direct thrombin inhibitor, significantly improved performance compared with the thrombin/vehicle group (P < 0.05) (Thrombin/PP2 and Thrombin/Hirudin vs Thrombin/Vehicle) (Figure 4).
Figure 4.
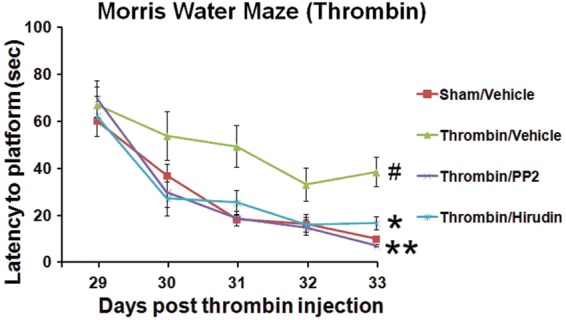
PP2 (SFK inhibitor) or hirudin (direct thrombin inhibitor) prevented thrombin-induced spatial memory loss in rats. Cognitive function was examined at 29 through 33 days after thrombin injections using the Morris Water Maze. The y-axis shows latency to finding the platform (s). Each point represents the mean ± standard error (n = 8). Red line: sham control (sham/vehicle); green line: vehicle-treated IVH (thrombin/vehicle); purple line: PP2-treated IVH (thrombin/PP2); blue line: hirudin-treated IVH (thrombin/hirudin). #P < 0.05 (thrombin/vehicle) vs (sham/vehicle); *P < 0.05 (thrombin/hirudin) vs (thrombin/vehicle); **P < 0.01 (thrombin/PP2) vs (thrombin/vehicle). (Repeated measures ANOVA followed by Dunnett’s post hoc test).
Delayed neuronal cell loss in hippocampus following intraventricular thrombin
NeuN immunostaining of neuronal nuclei in hippocampus showed little change in numbers of neurons at 1 (Figure 5(B)) and 7 (Figure 5(C)) days following intraventricular thrombin injections compared with sham controls (Figure 5(A)). There was a subtle loss of dorsal dentate granule cell neurons at 2 weeks (Figure 5(D)) and almost complete loss of all neurons in hippocampus by 5 weeks following intraventricular thrombin with survival of some medial dentate gyrus neurons (Figure 5(E)). Intraperitoneal PP2 following intraventricular thrombin completely protected all segments of hippocampus including CA1, CA2, CA3, dentate hilus, and dentate gyrus (Figure 5(F)).
Figure 5.

NeuN immunohistochemistry of hippocampus following intraventricular thrombin injections. Adult Sprague-Dawley rats had thrombin (20 U) injected into the left lateral ventricle (B–F) and then immediately injected with vehicle (B-E) or with the non-specific Src family kinase inhibitor PP2 (F, 1 mg/kg, i.p.). These animals were compared with sham operated animals (A). Animals were then sacrificed 1 day (B), 7 days (C), 14 days (D), or 5 weeks later (E,F). PFA fixed brains were sectioned through the hippocampus and immunostained for the neuronal nuclear marker NeuN (A-F). The data show marked loss of hippocampal neurons by 5 weeks after intraventricular thrombin (E), which was completely prevented by intraperitoneal PP2 (1 mg/kg, i.p.) (F).
Discussion
This study demonstrates that administration of the non-specific SFKs inhibitor PP2 or nanoparticle wrapped siRNA-Fyn (one SFK family member) improves spatial memory function after non-traumatic IVH in adult rats. The study confirms recent studies that intraventricular blood can produce spatial memory and other cognitive deficits in rodents23 and also demonstrates that intraventricular thrombin alone causes hippocampal neuronal cell death and spatial memory deficits via activation of SFKs. The current study found modest dentate gyrus cell loss at 14 days and severe neuronal cell throughout the hippocampus at 5 weeks following intraventricular thrombin at which time significant spatial memory loss was also demonstrated. The explanation for this very delayed neuronal cell death is unclear but may account for few studies reporting memory deficits after intraventricular blood or thrombin.
A large number of toxic molecules, such as thrombin, oxyHb, cytokines, ROS, and others, are released into CSF following IVH14,15 and any one might contribute to the cognitive deficits. As the CSF flows through the ventricular system in the brain, it exchanges molecules with the interstitial fluid of the brain parenchyma.24 Therefore, these toxic molecules in the CSF can diffuse throughout the ventricles and mediate secondary injury in structures adjacent to the ventricles including hippocampus. Indeed, our results show that intraventricular blood or intraventricular thrombin lead to severe neuronal cell death via SFK pathways in hippocampus, which likely accounts in large part for the spatial memory deficits.
Although it took 2–5 weeks to observe the cell death of hippocampal neurons after thrombin (20 U, i.c.v.) injection, the trigger for neuronal death via SFKs activation probably occurred shortly after the time of thrombin injection. This is because injection of SFK inhibitor immediately after the thrombin was sufficient to rescue the neurons. Thus, though the thrombin induced SFK pathways are rapidly activated it appears to take weeks for the neurons to die and to be removed. This very slow death could be via autophagy though we have not explored this possibility. Furthermore, the finding that a single acute administration of the SFK inhibitor prevented the delayed cell death argues for an acute “trigger” involving activation of SFK after intraventricular fresh blood or thrombin administration. The acute trigger phenomenon also occurs following cerebral ischemia in which hippocampal cell death is delayed for days after the insult, but can be blocked by acute administration of glutamate receptor antagonists.25
We performed water maze at 29–33 days after thrombin, because this is the time point at which the cell death in hippocampus was fully mature and complete. We were concerned that testing at earlier times would be complicated by ongoing cell death. We did not test at later times after 33 days because cell death was complete by this time and no further changes of memory performance would be expected. However, examining spatial memory function at several months following IVH would be needed prior to any human trials of SFK antagonists.
It is well documented that thrombin is released rapidly by proteolysis of its precursor prothrombin immediately after brain injury.11 Moreover, thrombin can be synthesized in brain microvascular endothelial cells, glia and neurons, and accumulates in the vessel walls and senile plaques in brains of patients with Alzheimer’s disease.26 Our previous studies showed that intraventricular thrombin was toxic, producing damage to astrocytes and endothelial cells as well as disruption of blood–brain barrier.21 The multiple sources and toxic characteristics of thrombin suggest that thrombin has the potential to serve as a major molecule that plays a substantial role in producing clinical features of IVH. Therefore, in this study, we infused thrombin as well as whole blood to model non-traumatic IVH.
Our results showed that both intraventricular blood and thrombin produced significant spatial memory deficits. We injected thrombin inhibitor hirudin to animals that received intraventricular thrombin injection, and found that hirudin attenuated thrombin-induced cognitive deficits. In contrast, hirudin was not administered in the intraventricular fresh whole blood model, because it could block the clotting process of fresh blood. Instead, we administered SFKs inhibitor in the intraventricular fresh whole blood model, because though SFKs are important downstream molecules of the thrombin signaling pathway,14,15 SFKs have no effect on hemostasis because they do not associate with integrin complexes until after platelet aggregation is mediated by αIIbβ3.27
Our results show that inhibiting SFKs was sufficient to attenuate the spatial memory deficits produced by both intraventricular thrombin and whole blood, because inhibiting SFKs completely eliminated the spatial memory deficits in both IVH models. It is important to note that SFKs can be activated by many trans-membrane receptors, such as adhesion receptors, tyrosine kinase receptors, G protein-coupled receptors, cytokine receptors, and others.9 This unique characteristic of the SFKs makes them a point of convergence for many toxic molecules (e.g. thrombin, oxyHb, ROS, cytokines, and others) that are released and mediate secondary injury after IVH. Therefore, inhibiting SFKs is likely to block multiple IVH-induced toxic pathways and have potent effects to attenuate the secondary brain injury after IVH. Of interest is that our data show that even the moderate siRNA knockdown of Fyn protein could completely protect against IVH and produce normal spatial memory function.
We have previously shown that intracerebral injections of blood or thrombin increase SFK activity over 3–5 fold, and that SFK inhibitor (PP1 or PP2) blocks this increase in SFK activity.22,28 Moreover, our previous studies have demonstrated that (a) thrombin activation of SFKs leads to activation of cell cycle genes and programmed neuronal cell death,22 and (b) thrombin activation of SFKs leads to damage of astrocytes and endothelial and BBB disruption.21 These mechanisms by which SFKs mediate thrombin-induced brain injury could be occurring in this IVH model. In light of the fact that the SFK genes belong to oncogenes that are associated with cell proliferation, neuronal differentiation, and other important cellular processes, chronic silencing of these SFK genes could lead to serious side effects.21 This was supported by our previous findings that delayed and lasting SFK inhibition diminished a SFK-mediated delayed repair processes after thrombin-induced blood–brain barrier injury.25 Therefore, it is essential to inhibit SFKs transiently, and thus we designed the experiments so that a single administration of PP2 or siRNA to Fyn, instead of chronic administration of PP2 or siRNA to Fyn, was given immediately following IVH.
PP2 is a non-specific SFK inhibitor, blocking multiple SFK family members (e.g. Fyn, c-Src, and Lck). To better define which SFK subtype is implicated in the injurious pathways post IVH, we knocked down a single SFK gene Fyn using the nanoparticle-based siRNA method rather than traditional knockouts or viral knock downs of the proposed gene. This is because the nanoparticle-siRNA, approved by FDA for human use,29,30 provides a transient effect and avoids the potential side effects caused by “long-term silencing” of SFK gene—like knock outs. We examined Fyn rather than other SFK subtypes in this study, partly because Fyn is ubiquitously expressed,31 with highest abundance in hippocampal neurons.32,33 In addition, Fyn is the SFK that selectively phosphorylates the GluN2B subunit of the NMDA receptor to increase calcium flux34 and Fyn phosphorylation of tyrosine 1070 of the GluN2B subunit is also critical for surface expression of NMDA receptors35—both of which can lead to excitotoxic cell death. Thus, the siRNA to Fyn that protects against intraventricular blood mediated cell death in hippocampus could be due in part to blocking NMDA receptor activation by decreasing calcium flux through NMDA receptors and decreasing surface localization of NMDA receptors. In addition, there are other possible mechanisms by which SFKs might mediate secondary brain injury via downstream molecules, such as RhoA-Rho-kinase1 (ROCK1), Jun N-terminal kinase (JNK), P38 and Erk mitogen-activated protein kinases (MAPKs).11–20 We have previously shown that PP2 decreases ROCK1 expression in a head trauma model.11
SFK inhibitors may be attractive candidates for future clinical translation to humans with IVH. SFK inhibitors have been shown to be safe in human cancer trials.36,37 The nanoparticle delivery methods used here for the SFKs are FDA approved for human use.29,30 SFK inhibitors block many injury pathways after IVH,11–20 and even a single dose produced extremely robust cognitive benefits post IVH.
There are several limitations to the current study. The dose responses of PP2 and siRNA-Fyn were not addressed in the specific models examined in this study. The time window was not addressed as well since it is unclear how long after IVH that SFK inhibition would still be effective. The spatial memory function at several months following IVH was not examined and will need future study. Sample size calculations were not performed since there were no previous data on which to estimate effect size.
In conclusion, we propose that SFKs mediate the hippocampal neuronal cell death and spatial memory deficits produced by intraventricular thrombin and intraventricular blood. Inhibiting SFKs including a single SFK family member Fyn can prevent the neuronal cell death and spatial memory deficits that occur following non-traumatic IVH in these animal models.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by American Heart Association Beginning Grant in Aid (DZL), NIH grants R01NS089901 (DZL) and R01NS066845 (FRS).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Da Zhi Liu, Ben Waldau, Bradley P Ander, Xinhua Zhan, Boryana Stamova, Glen C Jickling, Bruce G Lyeth, and Frank R Sharp participated in the conduct of the studies and have read and reviewed the manuscript. Da Zhi Liu and Frank R Sharp designed the experiments and wrote the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Badjatia N, Rosand J. Intracerebral hemorrhage. Neurologist 2005; 11: 311–324. [DOI] [PubMed] [Google Scholar]
- 2.Flaherty ML, Haverbusch M, Sekar P, et al. Long-term mortality after intracerebral hemorrhage. Neurology 2006; 66: 1182–1186. [DOI] [PubMed] [Google Scholar]
- 3.Fogelholm R, Murros K, Rissanen A, et al. Long term survival after primary intracerebral haemorrhage: A retrospective population based study. J Neurol Neurosurg Psychiatry 2005; 76: 1534–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanley DF. Intraventricular hemorrhage: Severity factor and treatment target in spontaneous intracerebral hemorrhage. Stroke 2009; 40: 1533–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nys GM, van Zandvoort MJ, de Kort PL, et al. Cognitive disorders in acute stroke: Prevalence and clinical determinants. Cerebrovasc Dis 2007; 23: 408–416. [DOI] [PubMed] [Google Scholar]
- 6.Wong GK, Wong R, Mok VC, et al. Clinical study on cognitive dysfunction after spontaneous subarachnoid haemorrhage: Patient profiles and relationship to cholinergic dysfunction. Acta Neurochirurg 2009; 151: 1601–1607. [DOI] [PubMed] [Google Scholar]
- 7.Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol 2002; 17: 1137–1152. [DOI] [PubMed] [Google Scholar]
- 8.Oda H, Kumar S, Howley PM. Regulation of the src family tyrosine kinase blk through e6ap-mediated ubiquitination. Proc Natl Acad Sci USA 1999; 96: 9557–9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatosyan AG, Mizenina OA. Kinases of the src family: Structure and functions. Biochemistry (Mosc) 2000; 65: 49–58. [PubMed] [Google Scholar]
- 10.Wang H, Reiser G. The role of the ca2+-sensitive tyrosine kinase pyk2 and src in thrombin signalling in rat astrocytes. J Neurochem 2003; 84: 1349–1357. [DOI] [PubMed] [Google Scholar]
- 11.Liu D, Sharp FR, Van KC, et al. Inhibition of src family kinases protects hippocampal neurons and improves cognitive function after traumatic brain injury. J Neurotrauma 2014; 31: 1268–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi AD, Dimitropoulou C, Thangjam G, et al. Hsp90 inhibitors prevent lps-induced endothelial barrier dysfunction by disrupting rhoa signaling. Am J Resp Cell Mol Biol 2013; 50: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubreuil CI, Marklund N, Deschamps K, et al. Activation of rho after traumatic brain injury and seizure in rats. Exp Neurol 2006; 198: 361–369. [DOI] [PubMed] [Google Scholar]
- 14.Jeon BT, Jeong EA, Park SY, et al. The rho-kinase (rock) inhibitor y-27632 protects against excitotoxicity-induced neuronal death in vivo and in vitro. Neurotox Res 2013; 23: 238–248. [DOI] [PubMed] [Google Scholar]
- 15.Liu DZ, Ander BP. Cell cycle phase transitions: Signposts for aberrant cell cycle reentry in dying mature neurons. J Cytol Histol 2011; 2: e101. [Google Scholar]
- 16.Liu DZ, Sharp FR. The dual role of src kinases in intracerebral hemorrhage. Acta Neurochirurg Suppl 2011; 111: 77–81. [DOI] [PubMed] [Google Scholar]
- 17.Ardizzone TD, Lu A, Wagner KR, et al. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke 2004; 35: 2587–2591. [DOI] [PubMed] [Google Scholar]
- 18.Liu DZ, Ander BP. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of aberrant cell cycle diseases: An update. Scientific World J 2012; 2012: 491737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu DZ, Ander BP, Sharp FR. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of central nervous system diseases. Neurobiol Dis 2010; 37: 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez PL, Sahay S, Olabisi OO, et al. Rock i-mediated activation of nf-kappab by rhob. Cell Signall 2007; 19: 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu DZ, Ander BP, Xu H, et al. Blood-brain barrier breakdown and repair by src after thrombin-induced injury. Ann Neurol 2010; 67: 526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu DZ, Cheng XY, Ander BP, et al. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol Dis 2008; 30: 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Q, Tang J, Tan L, et al. Intracerebral hematoma contributes to hydrocephalus after intraventricular hemorrhage via aggravating iron accumulation. Stroke 2015; 46: 2902–2908. [DOI] [PubMed] [Google Scholar]
- 24.Misra A, Ganesh S, Shahiwala A, et al. Drug delivery to the central nervous system: A review. J Pharm Pharm Sci 2003; 6: 252–273. [PubMed] [Google Scholar]
- 25.Diemer NH, Bruhn T, Christensen T, et al. Glutamate-mediated mechanisms in delayed neuronal death after cerebral ischemia. In: Ito U, Kirino T, Kuroiwa T, et al. (eds) Maturation Phenomenon in Cerebral Ischemia. 2nd ed. Berlin: Springer Berlin Heidelberg, 1997, pp. 53–56 . [Google Scholar]
- 26.Akiyama H, Ikeda K, Kondo H, et al. Thrombin accumulation in brains of patients with alzheimer’s disease. Neurosci Lett 1992; 146: 152–154. [DOI] [PubMed] [Google Scholar]
- 27.Thomas SM, Brugge JS. Cellular functions regulated by src family kinases. Annu Rev Cell Dev Biol 1997; 13: 513–609. [DOI] [PubMed] [Google Scholar]
- 28.Ardizzone TD, Zhan X, Ander BP, et al. Src kinase inhibition improves acute outcomes after experimental intracerebral hemorrhage. Stroke 2007; 38: 1621–1625. [DOI] [PubMed] [Google Scholar]
- 29.Eifler AC, Thaxton CS. Nanoparticle therapeutics: FDA approval, clinical trials, regulatory pathways, and case study. Methods Mol Biol 2011; 726: 325–338. [DOI] [PubMed] [Google Scholar]
- 30.Davis ME, Zuckerman JE, Choi CH, et al. Evidence of rnai in humans from systemically administered sirna via targeted nanoparticles. Nature 2010; 464: 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene 2004; 23: 7906–7909. [DOI] [PubMed] [Google Scholar]
- 32.Umemori H, Wanaka A, Kato H, et al. Specific expressions of fyn and lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res Mol Brain Res 1992; 16: 303–310. [DOI] [PubMed] [Google Scholar]
- 33.Ross CA, Wright GE, Resh MD, et al. Brain-specific src oncogene mrna mapped in rat brain by in situ hybridization. Proc Natl Acad Sci USA 1988; 85: 9831–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trepanier CH, Jackson MF, MacDonald JF. Regulation of nmda receptors by the tyrosine kinase fyn. FEBS J 2012; 279: 12–19. [DOI] [PubMed] [Google Scholar]
- 35.Lu W, Fang W, Li J, et al. Phosphorylation of tyrosine 1070 at the glun2b subunit is regulated by synaptic activity and critical for surface expression of n-methyl-d-aspartate (nmda) receptors. J Biol Chem 2015; 290: 22945–22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herold CI, Chadaram V, Peterson BL, et al. Phase ii trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of src inhibition to escalate dosing. Clin Cancer Res 2011; 17: 6061–6070. [DOI] [PubMed] [Google Scholar]
- 37.Gucalp A, Sparano JA, Caravelli J, et al. Phase ii trial of saracatinib (azd0530), an oral src-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer 2011; 11: 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.