

Abstract
The blood-brain barrier (BBB) and the glia limitans serve to prevent the migration of cells and other large molecules from the blood into the CNS. Neuroinflammatory diseases are characterized by disruption of the BBB and increased leukocyte infiltration into the CNS. In this issue of the JCI, Horng and colleagues demonstrate that astrocytes of the glia limitans induce tight junction formation in response to inflammatory cues, thereby tightening the border to limit the number of activated T cells infiltrating the CNS. Moreover, preventing the formation of this inducible barrier in mice increased disease severity in models of neuroinflammation. Together, the results of this study indicate that the inducible barrier of the glia limitans should be further explored as a therapeutic target.
Introduction
Glial cells are nonneuronal cells that play diverse roles in the CNS. Astrocytes are the predominant glial cell in the CNS and are important for the regulation of metabolism, for the modulation of neuronal transmission, and for CNS development and repair, among other functions (1–6). Astrocytes also are involved in the response to CNS injury and disease, and they are thought to participate in the pathogenesis of multiple sclerosis (MS) in humans and in the development of MS-like disease in animal models of experimental autoimmune encephalomyelitis (EAE) (7). Another essential function of astrocytes in health and disease is the control of the blood-brain barrier (BBB), which regulates the passage of blood leukocytes and molecules into the CNS (8, 9).
The BBB is a highly selective membrane formed by endothelial cells connected by tight junctions composed of claudin-5 (CLDN-5), occludin, and other molecules and separates the circulating blood from the brain extracellular fluid. BBB endothelial cells are surrounded by the endothelial basement membrane and interposed pericytes (9). This selective barrier allows the passage of water, gases, and nutrients such as glucose, while limiting the passage of cells, antibodies, neurotoxins, and other molecules present in the blood into the brain. Beyond the BBB lies the perivascular space, delimited between the endothelial basement membrane and a second barrier, termed the glia limitans, formed by astrocytic endfeet processes that surround the endothelial cells (9) (Figure 1).
Figure 1. The BBB and the glia limitans together limit the migration of inflammatory leukocytes from the blood into the CNS.
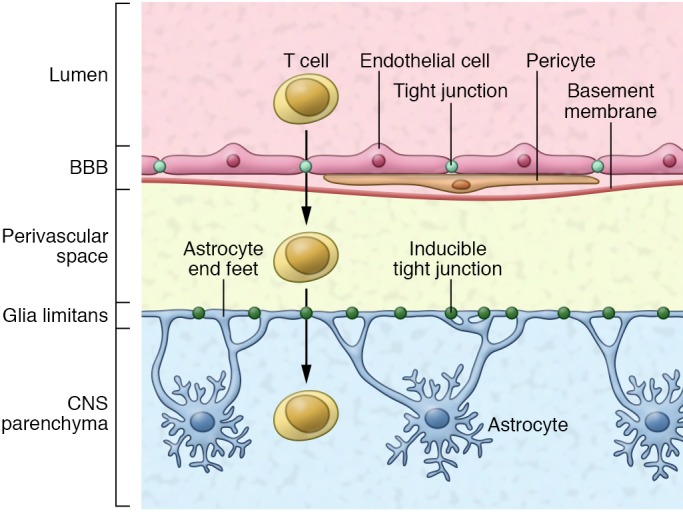
The BBB is formed by endothelial cells connected by tight junctions and surrounded by a basement membrane with pericytes interspersed. The glia limitans serves as a second barrier that is formed by astrocytic endfeet processes. In this issue, Horng et al. reveal that tight junction formation in the glia limitans is induced in response to inflammation and serves as a secondary barrier to limit infiltration of activated T cells into the CNS.
Astrocytes and pericytes establish and maintain the BBB and also participate in its disruption during CNS inflammation and infections. For example, following activation by proinflammatory cytokines, astrocytes can promote the downregulation of tight junction components in the BBB (10). Reactive astrocytes, however, can also form astroglial scars that limit access of inflammatory mediators to the CNS (11). The physiological relevance of reactive astrocytes in limiting CNS inflammation is highlighted by experiments in which astrocyte depletion worsens CNS pathology (12–14). Although astrocytes are known to play an important role in the control of BBB integrity, less is known about the regulation of the glia limitans and the role of this second barrier in CNS inflammation. In this issue, Horng et al. reveal that astrocytic endfeet at the glia limitans play an important role in regulating the influx of blood leukocytes and immune molecules into the CNS, consequently limiting immune-driven pathology.
The glia limitans controls CNS inflammation
While investigating the expression of tight junction proteins in reactive astrocytes, Horng et al. found that treatment astrocytes with the proinflammatory cytokine IL-1β induces the expression of CLDN-1, CLDN-4, and junction adhesion molecule-A (JAM-A), all of which are associated with tight junctions in skin and bladder epithelia but not expressed in the BBB (15). Coimmunoprecipitation studies showed that CLDN-1, CLDN-4, and JAM-A are anchored to the actin cytoskeleton in astrocytes through multiple protein complexes, suggesting that these proteins establish functional tight junctions. In a murine model, tight junction–like structures containing CLDN-1, CLDN-4, and JAM-A were induced at astrocytic endfeet in the glia limitans following virus-driven IL-1β expression in the CNS and also during the course of EAE.
Using an in vitro model to study interactions between astrocytes and T cells, Horng et al. found that expression of tight junction proteins in IL-1β–activated astrocytes results in formation of a network of processes that surround activated T cells. Of note, the corralling of the T cells was dependent on the activation of both astrocytes and T cells. Interestingly, activated T cells promoted the degradation of the tight junction proteins CLDN-1 and CLDN-4, expressed by reactive astrocytes through a mechanism mediated by the metalloproteinases 3, 7, and 9; kallikrein; and/or urokinase. These findings reveal a subtle ongoing battle between astrocytes that corral activated T cells and the T cells that try to break free to access the CNS parenchyma.
Finally, Horng and colleagues evaluated the physiological relevance of the tight junction–dependent corralling of T cells by astrocytic endfeet at the glia limitans. Specifically, the authors employed mice with a glial fibrillary acidic protein–Cre–driven (Gfap-Cre–driven) deletion of Cldn4 in astrocytes (Gfap-Cre Cldn4fl/fl mice). Of note, CLDN-4 is a structural component of tight junctions with no apparent signaling function. Following injection of an IL-1β–expressing adenovirus in the CNS or EAE induction, Gfap-Cre Cldn4fl/fl mice showed increased infiltration of CD4+, CD11b+, and CD45+ leukocytes into the CNS, as well as increased CNS antibody and fibrinogen deposition. Moreover, Cldn4 deletion in astrocytes led to increased CNS damage and worsened disease in EAE in terms of both clinical score and mortality.
Summary and future directions
Collectively, the findings of Horng et al. identify an inducible barrier that operates in the glia limitans to limit CNS infiltration by blood leukocytes and immune molecules. This is an exciting finding that suggests new research directions for the field of neuroimmunology. For example, it would be interesting to study whether TNFα and GM-CSF, cytokines known to contribute to disease pathology in MS and other neurologic disorders (16, 17), can also induce tight junction formation at the glia limitans. Moreover, based on the recently described regulation of astrocytes by microbial products via the aryl hydrocarbon receptor (18, 19), it will be interesting to investigate the regulation of glia limitans functionality by the commensal flora. These are important issues to address to further the understanding of the molecular mechanisms that regulate this inducible glial barrier in health and disease.
All cells are not created equal. Indeed, multiple T cell and astrocyte populations have been identified, spanning from effector and regulatory T cells to recent reports on A1 and A2 astrocytes (20). Thus, it is important to determine whether the astrocyte/T cell interaction at the glia limitans is influenced by the heterogeneity of its cellular components. Moreover, this interaction is likely to have bidirectional effects on both T cells and astrocytes. For example, although Horng et al. report that IL-17 does not affect the expression of tight junction proteins, it is possible that other cytokines secreted by Th17 cells may modulate the barrier function of the glia limitans, as previously reported for the BBB (21). In addition, IL-10 produced by type 1 Tregs has been recently shown to modulate astrocyte responses via IL-10 (22). Thus, it would be interesting to determine the effect of specific effector T cell and Treg populations on the functionality of the glia limitans. Conversely astrocytes produce cytokines that can potentially exert profound effects on the T cell response. Specifically, astrocytes have been shown to produce IL-27, which suppresses pathogenic Th17 cells and induces suppressive Tr1 cells (23, 24). Thus, cytokines and other molecules produced by astrocytes at the glia limitans have the potential to influence the phenotype and function of the T cells interacting with them.
Finally, the findings of Horng et al. raise additional questions. For example, what is the role of the inducible barrier at the glia limitans in the patrolling of the healthy CNS by T cells, an activity that is known to play an important role in preventing chronic viral infections, as evidenced by the development of JC virus–driven progressive multifocal leukoencephalopathy in some patients treated with blockers of T cell migration into the CNS (25)? In addition, what is the contribution of this astrocyte/T cell interaction to the suppression of the immune response directed against brain tumors (26). And finally, how can this barrier be targeted for drug delivery and for the therapeutic modulation of the T cell response in the CNS? The work of Horng et al. suggests that the further evaluation of this inducible barrier at the glia limitans will shed new light onto disease pathology and guide new therapeutic interventions for neurologic disorders.
Acknowledgments
FJQ would like to thank all members of the Quintana laboratory for helpful discussions and acknowledges support by the NIH (NS087867, ES025530, AI126880 and AI093903), the National MS Society (RG4111A1 and JF2161-A-5), and the International Progressive MS Alliance (PA-1604-08459).
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2017;127(8):2897–2899. https://doi.org/10.1172/JCI95769.
See the related article at Astrocytic tight junctions control inflammatory CNS lesion pathogenesis.
References
- 1.Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457(7230):675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- 2.Allen NJ, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486(7403):410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18(7):942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molofsky AV, et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature. 2014;509(7499):189–194. doi: 10.1038/nature13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai HH, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337(6092):358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothhammer V, Quintana FJ. Control of autoimmune CNS inflammation by astrocytes. Semin Immunopathol. 2015;37(6):625–638. doi: 10.1007/s00281-015-0515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez JI, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334(6063):1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 9.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argaw AT, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122(7):2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayo L, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med. 2014;20(10):1147–1156. doi: 10.1038/nm.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toft-Hansen H, Füchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 2011;59(1):166–176. doi: 10.1002/glia.21088. [DOI] [PubMed] [Google Scholar]
- 14.Voskuhl RR, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29(37):11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horng S, et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J Clin Invest. 2017;127(8):3136–3151. doi: 10.1172/JCI91301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Codarri L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 17.El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothhammer V, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586–597. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology. 2013;138(3):183–189. doi: 10.1111/imm.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kebir H, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13(10):1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayo L, et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain. 2016;139(Pt 7):1939–1957. doi: 10.1093/brain/aww113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mascanfroni ID, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat Med. 2015;21(6):638–646. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mascanfroni ID, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14(10):1054–1063. doi: 10.1038/ni.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wollebo HS, White MK, Gordon J, Berger JR, Khalili K. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann Neurol. 2015;77(4):560–570. doi: 10.1002/ana.24371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabriely G, Wheeler MA, Takenaka MC, Quintana FJ. Role of AHR and HIF-1α in Glioblastoma Metabolism. Trends Endocrinol Metab. 2017;28(6):428–436. doi: 10.1016/j.tem.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]