

Abstract
Microvascular endothelial cells at the blood–brain barrier exhibit a protective phenotype, which is highly induced by biochemical and biomechanical stimuli. Amongst them, shear stress enhances junctional tightness and limits transport at capillary-like levels. Abnormal flow patterns can reduce functional features of macrovascular endothelium. We now examine if this is true in brain microvascular endothelial cells. We suggest in this paper a complex response of endothelial cells to aberrant forces under different flow domains. Human brain microvascular endothelial cells were exposed to physiological or abnormal flow patterns. Physiologic shear (10–20 dyn/cm2) upregulates expression of tight junction markers Zona Occludens 1 (1.7-fold) and Claudin-5 (more than 2-fold). High shear stress (40 dyn/cm2) and/or pulsatility decreased their expression to basal levels and altered junctional morphology. We exposed cells to pathological shear stress patterns followed by capillary-like conditions. Results showed reversible recovery on the expression of tight junction markers. Flow protection of barrier phenotype commensurate with junctional signaling pathways decrease (Src, 0.25-fold, ERK, 0.77-fold) when compared to static conditions. This decrease was lost under high shear and pulsatile flow. In conclusion, abnormal shear stress inherent to systemic vascular disease leads to barrier impairment, which could be reverted by hemodynamic interventions.
Keywords: Blood–brain barrier, capillaries, cerebrovascular disease, hemodynamics, microcirculation, neurovascular unit, vascular biology
Introduction
In 2015, the American Heart Association estimated the incidence of stroke to be 15 million worldwide.1 Cerebrovascular disease is the fifth leading cause of death in the United States. Moreover, vascular dementia, the second most common form of dementia, is four times more likely in patients after stroke.2 The statistics linking vascular impairment and cerebral disease have motivated clinical studies to show how flow alterations caused by arterial stiffness3,4 and hypertension are extended to the brain and lead to microcirculation damage and the enhancement of cerebral disease.5–8
The blood–brain barrier (BBB) is a complex biological barrier that separates bloodstream at the capillary level and the central nervous system (CNS). It plays a key role in the transport of nutrients and other solutes through the brain and is characterized by a highly restrictive paracellular permeability that maintains and regulates brain homeostasis, metabolism, and neuronal activity.9 At the core of the BBB, microvascular endothelial cells line along brain capillaries and orchestrate molecular traffic, leveraging a distinctive and unique phenotype that distinguishes them from endothelial cells in other vascular beds. Complex tight junctions, mainly formed by claudins and Zona Occludens proteins, restrict paracellular transport, forcing substrates into transcellular routes. A specialized set of solute-like carriers and ATP-binding cassette transporters, such as glucose transporter 1 (GLUT1) and P-glycoprotein (Pgp) are responsible for molecular influx and efflux through the BBB.10
Different brain-related diseases, acute and chronic, such as cerebral reperfusion injury11 and posterior reversible encephalopathy syndromes (PRES)12 have been related to BBB dysfunction. Arterial hypertension is involved in hyperperfusion syndrome after arterial revascularization procedures, and is the most frequent cause of PRES. Increasing evidence supports BBB permeability as relevant in the pathogenesis of neurodegenerative diseases such as vascular dementia,13 leukoaraiosis,14 and Alzheimer’s disease.15 Vascular risk factors may be behind the underlying mechanisms that increase BBB permeability.16
In the present study, we hypothesized that brain microvascular endothelial cell state is affected by flow. Such cellular state plays a fundamental role in the performance of the BBB. Human brain microvascular endothelial cell (HBMEC) phenotype may be stimulated or silenced depending on the biomechanical conditions to which cells are exposed. We examined the impact of flow in vessel-like constructs mimicking HBMEC environment and a customized flow perfusion system. We analyzed protein expression of key components of BBB phenotype, junctional morphology, efflux activity, flow reversibility, and signaling phosphorylation to support our hypothesis.
Materials and methods
Materials and cell culture
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary human brain microvascular endothelial cells (HBMEC), purchased from Cell Systems Corporation (Kirkland, WA, USA), were maintained in Endothelial Growth Medium-2 (EGM-2) (Lonza, Walkersville, MD, USA) supplemented with 5% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY, USA), 1% penicillin-streptomycin (PS, Lonza) and growth factors (5 ng/mL human epidermal growth factor, 0.2 µg/mL hydrocortisone, 0.5 ng/mL vascular endothelial growth factor, 10 ng/mL human fibroblast growth factor, 20 ng/mL human recombinant insulin-like growth factor I, and 1 µg/mL ascorbic acid, all from Lonza). The vendor isolates these cells from donors and performs the necessary assays to confirm positive functional and structural features of brain microvascular endothelial cells (Positive VWF and ZO-1 immunostaining and uptake of Dil-Ac-LDL). In addition, prior to the experiments leading to our results, we characterized these cells. We verified proper endothelial cobblestone morphology in confluent monolayer cultures and we obtained positive immunostaining for CD31, as shown in Figure 1(a) and (b). Primary human astrocytes (HA) were purchased from ScienCell Research Laboratories (San Diego, CA, USA) and maintained in Astrocyte Medium (AM, ScienCell), supplemented with 2% FBS, 1% penicillin-streptomycin and growth supplements (ScienCell). Both cell types were expanded in treated tissue culture dishes (Corning, NY, USA) and were used for experiments between passages 4 and 7.
Figure 1.

Flow perfusion bioreactor application as a dynamic model of the blood–brain barrier. Sterile tubular constructs are prepared with silicone tubing and HBMEC are seeded to confluence prior to the experiments. Immunostaining of static (a) and dynamic (b) cultured HBMEC showed how cells align with flow direction in the dynamic case. ZO-1, green / nuclei, blue. Scale bar = 10 µm. White arrows indicate flow direction. (c) Cell seeded tubes are connected to a sterile closed flow perfusion system and stimulated with shear stress by perfusing culture media with a programmable peristaltic pump. (d) Shear stress is measured with continuous flow measurements using an ultrasonic flow probe. Cells can be stimulated with both steady and pulsatile shear stress.
Astrocyte-conditioned medium collection
HA were grown in tissue culture dishes with the above-mentioned culture medium. At confluency, cells were incubated in EGM-2 medium depleted with FBS and supplements. After 48 h, supernatant was collected and sterile-filtered to remove cellular debris. Astrocyte-conditioned medium (ACM) was aliquoted and stored at −80℃ until use. HA viability assays and immunofluorescent labeling of glial fibrillary acidic protein (GFAP) showed no significant growth or morphological differences in short-term cultures between the use of endothelial or astrocyte medium (supplementary Figure 1). The effect of ACM on BBB phenotype was evaluated in dynamic cultures of both human brain (HBMEC) and dermal (HDMEC) microvascular endothelial cells, confirming a significant junctional ZO-1 enhancement on HBMEC (supplementary Figure 2).
Dynamic cell culture
Tubular constructs were prepared with sterile silicone tubing (0.078” ID × 0.0125” OD, Dow Corning, Midland, MI, USA) and connectors (barbed fittings, reducing connector, 1/8” × 3/32” ID, Cole Parmer, Vernon Hills, IL, USA). SILASTIC® tubes (Dow Corning, Midland, MI, USA) were coated with 100 µg/mL fibronectin from bovine plasma (Alfa Aesar, Ward Hill, MA, USA) overnight at 4℃. Then, HBMEC were suspended (106 cells/mL), seeded in tubes and allowed growing for 48 h. In order to ensure homogeneous distribution of cells along the inner surface, tubes were placed in a cylindrical rotator turning at 3 r/min in a 37℃, 5% CO2 humidified incubator. Microscopic visualization of nuclei staining and CD31 immunostaining showed full coverage along the tubes inner surfaces. Tubes were discretized in 1 cm segments and observed consecutively under the microscope. CD31 imaging showed a continuous monolayer in all segments, as no evident gaps were observed between cells. In consequence, we considered that 100% cell confluence and tube inner surface coverage were achieved, with HBMEC displaying the typical cobblestone endothelial morphology and no gaps between cells (Figure 1(a) and (b)). After this process, tubes were connected to a perfusion bioreactor (Figure 1(c)), stimulated with flow and grown with EGM-2 (depleted of hEGF, VEGF and IGF, but supplemented with 5% FBS and 1% PS) and ACM in a 1:1 ratio. Cells were exposed to laminar flow with shear stress ranging from 0 to 40 dyn/cm2, under steady or pulsatile regime, using a peristaltic pump (MCP-Standard ISM 404, Ismatec, Vancouver, WA, USA). Flow measurements were performed using a data acquisition system coupled to ultrasound flow probes (Transonic Systems Inc., Ithaca, NY, USA). Measured profiles were integrated and compared to idealized profiles, showing equivalent mean shear stress (Figure 1(d)). Cell cultures were kept under dynamic conditions for 4 days prior to further analysis. Shear stress levels of 10 and 20 dyn/cm2 were considered in the physiological healthy range.17–19 Abnormal high shear stress level was established at 40 dyn/cm2. Using computational fluid dynamics, an increase of 5.4 mmHg in the pressure difference between arterioles and capillaries sufficed to raise shear stress from 20 to 40 dyn/cm2 (supplementary Figure 3). Carotid pulse pressure in hypertensive patients is at least 10 mmHg higher than normotensive patients.20 Since trans-endothelial electrical resistance (TEER) measurements were not measurable in this culture system, dynamic exposure time was determined with previous optimization of ZO-1 expression (supplementary Figure 4). HBMEC viability in dynamic cultures was not compromised during the course of experiments (supplementary Figure 5).
Western blotting
HBMEC were washed twice with ice-cold PBS and were lysed with radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitor cocktail. Lysates were centrifuged at 16,000 g for 30 min at 4℃ and supernatants were stored at −80℃ until use. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). About 5 µg of protein were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 60 min in phosphate-buffered saline (PBS, Lonza) containing 0.05% Tween-20 (PBS-T, Thermo Fisher Scientific, Waltham, MA, USA) and 5% non-fat dry milk (LabScientific Inc., Livingston, NJ, USA). Then, membranes were incubated overnight in primary antibody at 4℃. Primary antibodies were diluted in PBS-T supplemented with 10% starting block solution (Thermo Fisher Scientific): rabbit anti-Zona Occludens 1 (ZO-1, 1:1000, Life Technologies), mouse anti-Claudin-5 (1:2000, Life Technologies), rabbit anti-P-glycoprotein (1:1000, Abcam, Cambridge, MA, USA), rabbit anti-GLUT1 (1:1000, Abcam), rabbit anti-phosphorylated-ERK (1:2000, Cell Signaling, Beverly, MA, USA), rabbit anti-ERK (1:1000, Cell Signaling), rabbit anti-phosphorylated-Src (1:1000, Cell Signaling), rabbit anti-Src (1:1000, Cell Signaling), and rabbit anti-β-actin (1:1000, Cell Signaling). Washed membranes were incubated in horseradish peroxidase (HRP) conjugated goat anti-mouse or anti-rabbit IgG secondary antibodies (Abcam) diluted in PBS-T supplemented with 10% starting block solution for 45 min at room temperature (RT). Bands were detected after membrane incubation in Luminata Forte Western HRP substrate (Merck Millipore, Billerica, MA, USA) with a chemiluminescent image analyzer ChemiDoc XRS + System (BioRad, Hercules, CA, USA). Blots were quantified using Fiji imaging software21 (NIH) and β-actin served as loading control. Intensity of proteins of interest was first normalized to the respective β-actin blot in each band to ensure representative analysis of the data and comparison between samples. The resulting averaged ratios from each group were normalized against the static control group, which was present in all membranes analyzed.
Immunofluorescence
HBMEC were washed with PBS and fixed with 4% paraformaldehyde for 10 min at RT. Then, cells were permeabilized using 0.5% Triton X-100 in PBS for 20 min at RT and blocked with 5% goat serum (Life Technologies) in PBS for 60 min at RT. Cells were incubated overnight in rabbit anti-ZO-1 antibody (1:200 in blocking buffer, Life Technologies) at 4℃, followed by an incubation in secondary Alexa Fluor 488 goat anti-rabbit IgG antibody (1:200 in blocking buffer, Life Technologies). Nuclear staining with DAPI (1 µg/mL, Life Technologies) was also routinely conducted. Cells were visualized by fluorescent microscopy (Nikon Eclipse Ti, Tokyo, Japan). Antibody controls were included in all experiments.
Image analysis
Immunofluorescent images of tight junctions were analyzed using Fiji imaging software.21 Spline curves (4 pixel width) were traced along intercellular tight junctions (at least 5 curves were traced per image) and mean intensity was measured. Background fluorescent intensity was measured for each image and subtracted to junctional intensity.
Statistical analysis
All experiments were performed on triplicate or quadruplicate specimens (n = 3–4) and repeated at least in two independent experiments. Data from each experiment are expressed as mean ± standard error of the mean (SEM). Statistical differences were determined using Kruskal–Wallis test with Dunns’ post hoc analysis. Values of p < 0.05 were considered statistically significant.
Results
Pulsatility and high shear stress downregulate the expression of critical tight junction markers
HBMEC were cultured under static conditions (0 dyn/cm2) as a control, and up to six different flow conditions (10, 20 and 40 dyn/cm2 in steady and pulsatile flow patterns). After exposing HBMEC to laminar steady flow for 96 h, Western blot results support literature and confirm that, under physiological shear stress, expression of tight junction markers is upregulated (Figure 2(a)). The expression of ZO-1 (Figure 2(b)) increased significantly in steady flow at 10 dyn/cm2 (1.71 ± 0.23-fold) and 20 dyn/cm2 (1.68 ± 0.06-fold). Interestingly, ZO-1 dropped, and was not different from the static control, at 40 dyn/cm2 (1.03 ± 0.14-fold). Claudin-5 expression (Figure 2(c)) showed a similar trend, with significant increase at 10 and 20 dyn/cm2 (2.14 ± 0.05-fold and 2.45 ± 0.15-fold respectively). Again, 40 dyn/cm2 steady flow (1.20 ± 0.15-fold) brought Claudin-5 expression down to static basal expression. When cells were exposed to pulsatile flow, the expression of both junctional markers showed no significant difference as compared with the static control at any shear stress. It is important to remark that cells exposed to pulsatile flow patterns within the range of physiological shear stress levels revealed a significant downregulation of ZO-1 and Claudin-5 when compared to steady patterns with the same average shear stress of 10 (1.42 ± 0.32-fold and 1.88 ± 0.09-fold respectively) and 20 dyn/cm2 (1.69 ± 0.34-fold and 2.54 ± 0.20-fold respectively). In both steady and pulsatile flow patterns, the expression level of both ZO-1 and Claudin-5 at different shear stress showed linear correlation, of R2 = 0.97 according to Pearson’s correlation (Figure 2(d)). Regardless of individual expression variations, our results suggest that the whole junctional structure formed by transmembrane domains (Claudin-5) and cytosolic anchorage (ZO-1) is affected concurrently. Flow regulation of ZO-1 and Claudin-5 are intrinsically connected independently of flow type or shear.
Figure 2.

Effect of shear stress patterns on the expression of tight junction markers. (a) HBMEC were exposed to shear stress for 96 h. Protein was extracted and tight junction markers were analyzed with Western blotting. Normalized average intensities are indicated below each band. Densitometric analysis of the expression of ZO-1 (b) and Claudin-5 (c) showed that abnormal steady shear stress levels of 40 dyn/cm2 downregulated the expression of tight junction proteins as opposed to capillary-like levels (10–20 dyn/cm2). Pulsatile patterns induced a lower expression compared to the respective steady shear stress level. (d) Expression of ZO-1 and Claudin-5 induced by steady and pulsatile flow patterns showed linear correlation. *p ≤ 0.05 between indicated bars.
High shear stress disrupts tight junction morphology
HBMEC were exposed to steady and pulsatile shear stress for 96 h and stained with specific antibodies against ZO-1 in order to accurately study cell and junctional morphology under different scenarios. The static controls (Figure 3(a) and (e)) showed a disorganized junctional morphology. Tight junctions in static controls displayed discontinuous saw-tooth patterns. Such serrated patterns differed greatly from those observed in cells cultured under physiological steady shear stress (10–20 dyn/cm2, Figure 3(b) and (c)), which had linear junctions aligned with flow. ZO-1 was especially intense and localized in the intercellular junctions of cells exposed to 10 and 20 dyn/cm2. Junctional ZO-1 intensity measured by immunofluorescence (Figure 3(I)) was indeed significantly higher (1.21 ± 0.06-fold) than in the static control. Contrarily, ZO-1 was translocated to the cytoplasm in cells exposed to abnormally high shear stress (40 dyn/cm2, Figure 3(d)). ZO-1 junctional intensity decreased dramatically (0.41 ± 0.04-fold) in these cells. These results showed similar trends to those previously analyzed by Western blot (Figure (2)) and confirmed that abnormally high shear stress compromised barrier phenotype.
Figure 3.

Effect of steady shear stress on tight junction morphology. HBMEC were stimulated with different shear stress levels and ZO-1 was immunolabeled. While static control (a, e) showed disrupted tight junctions (yellow arrows), steady capillary-like shear stress (b, c) showed a continuous healthier morphology. At 40 dyn/cm2 (d), ZO-1 delocalized from the membrane and translocated to the cytoplasm. With pulsatile flow (f, g, h), junctional morphology was equivalent to the high steady shear case. Scale bar = 10 µm. White arrows indicate flow direction. (i) Fluorescent intensity quantified in junctional regions and normalized to cytosolic fluorescent intensity showed significant higher expression at 10 dyn/cm2 and reached a minimum expression at 40 dyn/cm2. Again, junctional expression at pulsatile flow was significantly lower than the corresponding steady analog. (j) Fluorescent intensity quantified in the cytosol and nuclei showed significant translocation of ZO-1 at any shear stress. (k) Junctional to cytosolic and nuclear ZO-1 ratios showed significant gradual decrease with increasing shear stress and significantly lower ratios at any pulsatile shear stress. *p ≤ 0.05 between indicated bars. #,&p ≤ 0.05 between steady (#) or pulsatile (&) shear stress group and static control.
Pulsatile flow (Figure 3(F) to (H)) caused ZO-1 delocalization independently of the applied shear stress. The fluorescence intensity of ZO-1 was significantly lower (0.30 ± 0.03-fold) than the static. In cells exposed to 10 dyn/cm2, ZO-1 junctional expression was 3.9 ± 0.4-fold lower when flow was pulsatile than when cells were exposed to steady flow. Similarly, there was a 3.1 ± 0.4-fold difference between cells exposed to pulsatile flow versus cells exposed to steady flow at 20 dyn/cm2. There was, however, no significant difference between cells exposed to pulsatile or steady 40 dyn/cm2 flow as ZO-1 was already low and translocated into the cytoplasm. Interestingly, the differences between steady and pulsatile shear stress at 10 and 20 dyn/cm2 were significantly higher in junctional ZO-1 (Figure 3(i)) than in total ZO-1 expression (Figure 2(b)), suggesting that pulsatility triggers ZO-1 delocalization. Cytoplasmic/nuclear ZO-1 intensity was quantified (Figure 3(j)), showing significantly higher non-junctional ZO-1 for all pulsatile shear stresses (2.32 ± 0.12-fold at 10 dyn/cm2, 2.03 ± 0.11-fold at 20 dyn/cm2 and 1.44 ± 0.11-fold at 40 dyn/cm2). The ratio between junctional and cytoplasmic/nuclear ZO-1 was measured (Figure 3(k)) for every shear stress, under steady or pulsatile flow. Under increasing steady shear stress, ZO-1 localization shifted gradually from junctional to internal, as the ratio decayed significantly (0.58 ± 0.05 and 0.27 ± 0.03 at 20 and 40 dyn/cm2 respectively). This ratio was significantly lower for all pulsatile shear stresses (0.10 ± 0.01 at 10 dyn/cm2, 0.09 ± 0.01 at 20 dyn/cm2 and 0.13 ± 0.01 at 40 dyn/cm2). In conclusion, pulsatile flow jeopardized microvascular junction independently of shear stress.
Shear stress and pulsatility induce different expression profiles in transporters P-gp and GLUT1
Western blotting of transport markers P-gp and GLUT1 was performed on cell lysates from HBMEC exposed to different shear stress values (Figure 4(a)). The expression of P-gp (Figure 4(b)) was significantly increased between 1.5 - and 1.6-fold in cells exposed to steady flow, independently of mean shear stress applied. No significant differences in expression of transporters were found between shear stress of 10, 20, and 40 dyn/cm2. Although total cellular expression was constant under steady flow, P-gp efflux activity was tested using Rhodamine 123 (Rho123) as a P-gp substrate.22 Results showed a significant activity peak at 10 dyn/cm2 and a significant minimum activity at 40 dyn/cm2 (supplementary Figure 6), indicating the activity of P-gp can be modulated by flow. The expression of P-gp in pulsatile cultures was in all cases lower than the steady analog, with a significant difference at 20 and 40 dyn/cm2. Thus, P-gp expression may be compromised when both pathological shear stress and pulsatile flow are present. Contrary to P-gp, the expression of GLUT-1 (Figure 4(c)) showed no significant difference between steady and pulsatile flow. Both flow patterns showed the same tendency, where GLUT1 expression peaked significantly in cells exposed to steady 10 dyn/cm2 (1.27 ± 0.03-fold) and decreased afterwards. No relevant significant differences were found compared to the static control, suggesting that GLUT1 expression is not governed by fluid dynamics.
Figure 4.
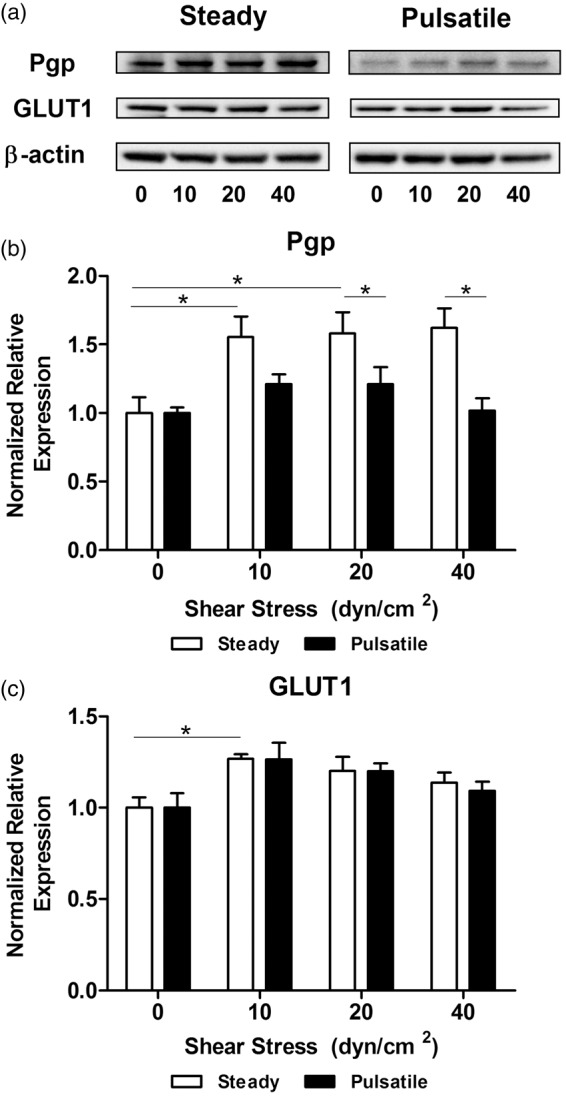
Effect of shear stress patterns on the expression of key transport markers. (a) HBMEC were exposed to shear stress for 96 h. Protein was extracted and transport proteins were analyzed with Western blotting. Densitometric analysis of the expression of P-glycoprotein (b) and GLUT1 (c) showed different responses. P-gp was significantly upregulated under steady shear stress, but pulsatile flow decreased the expression. GLUT1 showed only a statistical upregulation with steady flow at 10 dyn/cm2. *p ≤ 0.05 between indicated bars.
Tight junction markers can be recovered when physiological shear stress is reestablished
After analyzing the effect of shear stress on tight junction and transport markers, we investigated how microvascular endothelium responded to temporary modifications of fluid dynamics. HBMEC were exposed to steady shear stress of 10 or 40 dyn/cm2 for 3 days. Then, as illustrated in Figure 5(a), cells were either kept at their initial shear for three more days (Group A: 10 dyn/cm2; Group B: 40 dyn/cm2) or switched from one to the other (Group C: 10 → 40 dyn/cm2; Group D: 40 → 10 dyn/cm2). HBMEC were lysed and tight junction markers were analyzed by Western blotting (Figure 5(b)). As expected, the expression of ZO-1 and Claudin-5 (Figure 5(c) and (d)) showed a significant downregulation at 40 dyn/cm2 compared to 10 dyn/cm2 (0.41 ± 0.09 and 0.43 ± 0.12 respectively). Interestingly, when cells cultured at 40 dyn/cm2 were switched to 10 dyn/cm2, the expression of tight junction markers was significantly recovered and was comparable to the expression of cells maintained at 10 dyn/cm2 for the entire experiment (0.89 ± 0.14 and 0.99 ± 0.12 for ZO-1 and Claudin-5 respectively). This indicates that HBMEC may recover their BBB upregulated phenotype when the alteration of their normal fluid dynamic conditions is reversed. On the contrary cells exposed to 10 dyn/cm2 and switched to 40 dyn/cm2 showed a significant ZO-1 downregulation (0.32 ± 0.02) similar to the level of cells maintained at 40 dyn/cm2 (Figure 5(c)). Under the same circumstances, Claudin-5 showed resistance to pathological shear exposure and its expression decreased nonsignificantly (Figure 5(d)).
Figure 5.

Shear stress reversibility effects on tight junction markers. (a) HBMEC were exposed to steady flow for 6 days, maintaining shear stress at 10 and 40 dyn/cm2 or switching both levels at day 3. (b) Protein was extracted and transport proteins were analyzed with Western blotting. Densitometric analysis of the expression of ZO-1 (c) and Claudin-5 (d) showed that HBMEC are able to adapt their expression from a lower level at 40 dyn/cm2 to a significantly higher level at 10 dyn/cm2. *p ≤ 0.05 between indicated bars.
Effects induced by shear stress in HBMEC are modulated via the Src/ERK pathway
Phosphorylation analysis of Src and ERK1/2 was assessed by Western blotting after HBMEC exposure to flow (Figure 6(a)). Under physiological conditions, Src and ERK1/2 are inhibited and upregulate tight junction markers. Results showed that ERK1/2 (Figure 6(b)) and Src (Figure 6(c)) are inhibited in cells stimulated with steady shear stress of 10 and 20 dyn/cm2. Both phosphorylation ratios decreased significantly compared to the static control (ERK1/2, 0.25 ± 0.07; Src, 0.77 ± 0.03 at 20 dyn/cm2). Subsequent increase of shear stress to 40 dyn/cm2 activated the Src/ERK pathway showing a significant increase in phosphorylation compared to capillary-like shear stress levels (ERK1/2: 2.30 ± 0.60; Src, 1.20 ± 0.05). When cells were stimulated with pulsatile flow, phosphorylation ratios were equivalent to high steady shear stress levels independently of the pulsatile shear stress applied. Overall deviations from physiological dynamic conditions in HBMEC lead to the activation of stress signaling cascades Src and ERK, which in turn triggered a downregulation of tight junction markers (Figure 6(d)).
Figure 6.

Effect of shear stress on the activation of Src and ERK1/2. (a) HBMEC were exposed to shear stress for 96 h. Protein was extracted and signaling markers were analyzed with Western blotting. Densitometric analysis of the phosphorylation ratios of pSrc (b) and pERK1/2 (c) showed a minimal activation between 10 and 20 dyn/cm2 and steady flow. At 40 dyn/cm2, phosphorylation increased suggesting that signaling cascades were activated. Similarly, pulsatile flow lead to higher activation profiles. *p ≤ 0.05 between indicated bars. (d) Shear stress disruptions at the brain microvasculature are sensed by G-coupled protein receptors (GPCR). This leads to activation of tyrosine-protein kinases (Src) and extracellular signal-regulated kinases (ERK1/2), which directly induces downregulation of the expression of tight junction markers.
ERK: extracellular signal-regulated kinases; GPCR: G-coupled protein receptor; P: phosphorylation; Src: proto-oncogene tyrosine-protein kinase; TJ: tight junction; ZO-1: Zona Occludens 1.
Discussion
Our findings suggest that brain microvascular endothelial cells state is critical to cerebrovascular function. Abnormal flow patterns in the systemic vasculature may jeopardize endothelial behavior at the neurovascular unit, and impact transport at the BBB, resulting in a loss of functionality of the neurovascular unit. Our results show that high shear stress and pulsatile flow downregulated significantly the total expression of tight junction markers ZO-1 and Claudin-5. Moreover, abnormal flow induced ZO-1 delocalization from cell junctions towards cytoplasm and nuclei.
Flow waveform corrections are limited by ageing-associated arterial stiffness, which transports increased pulse pressure to peripheral organs.4 While shear stress in healthy brain capillaries is mostly steady and typically lower than 20 dyn/cm2 higher levels17 and pulsatile patterns could reach the endothelium in patients with increased arterial stiffness. Similarly, patients who suffer stroke or trauma lose the signaling mechanisms that regulate microvessel hemodynamics in brain microcirculation.23
Shear-induced effects on the endothelium have been widely studied in larger vessels,24–26 but less is known about the consequences of shear stress alterations in the brain microvascular endothelium. Our findings extend the dynamic vascular spectrum beyond capillary-like rates at the BBB and analyze hemodynamics at the cellular level in HBMEC. We show how critical markers of tight junctions (ZO-1 and Claudin-5) or efflux transport (P-gp) are upregulated under capillary-like shear stress. Conversely, high shear stress (40 dyn/cm2) and pulsatile profiles downregulate the expression of tight junction markers, translocate ZO-1 into the cytoplasm and nucleus and decrease the efflux activity of P-gp. To date, studies using in vitro dynamic models of the neurovascular unit have focused on capillary-like shear stress only. Our results using physiological conditions agree with previous reports as shear stress in such ranges promotes barrier phenotype. In a recent study, moderated shear stress (6.2 dyn/cm2) showed significant benefits in brain microvascular endothelial cells when compared to static cultures.19 Using a hollow-fiber based co-culture device, an upregulation was observed in genes related to tight junctions, drug transporters, ion channels, adhesion molecules and integrins. As a difference to this study, our in vitro system allows cellular visualization, local protein expression quantification and a wide range of hemodynamic parameterization. The lack of astrocytes in our system was corrected with the incorporation of human astrocyte-conditioned medium into the experimental setup, which enhanced tight junctional formation. Characterization of HBMEC in this series of experiments showed maximal total expression of BBB markers in static cultures after 96 h, a duration that falls inside the duration range of other dynamic published studies.17,19,27,28 We repeated the same experiment under dynamic physiological conditions and, in agreement with the static data, total expression of BBB markers was also highest at 96 h. Using this time point, we avoided the typical loss of phenotype due to long-term in vitro cultures. Although this model is not fully representative of the neurovascular unit, we reproduced some of the BBB hallmarks in a dynamic environment where barrier phenotype was altered by abnormal shear stress on the microvascular endothelium.
Barrier phenotype loss is critical to the etiology of different types of neurological diseases.29,30 Recent findings have shown how disruptive effects in HBMEC of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) can be relieved by capillary-like (8 dyn/cm2) shear stress.27,28 However, such protective effect may be lost under high shear stress and pulsatile flow patterns.
Special attention must be paid on pulsatile flow reaching the brain microvasculature. Cerebrovascular damage can be caused by arterial stiffening and hypertension, which provoke high shear and/or pulsatile flow in brain capillaries.5,6 We have now proven that pulsatile flow in the microvasculature can cause tight junctional impairment, ZO-1 translocation into the cytoplasm and nucleus and consequent loss of barrier phenotype in brain microvascular endothelial cells. In our experiments, steady high shear stress and all pulsatile shear stresses deteriorated the endothelial barrier functionality. Pulsatile shear stress induced ZO-1 translocation significantly from cell-cell junctions towards the cytoplasm and nucleus. It has been proved in the past that nuclear presence of ZO-1 can be caused by tight junction immaturity in in vitro cultures.31 In our study, endothelial cells were in culture for a total of 6 days, 2 days during the seeding process and 4 days under flow. Despite observing some nuclear presence of ZO-1 under static conditions, we only observed significant translocation when we exposed our cells to high shear or pulsatile shear stress. Our results indicate then that high and pulsatile shear stress may be an impediment in cell–cell contacts formation and tight junction maturation. Our findings also suggest an inverse correlation of phosphorylation ratio of Src and ERK1/2 with tight junction expression levels. This finding is in agreement with the literature that shows that this pathway is flow sensitive32–34 and how different insults to HBMEC lead to activation of Src,35,36 ERK1/2,37 and BBB alterations.38
A novel finding of our study is that shear-induced damage in the microvascular endothelium may be reversed if regular cerebral blood flow is recovered. Using nonmicrovascular endothelial cells, other authors proved that alterations of electrical impedance in endothelial monolayers were reversible between static and dynamic cultures.39,40 Here, we show how loss of tight junction markers due to high shear stress can be recovered if physiological conditions are restored. We have also observed that ZO-1 but not Claudin-5 was downregulated after imposing nonphysiological flow. This observation is consistent with the reported plasticity of ZO-1 as rapid trigger of barrier phenotype loss, which should eventually be followed by Claudin-5 downregulation.41 As the interface separating blood and the CNS, we believe that hemodynamic repair (pharmacological or interventional) could improve the performance of the neurovascular unit and mitigate the progression of neurological diseases through stabilizing shear stress at the BBB. A recent study showed how renal denervation treatment lead to permeability decrease and recovery of occludin expression in hypertensive rats.42
Our findings add insight into how shear stress governs barrier phenotype in microvascular endothelial cells. Physiological shear stress promotes the expression of tight junctions and transport markers. When such conditions are lost, mechano-transductors activate stress signaling pathways leading to a decrease in the expression of tight junction markers and a reduction of the efflux activity. We provide preliminary evidence supporting the hypothesis that shear-induced damage in the endothelium may be reversed if physiological cerebral blood flow is recovered. Correlations between flow patterns and endothelial cell state at the BBB might be of help for the diagnosis and treatment of neurological diseases. This manuscript opens a new frame to better understand and model cerebral diseases in which hemodynamic disorders and BBB disruption are involved.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Michael O’Rourke, Dr Vipul Chitalia, Dr Moshe Shashar, and Mario Lopez Moya for their insightful suggestions along the study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by a grant from Spain Ministerio de Economía y Competitividad (SAF2013-43302-R, MB), by a grant from NIH (R01 GM 49039, ERE), and by a grant from Spain’s Ministry of Health (Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III (FEDER, RD12/0042/0020) (JR, AO). MB was supported by Fundació Empreses IQS. FGP and PdRP were supported by Banco Santander fellowship programs. FGP was supported by a fellowship from the IQS School of Engineering. PML was supported by the Beatriu de Pinós Program, Modalitat-A awarded by AGAUR (fellowship number: 2013 BP_A 00051).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Conception and design of the experiments: FGP, JM, PML, COB, ERE, MB. Provision of clinical insight for design of experiments: JR, AO, AP. Performance of the experiments: FGP, PDRP. Analysis and interpretation of the data: FGP, JM, ERE, MB. Writing of the paper: FGP. Editing the paper: JM, PML, COB, JR, AO, AP, ERE, MB.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and Stroke Statistics—2016 Update: A Report From the American Heart Association. 2015. DOI: 10.1161/CIR.0000000000000350. [DOI] [PubMed]
- 2.Desmond DW, Moroney JT, Sano M, et al. Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke 2002; 33: 2254–2260. [DOI] [PubMed] [Google Scholar]
- 3.Hirata K, Yaginuma T, O’Rourke MF, et al. Age-related changes in carotid artery flow and pressure pulses: Possible implications for cerebral microvascular disease. Stroke 2006; 37: 2552–2556. [DOI] [PubMed] [Google Scholar]
- 4.O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: A clinical perspective. J Am Coll Cardiol 2007; 50: 1–13. [DOI] [PubMed] [Google Scholar]
- 5.Cooper LL, Woodard T, Sigurdsson S, et al. Cerebrovascular damage mediates relations between aortic stiffness and memory. Hypertension 2016; 67: 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell GF, van Buchem MA, Sigurdsson S, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: The age, gene/environment susceptibility - Reykjavik study. Brain 2011; 134: 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singer J, Trollor JN, Crawford J, et al. The association between pulse wave velocity and cognitive function: The Sydney memory and ageing study. PLoS One 2013; 8: 4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waldstein SR, Rice SC, Thayer JF, et al. Pulse pressure and cognitive decline pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore longitudinal study of aging. Hypertension 2008; 51: 99–104. [DOI] [PubMed] [Google Scholar]
- 9.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 2002; 200: 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohtsuki S, Terasaki T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 2007; 24: 1745–1758. [DOI] [PubMed] [Google Scholar]
- 11.Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: New insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke 2015; 10: 143–152. [DOI] [PubMed] [Google Scholar]
- 12.Marra A, Vargas M, Striano P, et al. Posterior reversible encephalopathy syndrome: The endothelial hypotheses. Med Hypotheses 2014; 82: 619–622. [DOI] [PubMed] [Google Scholar]
- 13.Schreiber S, Bueche CZ, Garz C, et al. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? - New insights from a rat model. Exp Transl Stroke Med 2013; 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topakian R, Barrick TR, Howe FA, et al. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry 2010; 81: 192–197. [DOI] [PubMed] [Google Scholar]
- 15.Di Marco LY, Venneri A, Farkas E, et al. Vascular dysfunction in the pathogenesis of Alzheimer’s disease — A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis 2015; 82: 593–606. [DOI] [PubMed] [Google Scholar]
- 16.Iadecola C. The pathobiology of vascular dementia. Neuron 2013; 80: 844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cucullo L, Hossain M, Tierney W, et al. A new dynamic in vitro modular capillaries-venules modular system: Cerebrovascular physiology in a box. BMC Neurosci 2013; 14: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong AD, Ye M, Levy AF, et al. The blood-brain barrier: An engineering perspective. Front Neuroeng 2013; 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cucullo L, Hossain M, Puvenna V, et al. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci 2011; 12: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benetos A, Laurent S, Hoeks AP, et al. Arterial alterations with aging and high blood pressure. A noninvasive study of carotid and femoral arteries. Arterioscler Thromb Vasc Biol 1993; 13: 90–97. [DOI] [PubMed] [Google Scholar]
- 21.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forster S, Thumser AE, Hood SR, et al. Characterization of Rhodamine-123 as a tracer dye for use in in vitro drug transport assays. PLoS One 2012; 7: e33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 2004; 5: 347–360. [DOI] [PubMed] [Google Scholar]
- 24.Martorell J, Santomá P, Kolandaivelu K, et al. Extent of flow recirculation governs expression of atherosclerotic and thrombotic biomarkers in arterial bifurcations. Cardiovasc Res 2014; 103: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamik A, Lin Z, Kumar A, et al. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem 2007; 282: 13769–13779. [DOI] [PubMed] [Google Scholar]
- 26.Chiu JJ, Usami S, Chien S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann Med 2009; 41: 19–28. [DOI] [PubMed] [Google Scholar]
- 27.Rochfort KD, Collins LE, McLoughlin A, et al. Shear-dependent attenuation of cellular ROS levels can suppress proinflammatory cytokine injury to human brain microvascular endothelial barrier properties. J Cereb Blood Flow Metab 2015; 35: 1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rochfort KD, Cummins PM. Thrombomodulin regulation in human brain microvascular endothelial cells in vitro: Role of cytokines and shear stress. Microvasc Res 2014; 97: 1–5. [DOI] [PubMed] [Google Scholar]
- 29.Taheri S, Gasparovic C, Huisa BN, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke 2011; 42: 2158–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg GA. Neurological diseases in relation to the blood–brain barrier. J Cereb Blood Flow Metab 2012; 32: 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottardi CJ, Arpin M, Fanning AS, et al. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc Natl Acad Sci USA 1996; 93: 10779–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarbell JM, Simon SI, Curry FE. Mechanosensing at the vascular interface. Annu Rev Biomed Eng 2014; 16: 505–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung B, Obinata H, Galvani S, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 2012; 23: 600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jalali S, Li YS, Sotoudeh M, et al. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol 1998; 18: 227–234. [DOI] [PubMed] [Google Scholar]
- 35.Liu D-Z, Ander BP, Xu H, et al. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol 2010; 67: 526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai Y, Xu G, Xu M, et al. Inhibition of Src phosphorylation reduces damage to the blood-brain barrier following transient focal cerebral ischemia in rats. Int J Mol Med 2014; 34: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Mrsny RJ. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J Cell Biol 2000; 148: 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta 2008; 1778: 729–756. [DOI] [PubMed] [Google Scholar]
- 39.DePaola N, Phelps JE, Florez L, et al. Electrical impedance of cultured endothelium under fluid flow. Ann Biomed Eng 2001; 29: 648–656. [DOI] [PubMed] [Google Scholar]
- 40.Seebach J, Donnert G, Kronstein R, et al. Regulation of endothelial barrier function during flow-induced conversion to an arterial phenotype. Cardiovasc Res 2007; 75: 596–607. [DOI] [PubMed] [Google Scholar]
- 41.Tornavaca O, Chia M, Dufton N, et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol 2015; 208: 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakagawa T, Hasegawa Y, Uekawa K, et al. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J Am Heart Assoc 2013; 2: e000375–e000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.