

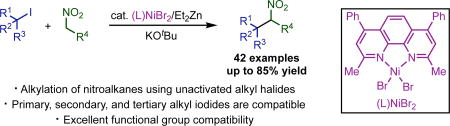
Abstract
Enabled by nickel catalysis, a mild and general catalytic method for C-alkylation of nitroalkanes with unactivated alkyl iodides is described. Compatible with primary, secondary and tertiary alkyl iodides, and tolerant of a wide range functional groups, this method allows rapid access to diverse nitroalkanes.
Nitroalkanes are one of the most versatile functional groups in organic synthesis.1 Their use in C-C bond formation, through conjugate addition, Henry reactions, palladium-catalyzed arylation and allylation, and related reactions, has been well established.1–2 In addition, the nitro group can be readily converted into a range of other functionalities, including amines, carbonyls, and alkanes.1 Despite this rich chemistry, however, C-alkylation of nitroalkanes with alkyl electrophiles has been historically challenging due to competing O-alkylation and formation of carbonyl byproducts.3 Early methods to overcome this inherent reactivity and favor C-alkylation suffered from lack of generality.4
In 2012, we reported a general method for benzylation of nitroalkanes using a simple copper catalyst.5 In subsequent studies, we have shown that this catalyst system is also capable of alkylating nitroalkanes using α-bromocarbonyls and α-bromonitriles.6 While these reactions provided a significant advance in nitroalkane synthesis compared to prior art, they all required a radical stabilizing group adjacent to the electrophilic center. Alkyl halides lacking such stabilization failed to provide more than trace amounts of the desired alkylated products.
Recognizing that a method capable of utilizing non-stabilized alkyl electrophiles would greatly expand the scope and utility of nitroalkane alkylation, we set out to identify catalysts that would enable this transformation. Herein, we report the nickel-catalyzed nitroalkane alkylation. For the first time, this system allows for the alkylation of nitroalkanes using primary, secondary, and tertiary alkyl halides without the requirement for stabilizing groups. The method allows for the preparation of a diverse array of complex nitroalkanes using simple starting materials.
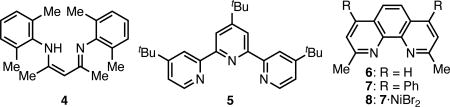
Our initial investigation focused on the reaction of primary alkyl iodide 1 and nitroalkane 2 (Table 1). Consistent with our previous reports, we observed no C-alkylation product 3 when the reaction was conducted in the presence of catalytic copper bromide and diketimine ligands (entry 1), or in the absence of catalyst. Other copper-based catalyst systems also failed to provide product.7 Early in our first studies of nitroalkane alkylation we had noted that nickel catalysts, although less reactive than the copper based systems we investigated at that time, did provide trace product in nitroalkane benzylation.5 Inspired by that result, and previous reports of nickel-catalyzed reactions with unactivated alkyl halides,8 we investigated the use of nickel catalysis in the present system. We were pleased to find that the use of catalytic Ni(COD)2 with an appropriate ligand led to the first appreciable production of 3. For example, with use of either tripyridyl ligand 5 (entry 2) or neocuproine (6, entry 3), single digit yields of the product were observed. Investigation of other nickel sources revealed that the combination of NiBr2.diglyme and Et2Zn (as reductant) was much more effective, providing 3 in 59% yield (entry 5).9 Further optimization revealed that the related ligand bathocuproine (7) was optimally effective, as was a mixed solvent of MTBE and dioxane (entries 6 and 7). Finally, the single-component catalyst 8 (prepared from 7 and NiBr2·diglyme) proved similarly effective and was selected for further study due to ease of handling and reaction setup (entry 8).10
Table 1.
Discovery of the Catalyst System.

| ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Additive | Yield of 3a |
|
| ||||
| 1 | 20 mol % CuBr/4b | hexanes | – | 0% |
| 2 | Ni(COD)2/5 | dioxane | – | 7% |
| 3 | Ni(COD)2/6 | dioxane | – | 8% |
| 4 | NiBr2·diglyme/6 | dioxane | – | 0% |
| 5 | NiBr2·diglyme/6 | dioxane | Et2Zn | 59% |
| 6 | NiBr2·diglyme/7 | dioxane | Et2Zn | 67% |
| 7 | NiBr2·diglyme/7 | MTBE/dioxane | Et2Zn | 74% |
| 8 | 8 | MTBE/dioxane | Et2Zn | 76% |
Determined via 1H NMR against internal standard;
60 °C.
As can be seen in Scheme 1, a broad range of primary alkyl iodides, including those bearing a high degree of functionality, are tolerated in the reaction. Particularly notable is the reaction’s tolerance of steric hindrance (product 10 is derived from a neopentyl iodide), common protecting groups (11, 16, 23), and aromatic halides (20–22). Biologically relevant heterocycles (both aromatic and non-aromatic) are particularly well tolerated (13–24). These include benzothiazoles, benzofurans, piperidines, thiophenes, indoles, pyridines, and pyrazoles, among others. Furthermore, alkyl chlorides and bromides (13 and 14) were unaffected, allowing for orthogonality with alkyl bis-electrophiles. Methyl iodide could also be used, but gave low yields.7
Scheme 1. Scope of Primary Alkyl Iodides.

a 15 mol % 8, 30 mol % Et2Zn; b 10 mol % 7 added.
We then turned our attention to more substituted alkyl iodides (Scheme 2). A variety of cyclic and acyclic secondary alkyl iodides were also well tolerated (25–32). As with the primary substrates, the reaction also displayed outstanding functional group tolerance. For example, aromatic (25, 28, 29), heteroaromatic (32) and aliphatic heterocycles (31 and 32) were all compatible.11 Unfortunately, little to no diastereoselectivity was observed with dissymmetric sec-alkyl iodide substrates (25, 29).
Scheme 2. Scope of Secondary and Tertiary Alkyl Iodides.

a 15 mol % 8, 30 mol % Et2Zn
We were very pleased to find that tertiary alkyl iodides can also be utilized in the reaction (33–36). These substrates provide very sterically encumbered nitroalkanes that are hard to access by other methods. Tert-butyl-, cyclohexylmethyl-, and adamantyl-iodide all participated in the reaction without incident.
A variety of functionalized nitroalkanes can also be used in the reaction (Scheme 3). Although secondary and β-branched nitroalkanes did not show satisfactory yields,7 various functional groups on the nitroalkane proved to be compatible; including alkenes (38), acetyl protected alcohols (41), esters (42), Boc-protected amines (43), phthalimides (44), and nitriles (45). Although nitroalkanes bearing unprotected ketone proved unfruitful as substrates, protecting the ketone as an acetal (39) allowed for good yields.
Scheme 3. Scope of Nitroalkanes.

a 15 mol % 8, 30 mol % Et2Zn; b 10 mol % 7 added.
With some substrates bearing ill-positioned Lewis basic groups (e.g., 18, 41, 44), poor reactivity was observed under the standard reaction conditions. We attribute this to competitive binding of the catalyst, possibly via chelation in a reactive intermediate. Gratifyingly, however, we found adding 10 mol % of bathocuproine (7, in addition to catalyst 8) to the reaction restores the reactivity.7
Finally, the use of nitromethane was also examined. With primary alkyl iodides, a mixture of mono- and di-alkylated products resulted.7 With secondary alkyl iodides, modest to good levels of monoalkylation product were observed (see Scheme 5 below, for an example).
Scheme 5. Stereoconvergence of the Reaction.

Prior studies have shown that nickel-catalyzed cross-couplings can proceed through diverse reaction mechanisms, including one and two electron pathways.12,13 To gain insight into the present alkylation reaction, several studies were conducted. First, the addition of one equivalent of TEMPO (a known radical scavenger)14 completely inhibits product formation (Scheme 4, top). Second, the reaction involving cyclopropylmethyliodide (46) with nitroalkane 47 resulted in ring-opening to provide product 48 in good yield (Scheme 4, middle).15 Conversely, the reaction using 1-iodohex-5-ene (49), resulted in 5-exotrig ring-closure to give product 50 (Scheme 4, bottom).
Scheme 4. Mechanistic Probes.

Third, we examined the stereospecificity of the alkylation using each diastereostereoisomer of 51 (Scheme 5). Although the yields in the reaction differed, both isomers led to the same mixture of isomers of 52 in the reaction with nitromethane (as determined by 1H and 19F NMR). Taken together, the results presented in Schemes 4 and 5 strongly support a radical-based mechanism of this transformation.,
Finally, to demonstrate the synthetic value of this alkylation reaction, the anti-viral drug adapromine (54) was prepared in two steps from commercially available materials.16 First, alkylation of 1-nitropropane with 1-admantyl iodide provides secondary nitroalkane 53 in good yield. Second, reduction of the nitro group to the primary amine under Raney nickel conditions provides the target compound in near quantitative yield. This two-step sequence is highly competitive with known routes to this pharmaceutical agent.17
In summary, enabled by the discovery of effective nickel catalysis, we have developed the first general catalytic system that achieves the C-alkylation of nitroalkanes with unactivated alkyl halides. The reaction proceeds under mild conditions using commercially available catalytic components and can be applied to primary, secondary and tertiary alkyl iodides. Moreover, it shows exceptional functional group tolerance. The simplicity of this reaction, combined with the rapid access to complex nitroalkanes that it provides, should find wide use in the preparation of nitrogen-containing molecules.
Supplementary Material
Figure 1.

General Method for Nitroalkane Alkylation.
Scheme 6. Synthesis of Adapromine.

Acknowledgments
The University of Delaware (UD), the University of Delaware Research Foundation, the Research Corp. Cottrell Scholars Program, and the NIH NIGMS (R01 GM102358) are gratefully acknowledged for support. Dr. Glenn Yap (UD) is acknowledged for assistant with X-ray crystallography. Data was acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF CHE0421224, CHE0840401, CHE1229234, CHE1048367; NIH S10 OD016267-01, S10 RR026962-01, P20GM104316, P30GM110758).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, crystallographic and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]
- 2.a Vogl EM, Buchwald SL. J. Org. Chem. 2002;67:106–111. doi: 10.1021/jo010953v. [DOI] [PubMed] [Google Scholar]; b Walvoord RR, Kozlowski MC. J. Org. Chem. 2013;78:8859–8864. doi: 10.1021/jo401249y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Padilla-Salinas R, Walvoord RR, Tcyrulnikov S, Kozlowski MC. Org. Lett. 2013;15:3966–3969. doi: 10.1021/ol401747u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d VanGelder KF, Kozlowski MC. Org. Lett. 2015;17:5748–5751. doi: 10.1021/acs.orglett.5b02793. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Aleksandrowicz P, Piotrowska H, Sas W. Tetrahedron. 1982;38:1321–1327. [Google Scholar]; f Tsuji J, Yamada T, Minami I, Yuhara M, Nisar M, Shimizu I. J. Org. Chem. 1987;52:2988–2995. [Google Scholar]; g Rieck H, Helmchen G. Angew. Chem. Int. Ed. Eng. 1995;34:2687–2689. [Google Scholar]; h Maki K, Kanai M, Shibasaki M. Tetrahedron. 2007;63:4250–4257. [Google Scholar]
- 3.a Weisler L, Helmkamp RW. J. Am. Chem. Soc. 1945;67:1167–1171. [Google Scholar]; b Hass HB, Bender ML. J. Am. Chem. Soc. 1949;71:1767–1769. [Google Scholar]; c Kornblum N. Angew. Chem. Int. Ed. Eng. 1975;14:734–745. [Google Scholar]
- 4.a Seebach D, Lehr F. Angew. Chem. Int. Ed. Eng. 1976;15:505–506. [Google Scholar]; b Seebach D, Henning R, Lehr F, Gonnermann J. Tetrahedron Lett. 1977;18:1161–1164. [Google Scholar]; c Seebach D, Henning R, Lehr F. Angew. Chem. Int. Ed. Eng. 1978;17:458–459. [Google Scholar]; d Katritzky AR, de Ville G, Patel RC. J. Chem. Soc., Chem. Commun. 1979:602–602. [Google Scholar]; e Katritzky AR, Kashmiri MA, De Ville GZ, Patel RC. J. Am. Chem. Soc. 1983;105:90–96. [Google Scholar]; f Russell GA, Hershberger J, Owens K. J. Am. Chem. Soc. 1979;101:1312–1313. [Google Scholar]; g Russell GA, Khanna RK. Tetrahedron. 1985;41:4133–4145. [Google Scholar]; h P Branchaud B, Yu G-X. Tetrahedron Lett. 1988;29:6545–6548. [Google Scholar]
- 5.Gildner PG, Gietter AAS, Cui D, Watson DA. J. Am. Chem. Soc. 2012;134:9942–9945. doi: 10.1021/ja304561c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Gietter AAS, Gildner PG, Cinderella AP, Watson DA. Org. Lett. 2014;16:3166–3169. doi: 10.1021/ol5014153. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shimkin KW, Gildner PG, Watson DA. Org. Lett. 2016;18:988–991. doi: 10.1021/acs.orglett.6b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.See Supporting Infomation
- 8.a Netherton MR, Fu GC. Adv. Synth. Cat. 2004;346:1525–1532. [Google Scholar]; b Rudolph A, Lautens M. Angew. Chem. Int. Ed. 2009;48:2656–2670. doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]; c Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For additional optimization details, including the use of other bases and reductants, see Supporting Information.
- 10.The reaction does not require light, see Supporting Information for details.
- 11.In cases where lower product yield was observed, the mass balance for the reaction was primarily starting materials, along with traces of alkene and hydrocarbon derived from elimination or reduction of the alkyl iodide.
- 12.a Hu X. Chem. Sci. 2011;2:1867–1886. [Google Scholar]; b Biswas S, Weix DJ. J. Am. Chem. Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Breitenfeld J, Ruiz J, Wodrich MD, Hu X. J. Am. Chem. Soc. 2013;135:12004–12012. doi: 10.1021/ja4051923. [DOI] [PubMed] [Google Scholar]; d Schley ND, Fu GC. J. Am. Chem. Soc. 2014;136:16588–16593. doi: 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan C-M, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J. Am. Chem. Soc. 2016;138:2174–2177. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mohadjer Beromi M, Nova A, Balcells D, Brasacchio AM, Brudvig GW, Guard LM, Hazari N, Vinyard DJ. J. Am. Chem. Soc. 2017;139:922–936. doi: 10.1021/jacs.6b11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For related radical reactions involving palladium-catalysts, see: Bloome KS, Alexanian EJ. J. Am. Chem. Soc. 2010;132:12823–12825. doi: 10.1021/ja1053913.Monks BM, Cook SP. Angew. Chem. Int. Ed. 2013;52:14214–14218. doi: 10.1002/anie.201308534.Sargent BT, Alexanian EJ. J. Am. Chem. Soc. 2016;138:7520–7523. doi: 10.1021/jacs.6b04610.
- 14.a Beckwith ALJ, Bowry VW, Ingold KU. J. Am. Chem. Soc. 1992;114:4983–4992. [Google Scholar]; b Bowry VW, Ingold KU. J. Am. Chem. Soc. 1992;114:4992–4996. [Google Scholar]
- 15.a Griller D, Ingold KU. Acc. Chem. Res. 1980;13:317–323. [Google Scholar]; b Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 16.Secondary nitronate anions have been shown to undergo reaction with free-radicals (see references 3c, 4f, and 4g for examples). The fact that secondary nitroalkanes provide poor yield in this alkylation may suggest that a complex mechanism is at play. Further studies will be directed at elucidating the mechanistic details.
- 17.a Aldrich PE, Hermann EC, Meier WE, Paulshock M, Prichard WW, Synder JA, Watts JC. J. Med. Chem. 1971;14:535–543. doi: 10.1021/jm00288a019. [DOI] [PubMed] [Google Scholar]; b Spasov AA, Khamidova TV, Bugaeva LI, Morozov IS. Pharm. Chem. J. 2000;34:1–7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.