

Abstract
Stroke-induced blood–brain barrier breakdown promotes complications like cerebral edema and hemorrhagic transformation, especially in association with therapeutical recanalization of occluded vessels. As arteries, capillaries and veins display distinct functional and morphological characteristics, we here investigated patterns of blood–brain barrier breakdown for each segment of the vascular tree in rodent models of embolic, permanent, and transient middle cerebral artery occlusion, added by analyses of human stroke tissue. Twenty-four hours after ischemia induction, loss of blood–brain barrier function towards FITC-albumin was equally observed for arteries, capillaries, and veins in rodent brains. Noteworthy, veins showed highest ratios of leaky vessels, whereas capillaries exhibited the most and arteries the least widespread perivascular tracer extravasation. In contrast, human autoptic stroke tissue exhibited pronounced extravasations of albumin around arteries and veins, while the pericapillary immunoreactivity appeared only faint. Although electron microscopy revealed comparable alterations of the arterial and capillary endothelium throughout the applied animal models, structural loss of arterial smooth muscle cells was only observed in the translationally relevant model of embolic middle cerebral artery occlusion. In light of the so far available concepts of stroke treatment, the consideration of a differential vascular pathophysiology along the cerebral vasculature is likely to allow development of novel effective treatment strategies.
Keywords: Blood–brain barrier, stroke, focal ischemia, neurovascular unit, endothelium
Introduction
Adequate neuronal function of the central nervous system strictly depends on the maintenance of the unique neuronal microenvironment, which is orchestrated by different cell populations, commonly described as neurovascular unit (NVU).1,2 This perspective further emphasizes the permanent bidirectional interactions of the vasculature with the associated blood–brain barrier (BBB) on the one hand and the parenchymal compartment comprising neurons and glial cells on the other.3,4 Therefore, vascular function and dysfunction have recently turned into focus in understanding the pathophysiology of several diseases such as acute ischemic stroke.5,6 In the setting of stroke, the underlying occlusion of supplying arteries and the resulting energy failure typically affect neuronal function, but also the vasculature itself.2,7 Therefore, loss of BBB integrity with extravasation of water and blood-borne molecules into the ischemia-affected brain tissue is considered as a major factor for edema formation, consecutively associated with poor clinical outcome.8–11 Further, extravasation of blood-borne albumin is assumed to trigger stroke-related complications like astrocyte-mediated epileptogenesis.12,13
Although stroke represents one of the leading causes of death and accounts for a relevant number of newly acquired disabilities,14 treatment options during the early, highly vulnerable phase of stroke evolution are still limited. In light of data from multicenter trials, therapeutic strategies currently focus on recanalizing approaches, either by intravenous application of recombinant tissue plasminogen activator (rtPA),15 or the very recently added mechanical thrombectomy in terms of catheter-based interventions.16,17 However, with increasing time from ischemia onset, acute vessel re-opening is assumed to be associated with an increased risk of hemorrhagic transformation and intracerebral bleeding, classically attributed to a so-called reperfusion injury.18,19 Therefore, ongoing attempts in stroke research include a more detailed understanding of cellular alterations responsible for tissue damage caused by the ischemic stimulus itself and added approaches for acute vessel recanalization. In light of the primarily intended recanalization of occluded vessels, especially affections of the vascular constituent within the NVU, i.e. the BBB, are of increasing interest.20
In the past, the associated loss of BBB integrity following stroke was predominantly ascribed to altered endothelial tight junction (TJ) complexes.21–24 However, early reports rather suggested a transendothelial leakage pattern for ischemia-induced BBB breakdown, which leaves TJs intact.8 Although molecular alterations of TJ proteins such as phosphorylations cannot be ruled out, novel data further support the view of a predominantly transendothelial leakage mechanism after stroke,25,26 while electron microscopy revealed detectable endothelial TJs.27,28 Using different models of focal cerebral ischemia, our group has recently demonstrated that vessels showing BBB breakdown exhibited different stages of endothelial degeneration, ranging from endothelial edema and increased endothelial vesicles to impaired cellular integrity and loss of the endothelial layer.27,29 Overall, these data strongly support the perspective that structural alterations of the endothelial layer may play a pivotal role during the evolution of the ischemic lesion and associated treatment-related complications like hemorrhagic transformation and intracerebral bleeding.
As more than one thousand experimental treatments of stroke failed to provide successful translation into the clinics,30 detailed knowledge of potential vascular features underlying secondary injuries is a prerequisite to develop advanced treatment concepts in terms of potential co-medications to rtPA or mechanical thrombectomy. Aiming on stroke-induced alterations of the endothelium as part of the BBB, differing features of the brain’s vasculature need to be considered for each segment of the vascular tree.31 In fact, precapillary arterial segments, in contrast to capillaries, exhibit the highest pressure drop in the vascular bed and thereby primarily contribute to the vascular resistance.32 In the setting of ischemia and reperfusion, the combination of higher pressures and higher blood flow velocities in arterial compared to capillary or venous vessels might therefore cause mechanical stress which is more pronounced in the arterial endothelium than in other segments of the vascular tree.33
The present study was consequently aimed to address differences of vascular affection for arteries, capillaries, and veins. For this purpose, we used fluorescence microscopy to systematically analyze the patterns of BBB breakdown for different segments of the vascular tree using three different models of experimental focal cerebral ischemia and human autoptic brain tissue. Further, the differential endothelial affection of arterial and non-arterial vessels was addressed by electron microscopy in areas of apparent BBB breakdown.
Materials and methods
Experimental study setup
All experiments involving animals were performed along the ARRIVE guidelines and according to the European Communities Council Directive (86/609/EEC) and were approved by local authorities (Landesdirektion Leipzig, Germany). Animals were housed with a light/dark cycle of 12 h with free access to food and water at a temperature of 21℃ to 22℃ and a humidity of 45–60%.
Adult male and female wildtype mice (129Sv/B6) weighing about 25 g, bred by the Medizinisch-Experimentelles Zentrum (Leipzig, Germany), were subjected to right-sided middle cerebral artery occlusion (MCAO) either with permanent occlusion (pMCAO) or with transient occlusion (tMCAO) for 90 min. Sufficient stroke-induced cerebral affection was confirmed by a relevant neurological deficit at 24 h after ischemia induction, which had to comply with at least two points on the standardized scoring system described by Menzies et al.,34 representing a pre-defined study inclusion criterion. After neurological evaluation, the permeability marker FITC-albumin (Sigma, Taufkirchen, Germany, 2 mg dissolved in 0.1 ml saline) was intravenously administered with a circulation period of 60 min to allow demarcation of areas with impaired BBB integrity. Animals were then sacrificed and transcardially perfused with saline, followed by snap freezing of the carefully removed brains for fluorescence microscopy (pMCAO, n = 4; tMCAO, n = 3). For electron microscopy, animals were perfused with saline, followed by a fixative containing 4% paraformaldehyde (PFA) and 0.5% GA in phosphate-buffered saline (PBS) (pMCAO, n = 3; tMCAO, n = 3).
Additional analyses for fluorescence microscopy further included adult male wildtype mice (about 25 g) which underwent pMCAO (n = 4) for 4 h to address patterns of BBB breakdown at an earlier time point.
Further, we analyzed brains from adult male Wistar rats weighing about 300 g, bred by Charles River (Sulzfeld, Germany), which underwent the translationally relevant model of embolic middle cerebral artery occlusion (eMCAO). Twenty-four hours after ischemia onset, animals were neurologically evaluated using the Menzies score34 and had to demonstrate a pre-defined score of at least 2 to enter the study. FITC-albumin as permeability marker (20 mg dissolved in 1 ml saline) was intravenously administered, followed by a circulation period of 60 min prior to sacrifice. For fluorescence microscopy (eMCAO, n = 4), animals were sacrificed and transcardially perfused with saline followed by snap freezing of the brains, while the tissue for electron microscopy (eMCAO, n = 3) was perfused with saline followed by the fixative containing 4% PFA and 0.5% GA in PBS, analogous to mice.
Induction of focal cerebral ischemia
Surgical procedures in mice were generally performed in deeply anesthetized animals using intraperitoneally applied etomidate (33 mg/kg body weight, Hypnomidate, Janssen-Cilag, Neuss, Germany; in some animals with an additional dose for filament removal). For the models of pMCAO and tMCAO, ischemia was induced according to Longa et al.35 with some minor modifications as described before in detail.36 Briefly, a standardized and silicon-coated 6–0 monofilament (Doccol Corporation, Redlands, CA, USA) was inserted into the right external or common carotid artery, moved forward into the internal carotid artery to approach the middle cerebral artery until bending was observed or resistance felt. For the model of tMCAO, the filament was removed 90 min after ischemia induction, whereas the filament was left in position for pMCAO.
The model of eMCAO in rats was performed as reported before.27 In brief, one day prior to ischemia induction rat blood was collected to prepare standardized clots in polyethylene tubes with a medium length of 50 mm. The polyethylene tube with collected blood clot was inserted into the right external or common carotid artery and moved forward into the distal part of the internal carotid artery. Here, the blood clot was released with a small bolus of saline to occlude the origin of the middle cerebral artery. Anesthesia in rats was achieved using 2.0–2.5% isoflurane (Isofluran Baxter, Baxter, Unterschleißheim, Germany; mixture: 70% N2O and 30% O2; vaporisator: VIP 3000, Matrix, New York, NY, USA).
In general, during anesthesia and the surgical procedure, the body temperature was monitored and adjusted to 37℃ using a rectal probe combined with a thermostatically controlled warming pad (Fine Science Tools, Heidelberg, Germany) for all the models applied.
Human brain tissue
Autoptic tissue from human stroke cases was provided by the Department of Neuropathology, University of Leipzig, Germany (approved by written consent from the University Hospital Leipzig). The tissue was fixed using 4% PFA, dehydrated and embedded in paraffin. After histopathological screening of the tissue and verification of infarcted areas, microtome sections of 11 µm thickness were prepared for fluorescence microscopy. Further, non-affected tissue from corresponding contralateral areas or non-affected ipsilateral areas served as controls. Therefore, paraffin sections of seven human stroke cases were analyzed (Table 1).
Table 1.
Characterization of human brain tissue.
| Case | Age | Gender | Localization | Histopathological stage |
|---|---|---|---|---|
| #1 | 63 | Male | MCA right, right hemisphere, occipital | II |
| #2 | 83 | Female | MCA left, basal ganglia | I-II, III |
| #3 | 72 | Male | MCA right, occipital | III |
| #4 | 70 | Female | MCA left, basal ganglia | II |
| #5 | 59 | Male | MCA right, Sylvian fissure | III |
| #6 | 69 | Male | MCA left, basal ganglia | III |
| #7 | 62 | Female | MCA right, frontal | II-III |
MCA: middle cerebral artery.
Fluorescence microscopy and quantification
Brains were collected and snap frozen in cryo embedding medium (Sakura Finetek, Torrance, CA, USA) using isopentane on dry ice. Cryostat sections of 10 µm thickness were prepared with a cryostat (Leica, Wetzlar, Germany). Cryostat sections were post-fixed using ethanol, rinsed in PBS followed by blocking of unspecific binding sites with 1% normal goat serum (NGS) in PBS containing 0.3% Triton X-100 for permeabilization. After that, primary antibodies were incubated at 4℃ overnight, rinsed in PBS and incubated with appropriate fluorochromated antibodies at room temperature for 2 h at a dilution of 1:250. To demark the vasculature, immunolabeling of laminin (rabbit anti pan-laminin, 1:200; Dako, Hamburg, Germany) was applied, while mouse anti-smooth muscle actin (SMA, 1:500; Sigma) was used to further differentiate arteries from veins. Capillaries were identified by diameter. Pericytes were identified by immunolabeling for CD13 (pericytic aminopeptidase N [pAPN], rat anti CD13, 1:200, Acris, Herford, Germany), while occludin (guinea pig anti occludin, 1:200; Acris) and claudin-5 (rabbit anti claudin-5, 1:200; Abcam, Cambridge, UK) were used to demark endothelial TJs. Areas of ischemia-induced BBB breakdown were identified by extravasation of intravenously applied FITC-albumin as described before.27,29
In human autoptic brain tissue, BBB breakdown in ischemia-affected areas of human brain tissue was visualized by labeling of extravasated albumin using rabbit anti-albumin (1:200, Synaptic Systems, Göttingen, Germany). Goat anti-collagen IV (1:50; Merck Millipore, Billerica, MA, USA) was applied as a pan-vascular marker in humans. Autofluorescence of human brain tissue was quenched with 0.3% Sudan Black B dissolved in 70% ethanol at 60℃ for 1 min to achieve an optimal signal to background ratio. Therefore, tissue-specific autofluorescence was nearly abolished with the fluorophores of secondary antibodies being preserved. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, 1:10,000; Sigma-Aldrich, Hamburg, Germany).
For quantitative analysis, in areas of tracer extravasation the total number of cross-sectioned vessels as well as the number of leaky and non-leaky arteries, capillaries and veins were counted in at least 20 fields of view (20 × objective) of different, non-consecutive sections of each animal to obtain the ratio of leaky arteries, capillaries and veins. In total, these analyses included 160–200 arteries, 170–250 veins, and 3300–3700 capillaries per group.
In addition, the spreading of the tracer indicating BBB breakdown was addressed around arteries, capillaries, and veins by measuring the area covered by extravascular FITC-albumin and the area covered by the respective vessel using Image J (version 1.48, NIH, Bethesda, MD, USA) in representative fields of view with apparent tracer extravasation. As the respective sections usually exhibit cross- and longitudinally sectioned vessels, a ratio of the area of extravasated FITC-albumin to the area covered by the cross-sectioned vessel was created, thereby normalizing differences of the cross-sectioned length and diameter of the respective vessels. These measurements included around 300 vessels per group and at least 20 arteries, capillaries, and veins of each of the animals for all three models applied (eMCAO, n = 4; pMCAO, n = 4; tMCAO, n = 3).
Electron microscopy and quantification
Brains were post-fixed in 4% PFA and 0.5% GA overnight at 4℃ followed by preparation of 60 µm sections using a vibrating microtome (Leica). After blocking in Tris-buffered saline with 1% of bovine serum albumin (TBS-BSA), the tissue was incubated with peroxidase-conjugated anti-FITC IgG (1:2000 = 0.5 µg/ml; Dianova, Hamburg, Germany) over night at 4℃. Subsequently, the tissue was stained using diaminobenzidine (DAB) to gain an electron dense adduct allowing detection of extravasated FITC-albumin by light- and electron microscopy. Further, the sections were stained using 0.5% osmium tetroxide in PBS for 30 min and 1% uranyl acetate for 60 min. Dehydration was followed by embedding of the sections in Durcupan (Sigma-Aldrich) between coated microscope slides and cover glasses and polymerization of the resin for 48 h at 56℃. Thus, areas of BBB breakdown indicated by extravasation of FITC-albumin can be identified and precisely trimmed before ultra-thin sections of 55 nm were acquired using an ultra-microtome (Leica Ultracut R, Leica). Ultra-thin sections were stained with lead citrate for 6 min. Ultrastructural analysis was performed with a Zeiss SIGMA electron microscope equipped with an STEM detector (Zeiss, Oberkochen, Germany).
For quantification, areas showing BBB breakdown indicated by DAB were identified in blocks of resin by light microscopy, trimmed and sectioned. In non-consecutive ultra-thin sections, the vessels contained were evaluated to assess the stage of endothelial ultrastructural affection as previously described.29 Mean values of ultrastructural damage of endothelial cells and arterial smooth muscle cells were compared for the model of eMCAO, pMCAO, and tMCAO. For each model 160–300 vessels were staged with no less than 20 arterial vessels per group (eMCAO, pMCAO, tMCAO; each n = 3).
Statistical analyses
The data obtained were processed and evaluated using Graph Pad Prism 5.01v (GraphPad Software Inc., La Jolla, CA, USA). As the presented data were generally arranged in three or more groups, the non-parametric Kruskal–Wallis test was used to check for statistically significant differences between groups, added by Dunn’s multiple comparison test. In general, a p < 0.05 was considered statistically significant.
Results
Ischemia-induced extravasation of FITC-albumin is observed in all segments of the cerebral vasculature with no particular limitation to arterial vessels
In order to investigate whether arterial vessels are preferentially affected by ischemia or reperfusion, cryostat sections of ischemic hemispheres from animals which underwent eMCAO, pMCAO, and tMCAO were analyzed for presence of BBB breakdown as indicated by FITC-albumin extravasation. Further, immunolabeling of laminin was applied to demark all sectioned vessels irrespective of their position in the vascular tree according to Sixt et al.37 Arteries and veins were differentiated either by immunoreactivity for SMA (arteries) or lack of immunoreactivity (veins), while vessels with a diameter of less than 8 µm were regarded as capillaries (adapted from Hawkes et al.38). Although CNS pericytes have been reported to express SMA in vitro and distinct animal models,39 the authors ensured that SMA immunoreactivity is restricted to arterial vessels, whereas cerebral pericytes surrounding capillaries and veins do not show detectable levels of SMA immunoreactivity (Supplementary Figure 1(a) and (b)), which is in line with previous reports.40,41 While contralateral extravasation of FITC-albumin was not observed, the ipsilateral striatum and cortical areas were regularly affected throughout the applied models (Supplementary Figure 1(c)). Remarkably, extravasation of the tracer was apparently not confined to a particular vascular segment, but exhibited comparable patterns of extravasations around arteries, capillaries, and veins throughout the models applied (Figure 1(a)). Although endothelial cells of arterial vessels are believed to face higher pressures and blood flow velocities,33 lower magnification (Figure 1(b)) repeatedly revealed that particular arteries (arrow) did not show extravasation of FITC-albumin while directly neighboring capillaries or veins regularly exhibited leakage into the adjacent neuropil beyond the glial basal lamina as demarked by laminin staining (Figure 1(b)).
Figure 1.

(a) The models of tMCAO, pMCAO, and eMCAO exhibit comparable patterns of BBB breakdown around arteries, capillaries, and veins at 24 h after ischemia induction. Arteries are demarked by SMA (red), while veins are lacking a continuous layer of vascular smooth muscle cells. Capillaries were identified by diameter, smaller than 8 µm in mice and rats. Scale bar: 10 µm. (b) Low magnification showing representative area of BBB breakdown after 24 h of experimental cerebral ischemia indicated by FITC-albumin (FITC, green) extravasation. Blood vessels are demarked by laminin. Noteworthy, the sectioned SMA-positive artery (arrow) does not show leakage of FITC-albumin, while adjacent capillaries and veins (arrow head) are surrounded by extravascular accumulations of the tracer. Thus, arterial vessels are not preferentially affected by ischemia-induced BBB breakdown. Scale bar: 50 µm.
Since our previous reports on detectable TJ-proteins in areas of BBB breakdown were not assigned to distinct segments of the vascular tree,29 we here investigated the expression of critical TJ proteins in arteries, capillaries, and veins showing FITC-albumin extravasation. Here, immunoreactivity for TJ proteins was preserved throughout the vascular tree in areas of BBB breakdown in all the models applied as exemplarily shown for claudin-5 and occludin (Figure 2). Moreover, subsequent experiments indicated that these patterns of BBB breakdown along arteries, capillaries, and veins are not restricted to 24 h after ischemia onset, but can also be observed after 4 h of pMCAO (Supplementary Figure 2).
Figure 2.

Representative images of multiple fluorescence labeling reveal detectable immunoreactivity for the critical TJ proteins claudin-5 (a) and occludin (b) in arterial, capillary, and venous vessels in areas showing FITC-albumin (FITC) extravasation 24 h after ischemia induction in all the applied models. Arteries are demarked by SMA (blue), while nuclei were counterstained with DAPI (white). A differential distribution of TJ proteins compared to contralateral areas was not observed. Scale bar: 10 µm.
Veins consistently exhibited highest ratio of leaky vessels while capillaries showed most widespread extravasations of FITC-albumin
As clear-cut morphological differences between the patterns of BBB breakdown along arterial, capillary, and venous segments were not detectable, the ratio of vessels showing extravasation of FITC-albumin was calculated and compared in regions of apparently impaired BBB. Therefore, the total number of cross-sectioned arteries, capillaries, and veins with and without detectable FITC-albumin extravasation was investigated in areas of BBB breakdown to calculate the ratio of leaky vessels. Of note, the highest ratio of leaky vessels was consistently observed for veins in all the models applied (Figure 3(a)). Here, 79% (eMCAO), 78% (pMCAO), and 72% (tMCAO) of the cross-sectioned veins showed tracer extravasation beyond the parenchymal basal lamina, with statistically significant differences compared to capillary vessels in the model of eMCAO (p = 0.0308, n = 4, Kruskal–Wallis test, followed by Dunn’s multiple comparison test) and pMCAO (p = 0.0183, n = 4). Further, a robust trend towards a more frequent affection of cross-sectioned veins was observed in the model of tMCAO, but failed to reach statistical significance (p = 0.0794, n = 3). To ensure robust calculations, an overall number of 160–200 arteries, 170–250 veins, and 3300–3700 capillaries were analyzed per group.
Figure 3.
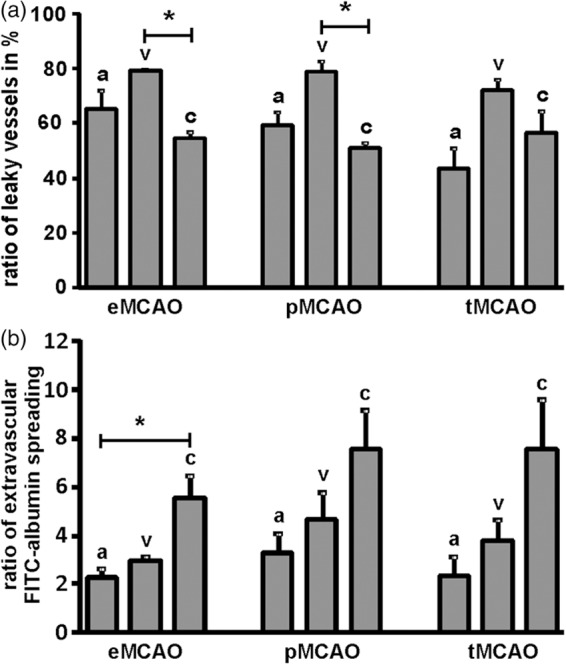
(a) The relative contribution to FITC-albumin extravasation was addressed by calculating the ratio of leaky arteries (a), veins (v) and capillaries (c) in areas showing FITC-albumin extravasation in the models of eMCAO, pMCAO, and tMCAO at 24 h after ischemia induction. (b) Further, the ratio of extravascular FITC-albumin spreading for arterial, capillary, and venous vessels was compared by calculating a ratio reflecting the area showing extravascular tracer accumulation per area of the respective cross-sectioned vessel in µm2 to normalize for differences in diameter and length of analyzed vessels. Bars are given as means and added lines represent the standard error of the mean (Kruskal–Wallis, added by Dunn’s multiple comparison test; eMCAO n = 4, pMCAO n = 4, tMCAO n = 3).
To investigate whether veins also quantitatively exhibited a more pronounced vascular leakage, the ratio of FITC-albumin spreading was calculated around arteries, capillaries, and veins, thereby accounting and normalizing for differences of cross-sectioned length and diameter of individual vessels analyzed. Here, robust trends to higher ratios of FITC-spreading were consistently observed in capillary vessels throughout the models applied (Figure 3(b)), reaching statistical significance in the model of eMCAO (p = 0.0183, n = 4, Kruskal–Wallis test, followed by Dunn’s multiple comparison test). Notably, the patterns of BBB breakdown proved comparable for all the applied models including the reperfusion scenario reflected by the model of tMCAO with around two times higher ratios of FITC-albumin spreading for capillaries compared to arterial vessels (Figure 3(b)). These measurements included around 300 vessels per group.
Ultrastructural analysis reveals comparable patterns of endothelial degeneration in arterial and non-arterial vessels
As fluorescence microscopy consistently revealed smaller ratios of FITC-albumin spreading around leaky arteries compared to capillaries, electron microscopy was applied to investigate whether this observation correlates with a less affected endothelial layer in arterial vessels. Therefore, the ultrastructural pattern of endothelial degeneration in arterial vessels was compared with the morphology of endothelial cells of closely associated capillaries. For this purpose, areas showing BBB breakdown as indicated by FITC-albumin extravasation were identified by light microscopy on resin-embedded samples followed by preparation of ultra-thin sections and ultrastructural evaluation of the endothelial layer of each arterial and non-arterial vessel contained. With an average surface area of 0.5 mm2, the sectioned vessels within the ischemic areas are thus assumed to be equally affected. As described before,27,29 the endothelial layer on contralateral control areas exhibited no signs of degeneration with a continuous cellular surface, intact organelles and no signs of tracer extravasation (Figure 4). Furthermore, vascular smooth muscle cells (M) of arterial vessels showed a normal morphology, while the cytoplasm was found to be comparably electron dense to the endothelial cells.
Figure 4.

Representative electron micrographs illustrating ultrastructural alterations of ischemia-affected arteries and adjacent capillaries. In contralateral control areas, the endothelium of capillary as well as arterial vessels appears smooth and continuous (arrow heads). The cytoplasm is dense and organelles are unaffected, while arterial smooth muscle cells (M), delineated by vascular basal laminae (transparently highlighted in red), show an isodense cytoplasm compared to the underlying endothelial cells. Extravasations of FITC-albumin as indicated by electron dense grains of DAB or structural alterations of the adjacent neuropil were not observed. In contrast, ischemia-affected areas of FITC-albumin extravasation exhibit degeneration of endothelial cells in arterial vessels as well as in adjacent capillaries. The luminal plasma membrane (arrow heads) is discontinuous and cellular integrity is lost. Thus, barrier function to FITC-albumin is lost and endothelium or debris is often filled with grains of DAB throughout the applied models. However, despite of an impaired endothelial integrity, vascular smooth muscle cells (M) in the models of tMCAO and pMCAO appear to be much less affected, showing an intact cellular structure and a less dense cytoplasm as signs of an intracellular edema, only. In contrast, after eMCAO, the cellular integrity of arterial smooth muscle cells (M) is lost. Here, cellular fragments (asterisks) and DAB-filled debris are often found between the basal laminae of the arterial vascular wall. E: erythrocyte; L: vascular lumen, Scale bars: 1 µm.
In areas of tracer extravasation, the endothelial surface regularly appeared to be discontinuous in all the three models applied (Figure 4). Therefore, cellular integrity is lost and endothelial cells exhibited electron dense grains of DAB in the cytoplasmic remnants or cellular debris as a sign of impaired BBB integrity towards FITC-albumin. Further, presence of DAB grains beyond the vascular basal lamina indicated extravasation of FITC-albumin into the adjacent parenchyma around affected vessels. Of note, the endothelial layer of both, arteries and capillaries displayed comparable signs of degeneration (Figure 4). In the mouse models of pMCAO and tMCAO (Figure 4), the vascular smooth muscle cells (M) of arteries appeared to be much less affected than the respective endothelium, showing a less dense cytoplasm as a sign of intracellular edema, only. However, in the model of eMCAO (Figure 4), arterial smooth muscle cells exhibited a more severe structural affection which was regularly comparable to the underlying endothelial cells with loss of cellular integrity and accumulation of DAB grains between vascular basal laminae.
Arterial and capillary vessels exhibit equal patterns of endothelial degeneration, while the arterial wall is more severely affected in the model of eMCAO
To further quantify the observed ultrastructural alterations of the arterial endothelial layer and the vascular smooth muscle cells, arteries and adjacent capillary vessels were evaluated as previously described. Here, areas of tracer extravasation were found to regularly exhibit comparable patterns of vascular affection ranging from cellular edema to endothelial loss, whereas border zones showing no tracer extravasation exhibited signs of an endothelial edema, only.29 In brief, non-affected vessels and endothelial cells were classified as ‘stage 0’, endothelial edema combined with no signs of tracer extravasation as ‘stage 1’, distinct uptake of FITC-albumin into endothelial cells as ‘stage 2’, endothelial cell loss or loss of endothelial integrity and extravasation beyond the vascular basement membrane as ‘stage 3’, whereas a further loss of basement membrane integrity and extravasation of erythrocytes was classified as ‘stage 4’. Increasing cellular and vascular affection are thus mirrored by increasing scores. Again, each vessel of different non-consecutive sections of 0.5 mm2 obtained from areas with apparent tracer extravasation was systematically analyzed. For robust data generation, per group not less than 20 to 30 arteries were compared with adjacent capillaries thereby including 160–300 vessels per group (eMCAO, pMCAO, tMCAO; each n = 3; Kruskal–Wallis test, added by Dunn’s Multiple comparison test). Further, mean values for the level of ultrastructural affection were calculated and compared in between groups and for arterial endothelial cells, arterial smooth muscle cells, and adjacent capillary endothelial cells. Of note, the pattern of endothelial damage proved comparable for arteries and adjacent capillaries in all applied models (Figure 5). However, mean values for arterial smooth muscle cells were significantly lower after pMCAO and tMCAO compared to the respective endothelial cells, while the smooth muscle cells of arteries following eMCAO exhibited values of ultrastructural affection that were comparable to the respective endothelial cells and the surrounding capillaries. Further, the ratio of arteries showing less affected smooth muscle cells compared to the directly underlying endothelium was calculated and compared for all the models applied. Noteworthy, the majority of arterial vessels in the models of pMCAO and tMCAO exhibited less affected smooth muscle cells. In contrast, in the model of eMCAO only 13% of the analyzed arteries exhibited smooth muscle cells which were less affected than the respective endothelium. Here, the majority of smooth muscle cells exhibited severe structural alterations which were comparable to the endothelial layer (Figure 5).
Figure 5.

Endothelial cells of arteries and adjacent capillaries as well as arterial smooth muscle cells were evaluated according to the stage of ultrastructural damage25 to create a mean value of structural affection for the models of eMCAO, pMCAO, and tMCAO. Comparable signs of endothelial degeneration were observed for capillaries and arteries throughout the models. Arterial smooth muscle cells showed less severe structural affections in the models of tMCAO and pMCAO. Of note, in the model of eMCAO, arterial smooth muscle cells exhibited equal patterns of structural affection compared to the respective endothelial cells. Further, the ratio of arteries showing less affected smooth muscle cells compared to the underlying endothelium was calculated in areas of BBB breakdown for the models of eMCAO, pMCAO, and tMCAO. Bars are given as means and added lines represent the standard error of the mean (Kruskal–Wallis test, Dunn’s multiple comparison test; each n = 3, 20–30 arteries per group).
Human autoptic stroke tissue reveals localized extravasation of endogenous serum albumin in ischemic areas with pronounced paravascular accumulation around arteries and veins
Since fluorescence microscopy revealed BBB breakdown for each segment of the vascular tree in all three models of experimental focal cerebral ischemia, human autoptic brain tissue from seven different patients suffering from ischemic stroke (Table 1) was analyzed to determine the characteristics of BBB breakdown in human tissue. Therefore, immunofluorescence labeling of albumin was used to demark areas of albumin extravasation indicative for BBB breakdown. Here, vessels were identified by visualization of nuclei using DAPI, while arterial vessels were identified by presence of SMA expression of vascular smooth muscle cells.38,40 In line with the animal models, ischemic areas of human stoke tissue are characterized by extravasation of albumin leading to an enhanced parenchymal immunoreactivity (Figure 6(a)) in histopathologically verified regions of cerebral ischemia compared to adjacent, non-affected areas, or control regions. Further, higher magnification revealed a pronounced extravasation of albumin around SMA-positive arteries and SMA-negative veins (Figure 6(b)). However, identification of capillary endothelial cells by DAPI labeling alone is rather challenging, while the general immunoreactivity of albumin in the ischemic parenchyma rendered a clear-cut detection of extravasated albumin around capillaries even more difficult (not shown).
Figure 6.

(a) Autoptic human tissue with histopathologically verified stroke shows enhanced albumin-immunoreactivity in the parenchyma of ischemic regions. (b) Further, extravasations of albumin (arrow heads) are found to be most pronounced around arterial vessels and veins, the latter of which do not exhibit a continuous layer of SMA-expressing smooth muscle cells. (c) Triple immunofluorescence labeling was applied using anti-collagen IV to demark capillary, arterial and venous basement membranes (blue) combined with labeling for SMA (red) to reveal arterial smooth muscle cells and anti-albumin (green) to visualize BBB breakdown. Nuclei were counterstained with DAPI (white). Pronounced extravasation of albumin (arrow heads) was regularly detected around arterial and venous vessels within ischemic regions. However, immunoreactivity of albumin appeared to be only faint around capillaries (inset). In control regions, if detectable at all, albumin immunoreactivtiy was confined to the vascular lumen (arrows). Each scale bar: 50 µm.
In order to clearly identify capillary segments, triple immunofluorescence labeling was applied in addition to the visualization of endogenous albumin. Here, immunolabeling for collagen IV was used to demark vascular basement membranes, which surround arterial, capillary, and venous vessels, while SMA demarks arteries. Again, the parenchymal immunoreactivity for endogenous albumin was higher in the ischemic regions compared to the unaffected control areas (Figure 6(c)). Moreover, the detectable extravasation of albumin appeared to be more pronounced around arterial and venous vessels, whereas pericapillary extravasations were only faint. However, capillary vessels are likely to contribute to the diffuse albumin reactivity of the ischemic parenchyma (Figure 6(a)). In control areas, extravasation of endogenous albumin was neither observed around arterial, nor venous or capillary vessels and its immunoreactivity, if detectable at all, appeared to be confined to the vascular lumen (arrows, Figure 6(c)).
Discussion
In the setting of stroke, ischemia not only affects neuronal function but also the cerebral vasculature, thus contributing to an aggravation of the ischemic lesion size and the formation of cerebral edema by an impaired BBB function.11 However, the NVU is characterized by striking differences along the segments of the vascular tree,31 which not only concern the cellular composition42 but also the extracellular matrix,43 thereby provoking distinct functional as well as mechanical characteristics along the different segments of the cerebral circulation. While BBB properties are considered to be most pronounced in the capillary segments, extravasation of leukocytes preferentially takes place in post-capillary veins,37 whereas precapillary arterioles are largely contributing to vascular resistance and are exposed to higher blood flow velocities.32,33 These differing characteristics of the cerebral vasculature need consideration in the field of translational stroke research, as abrupt re-opening of occluded vessels represents further strains to the ischemia-affected vasculature which is assumed to increase the risk of hemorrhagic transformation and intracerebral bleeding in the clinical setting.18,19 Since the current direction of therapeutic approaches for acute ischemic stroke is aiming at the revascularization of occluded vessels,44,45 detailed knowledge on ischemia-associated alterations of the brain’s vasculature becomes more and more important.
Therefore, the present study was focused to investigate the impact of focal cerebral ischemia on different segments of the brain’s vascular tree in three different animal models by fluorescence and electron microscopy at the 24-h time point after ischemia onset. In addition to pMCAO, reflecting the scenario of ongoing occlusion, a model of transient ischemia (tMCAO) was used to mimic recanalization 90 min after ischemia onset. Furthermore, we added the model of embolic middle cerebral artery (eMCAO) occlusion to consider translational issues since this model is believed to best mimic the human situation of stroke evolution.46
Although a biphasic time course of BBB breakdown has been demonstrated before,47–49 there is also data reporting on a continuous BBB breakdown with increased endothelial permeability over weeks after the ischemic event.50 In the context of edema formation, which is associated with poor clinical outcome,10,11 it is important to note that the given time point at 24 h after ischemia induction not only correlates with an increased BBB permeability,48,51 but was further shown to coincide with a peak of edema formation.52
In line with our previous work, intravenously applied FITC-albumin proved as an elegant tool for clear-cut detection of areas exhibiting BBB breakdown by fluorescence microscopy and, upon immunohistochemical conversion with DAB, at the level of light- and electron microscopy in embedded vibratome sections. In addition to easy detection at the level of fluorescence microscopy, it proves extremely well fixable within the tissue and allows high concentration of glutaraldehyde (GA) as a prerequisite for optimal preservation of the ultrastructure. As described before,27,29 ischemic areas regularly exhibited BBB breakdown as indicated by extravasation of FITC-albumin 24 h after ischemia induction in all the models applied (Figure 1), while corresponding contralateral areas did not show leakage of the tracer into the neuropil (Supplementary Figure 1(c)). Considering the concept of an ischemic core and a shell-like surrounding penumbra,53,54 it is important to note that focusing on vessels showing FITC-albumin extravasation offers the advantage to precisely identify vessels contributing to the formation of a vasogenic edema.11 Therefore, it is remarkable that BBB breakdown as indicated by FITC-albumin extravasation was demonstrated to exceed the outer layers of the so-called “molecular penumbra”54 in the applied models of experimental stroke.29 Using these models in conjunction with the chosen tracer, future translationally oriented studies are needed to address the long-term course of edema formation and concomitant alterations of BBB integrity.
In the present study, fluorescence microscopy revealed comparable patterns of BBB breakdown in the arterial, capillary, and venous segments of the brain’s vascular tree in all the applied animal models including the transient model reflecting the reperfusion injury. Although arterial vessels are facing a higher endothelial shear stress, especially in the setting of tMCAO, arteries were not found to be preferentially affected as highest ratios of leaky vessels were consistently observed for veins (Figure 3(a)). Moreover, arteries consistently showed a less widespread distribution of extravasated FITC-albumin than capillaries showing BBB breakdown (Figure 3(b)). Therefore, our data obtained from the animal models suggest that capillaries are predominantly contributing to the cerebral edema after stroke although the arterial branch of the vascular tree is facing higher intravascular pressures.
However, the question remains whether or not these differences can be explained by a differential pattern of endothelial affection throughout arterial, capillary, and venous branches of the brain’s vascular tree, or simply derive from an additional ‘mechanical barrier’ around arterial vessels, which is constituted by the layer of smooth muscle cells. In fact, site-specific cellular interactions are likely to the impact on BBB function. Astrocyte–endothelial interactions involving sonic hedgehog55,56 and Wnt/beta-catenin signaling and their crucial roles for the maintenance of BBB characteristics by up-regulation of the typical BBB markers claudin-3 and Glut-1 are well described at the level of capillaries.57,58 Further, pericytes were shown to contribute to BBB formation and maintenance.58–60 Of note, capillary as well as post-capillary endothelial cells are in direct contact to pericytes and astrocytes, being separated from a shared basement membrane, only.41 In contrast, in larger veins such direct astrocyte–endothelial interactions can be impeded by the presence of perivascular spaces or, in arteries, additionally by smooth muscle cells of the tunica media.
Therefore, the patterns of ultrastructural affection of arterial endothelial cells, which are lacking direct contact to astrocytes and pericytes, were analyzed and compared with endothelial cells of adjacent capillaries for all the experimental models applied in mice and rats. By trimming the respective sections to considerably small sizes of only 0.5 mm2, analyses were restricted to directly neighboring vessels, thereby assuring equal ischemia-induced affection, while the general pattern of BBB breakdown is found to be comparable for the applied models.29
Importantly, arterial endothelial cells exhibited comparable signs of degeneration throughout the applied models which matched the adjacent capillary endothelium in areas of tracer extravasation (Figure 4). Here, the majority of the endothelial cells exhibited loss of cellular integrity, thereby resulting in accumulation of the tracer within endothelial debris and extravasation into adjacent compartments. However, although arterial smooth muscle cells were shown to be much less affected in the models of pMCAO and tMCAO, loss of smooth muscle cell integrity was frequently observed in the model of eMCAO, being comparable to the underlying endothelium (Figure 4). These findings were further quantified according to patterns of cellular degeneration, thereby confirming an identical affection of arterial and capillary endothelial cells throughout the applied models, whereas arterial smooth muscle cells were consistently found to be more severely affected in the model of eMCAO (Figure 5). If these differences in fact derive from different approaches of ischemia induction or rather mirror characteristics of different species, using rats for the model of eMCAO and mice for the model of pMCAO and tMCAO, needs to be addressed by further studies. As arterial and capillary endothelial cells displayed comparable patterns of degeneration in equally affected regions, the observed trend towards lower rates of tracer extravasation around arterial vessels (Figure 3(b)) is likely to result from the additional mechanical stabilization by the continuous layer of vascular smooth muscle cells and their surrounding basal laminae (Figure 4).
Although stroke-related BBB breakdown has been widely attributed to distinct modifications of endothelial TJs21–24 leading to a paracellular leakage of blood-borne molecules into the parenchyma, more and more experimental evidence is highlighting the role of a transendothelial leakage mechanism.61,62 In this context, caveolin-dependent mechanisms of endocytosis and the role of connexin hemichannels,25,26,63 which lead to an endothelial edema, may ultimately result in more severe vascular damage as described in the manuscript and other reports before.29 Noteworthy, since important TJ proteins are equally distributed throughout arteries, capillaries, and veins of CNS vessels under physiological conditions,64 in this setting, a loss of immunoreactivity for TJ proteins in arteries, capillaries, and veins was not observed (Figure 2), which is line with previous findings, demonstrating detectable TJs by fluorescence and electron microscopy.28,29,61
In addition to the animal models applied, we further analyzed human autoptic brain tissue of histopathologically confirmed stroke victims to investigate whether the presented patterns of BBB breakdown in fact hold true for the clinical situation.
Studying aged human brain tissue, in contrast to adult rodents, is inflicted by the considerable background fluorescence, which renders identification of areas of albumin extravasation difficult by fluorescence microscopy. However, application of Sudan Black B nearly completely abolished tissue-specific autofluorescence, thereby allowing detection of even faint immunoreactivity for parenchymal albumin extravasates (Figure 6).
Noteworthy, accumulations of parenchymal albumin were not restricted to arterial vessels, but also pronounced around veins. However, in contrast to the models of eMCAO, pMCAO, and tMCAO showing clear-cut extravasations of FITC-albumin at the capillary level (Figure 1), immunoreactivity for peri-capillary albumin was only faint in ischemic areas of both human cases analyzed (Figure 6(c)). Nevertheless, the capillary contribution to overall BBB breakdown in human tissue is likely to be responsible for the generally increased albumin-immunoreactivity in ischemic regions.
Although the present study is rather descriptive and the analyses of animal models are confined to 24 h after ischemia induction, we here for the first time addressed distinct segments of the brain’s vascular tree and their contribution to BBB breakdown in threee different animal models and histopathologically confirmed human stroke tissue. Moreover, the applied tools can easily be adapted to various experimental setups to address site-specific alterations of the vascular tree in different neuropathologies, especially scenarios of occluded and re-opened cerebral vessels at different time points after ischemia onset, and with potential co-medications. While the pathophysiology underlying BBB breakdown was rated as high priority for stroke research,65,66 deciphering differences of vascular pathophysiology along arterial, capillary, and venous segments of the cerebral vasculature are likely to contribute to more complex therapeutic strategies, and, therefore, might help to overcome the translational roadblock in stroke research.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Deutsche Forschungsgemeinschaft (DFG-FOR 1336) and the Helmholtz Alliance ICEMED (Imaging and Curing Environmental Metabolic Diseases) to IB, as well as by the Europäischer Sozialfonds (ESF, reference number 100270131) to DM.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Animal experiments and study design: DM, MK. Manuscript preparation: MK, DM. Regulatory affairs on animal experiments: DM, WH, MK. Histopathological evaluation and provision of human tissue: CF, WM. Immunofluorescence microscopy and electron microscopy: MK. Critical Revision: WH, AR, and IB.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 2005; 57: 173–185. [DOI] [PubMed] [Google Scholar]
- 2.del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009; 158: 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lo EH, Rosenberg GA. The neurovascular unit in health and disease: introduction. Stroke 2009; 40: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Z, Nelson AR, Betsholtz C, et al. Establishment and dysfunction of the blood-brain barrier. Cell 2015; 163: 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010; 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Zhang ZG, Chopp M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol Sci 2012; 33: 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999; 22: 391–397. [DOI] [PubMed] [Google Scholar]
- 8.Dietrich WD, Busto R, Watson BD, et al. Photochemically induced cerebral infarction. II. Edema and blood-brain barrier disruption. Acta Neuropathol 1987; 72: 326–334. [DOI] [PubMed] [Google Scholar]
- 9.Hom J, Dankbaar JW, Soares BP, et al. Blood-brain barrier permeability assessed by perfusion CT predicts symptomatic hemorrhagic transformation and malignant edema in acute ischemic stroke. AJNR Am J Neuroradiol 2011; 32: 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Battey TW, Karki M, Singhal AB, et al. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 2014; 45: 3643–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 2016; 36: 513–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivens S, Kaufer D, Flores LP, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007; 130: 535–547. [DOI] [PubMed] [Google Scholar]
- 13.Weissberg I, Wood L, Kamintsky L, et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis 2015; 78: 115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 2016; 133: e38–e360. [DOI] [PubMed] [Google Scholar]
- 15.Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359: 1317–1329. [DOI] [PubMed] [Google Scholar]
- 16.Campbell BC, Donnan GA, Lees KR, et al. Endovascular stent thrombectomy: the new standard of care for large vessel ischaemic stroke. Lancet Neurol 2015; 14: 846–854. [DOI] [PubMed] [Google Scholar]
- 17.Balami JS, Sutherland BA, Edmunds LD, et al. A systematic review and meta-analysis of randomized controlled trials of endovascular thrombectomy compared with best medical treatment for acute ischemic stroke. Int J Stroke 2015; 10: 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pundik S, Xu K, Sundararajan S. Reperfusion brain injury: focus on cellular bioenergetics. Neurology 2012; 79: 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whiteley WN, Thompson D, Murray G, et al. Effect of alteplase within 6 hours of acute ischemic stroke on all-cause mortality (third International Stroke Trial). Stroke 2014; 45: 3612–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kassner A, Merali Z. Assessment of blood-brain barrier disruption in stroke. Stroke 2015; 46: 3310–3315. [DOI] [PubMed] [Google Scholar]
- 21.Fischer S, Wobben M, Kleinstück J, et al. Effect of astroglial cells on hypoxia-induced permeability in PBMEC cells. Am J Physiol Cell Physiol 2000; 279: 935–944. [DOI] [PubMed] [Google Scholar]
- 22.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008; 32: 200–219. [DOI] [PubMed] [Google Scholar]
- 23.Jiao H, Wang Z, Liu Y, et al. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci 2011; 44: 130–139. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Jin X, Liu KJ, et al. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci 2012; 32: 3044–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Bock M, Culot M, Wang N, et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J Cereb Blood Flow Metab 2011; 31: 1942–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danesh-Meyer HV, Kerr NM, Zhang J, et al. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012; 135: 506–520. [DOI] [PubMed] [Google Scholar]
- 27.Krueger M, Härtig W, Reichenbach A, et al. Blood-brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS One 2013; 8: e56419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab 2016; 36: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger M, Bechmann I, Immig K, et al. Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab 2015; 35: 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Collins VE, Macleod MR, Donnan GA, et al. 1,026 experimental treatments in acute stroke. Ann Neurol 2006; 59: 467–477. [DOI] [PubMed] [Google Scholar]
- 31.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol 2007; 28: 5–11. [DOI] [PubMed] [Google Scholar]
- 32.Stromberg DD, Fox JR. Pressures in the pial arterial microcirculation of the cat during changes in systemic arterial blood pressure. Circ Res 1972; 31: 229–239. [DOI] [PubMed] [Google Scholar]
- 33.Mayrovitz HN, Tuma RF, Wiedeman MP. Relationship between microvascular blood velocity and pressure distribution. Am J Physiol 1977; 232: 400–405. [DOI] [PubMed] [Google Scholar]
- 34.Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery 1992; 31: 100–106. [DOI] [PubMed] [Google Scholar]
- 35.Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20: 84–91. [DOI] [PubMed] [Google Scholar]
- 36.Michalski D, Härtig W, Krügel K, et al. Region specific expression of vesicular glutamate and GABA transporters under various ischaemic conditions in mouse forebrain and retina. Neuroscience 2013; 231: 328–344. [DOI] [PubMed] [Google Scholar]
- 37.Sixt M, Engelhardt B, Pausch F, et al. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 2001; 153: 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawkes CA, Härtig W, Kacza J, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 2011; 121: 431–443. [DOI] [PubMed] [Google Scholar]
- 39.Attwell D, Mishra A, Hall CN, et al. What is a pericyte? J Cereb Blood Flow Metab 2016; 36: 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill RA, Tong L, Yuan P, et al. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 2015; 87: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krueger M, Bechmann I. CNS pericytes: concepts, misconceptions, and a way out. Glia 2010; 58: 1–10. [DOI] [PubMed] [Google Scholar]
- 42.Dyrna F, Hanske S, Krueger M, et al. The blood-brain barrier. J Neuroimmune Pharmacol 2013; 8: 763–773. [DOI] [PubMed] [Google Scholar]
- 43.Wu C, Ivars F, Anderson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 2009; 15: 519–527. [DOI] [PubMed] [Google Scholar]
- 44.Bendszus M, Hacke W. Acute endovascular recanalization: lessons from randomized controlled trials. Curr Opin Neurol 2016; 29: 30–36. [DOI] [PubMed] [Google Scholar]
- 45.Muir KW. Stroke in 2015: the year of endovascular treatment. Lancet Neurol 2016; 15: 2–3. [DOI] [PubMed] [Google Scholar]
- 46.Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009; 40: 2244–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroiwa T, Ting P, Martinez H, et al. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol 1985; 68: 122–129. [DOI] [PubMed] [Google Scholar]
- 48.Huang ZG, Xue D, Preston E, et al. Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci 1999; 26: 298–304. [DOI] [PubMed] [Google Scholar]
- 49.Belayev L, Busto R, Zhao W, et al. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res 1996; 739: 88–96. [DOI] [PubMed] [Google Scholar]
- 50.Strbian D, Durukan A, Pitkonen M, et al. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience 2008; 153: 175–181. [DOI] [PubMed] [Google Scholar]
- 51.Durukan A, Marinkovic I, Strbian D, et al. Post-ischemic blood-brain barrier leakage in rats: one-week follow-up by MRI. Brain Res 2009; 1280: 158–165. [DOI] [PubMed] [Google Scholar]
- 52.Pillai DR, Dittmar MS, Baldaranov D, et al. Cerebral ischemia-reperfusion injury in rats – a 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J Cereb Blood Flow Metab 2009; 29: 1846–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia – the ischemic penumbra. Stroke 1981; 12: 723–725. [DOI] [PubMed] [Google Scholar]
- 54.Sharp FR, Lu A, Tang Y, et al. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab 2000; 20: 1011–1032. [DOI] [PubMed] [Google Scholar]
- 55.Haseloff RF, Blasig IE, Bauer HC, et al. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol 2005; 25: 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011; 334: 1727–1731. [DOI] [PubMed] [Google Scholar]
- 57.Liebner S, Corada M, Bangsow T, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 2008; 183: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daneman R, Agalliu D, Zhou L, et al. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 2009; 106: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 2003; 314: 15–23. [DOI] [PubMed] [Google Scholar]
- 60.Daneman R, Zhou L, Kebede AA, et al. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010; 468: 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reeson P, Tennant KA, Gerrow K, et al. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J Neurosci 2015; 35: 5128–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Bock M, Van Haver V, Vandenbroucke RE, et al. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016; 64: 1097–1123. [DOI] [PubMed] [Google Scholar]
- 63.Davidson JO, Green CR, Bennet L, et al. Battle of the hemichannels – connexins and pannexins in ischemic brain injury. Int J Dev Neurosci 2015; 45: 66–74. [DOI] [PubMed] [Google Scholar]
- 64.Hanske S, Dyrna F, Bechmann I, et al. Different segments of the cerebral vasculature reveal specific endothelial specifications, while tight junction proteins appear equally distributed. Brain Struct Funct. 2016; 222(3): 1179–1192. [DOI] [PubMed] [Google Scholar]
- 65.Meairs S, Wahlgren N, Dirnagl U, et al. Stroke research priorities for the next decade – a representative view of the European scientific community. Cerebrovasc Dis 2006; 22: 75–82. [DOI] [PubMed] [Google Scholar]
- 66.Endres M, Engelhardt B, Koistinaho J, et al. Improving outcome after stroke: overcoming the translational roadblock. Cerebrovasc Dis 2008; 25: 268–278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.