

Abstract
Nonribosomal peptide synthetases (NRPSs), responsible for synthesizing many medicinally important natural products, frequently use adenylation domain activators (ADAs) to promote substrate loading. Although ADAs are usually MbtH-like proteins (MLPs), a new type of ADA appears to promote an NRPS-dependent incorporation of a dihydropyrrole unit into sibiromycin. The adenylation and thiolation didomain of the NRPS SibD catalyzes the adenylation of a limited number of amino acids including L-tyr, the precursor in dihydropyrrole biosynthesis, using a standard radioactivity exchange assay. LC-MS/MS analysis confirmed loading of L-tyr onto the thiolation domain. SibB, a small protein with no prior functional assignment nor sequence homology to MLPs, was found to promote the exchange activity. MLPs from bacteria expressing homologous biosynthetic pathways were unable to replace this function of SibB. Discovery of this new type of ADA demonstrates the importance of searching beyond the conventional MLP standard for proteins affecting NRPS activity.
Keywords: adenylation domain activators, MbtH, biosynthesis, nonribosomal peptide synthetases, natural products
Graphical Abstract
Nonribosomal peptide synthetases (NRPSs) frequently use MbtH-like proteins to promote activity of their adenylation domains. Sibiromycin biosynthesis provides an example of a pathway that lacks such a protein and instead uses an atypical adenylation domain activator, SibB, to promote the activity of the NRPS SibD.
Nonribosomal peptide synthetases (NRPSs) are multidomain proteins organized into initiation, elongation, and termination modules that work successively to synthesize small peptides of pharmacological importance.[1] Each module contains an adenylating (A) domain that activates its substrate amino or hydroxy acid by forming an acyl adenylate intermediate. A thiolation (T) domain then serves as an acyl acceptor by forming a thioester. Elongation modules include an additional condensation (C) domain for coupling two NRPS-conjugates through a peptide bond. Termination modules contain yet another domain most typically a thioesterase (TE) domain or infrequently a reductase (R) domain that promotes hydrolysis of the nascent peptide from the NRPS. These modules often are complemented by a protein known as an A domain activator (ADA).[2] ADAs are small proteins that stimulate A domain activity and were first found necessary for successful biosynthesis of capreomycin and viomycin.[3] In a few cases, NRPSs were not even observed or isolated in a soluble form after heterologous expression without coexpression of an appropriate ADA.[4] The first ADAs reported were all MbtH-like proteins (MLPs) named after a homolog (MbtH) in Mycobacterium tuberculosis and identifiable by a Pfam domain (PF03621).[5] More recently, two new ADAs have been discovered that are devoid of the MLP signature sequences. One of these resembles an incomplete C domain and the other shares very low sequence similarity to known NRPSs (E > 0.002).[6] Both of these ADAs as well as one example of an MLP are covalently fused to their A domains.[6–7] In contrast, the vast majority of ADAs exist as independent proteins encoded by genes within their associated biosynthetic gene clusters[3,4a–c,4d–f,8] or elsewhere in the genome.[4c,8a,8b,8d,8g,8i,9] The frequency for which A domains require ADAs is now sufficient that their potential participation should be checked routinely. However, this can be difficult when genomic DNA has not been fully sequenced or the ADA gene lacks the sequence expected for coding an MLP.
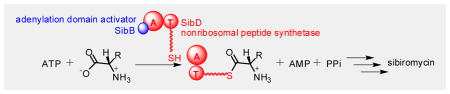
Sibiromycin represents one of many pyrrolo[1,4]benzo-diazepines (PBDs) that are assembled by NRPSs. This natural product has attracted attention based on its ability to alkylate DNA selectively.[10] The combination of high potency and poor pharmacology as an antitumor agent makes this PBD a particularly interesting target for generating analogues through manipulation of its biosynthetic pathway.[11] For example, the organism producing sibiromycin (Streptosporangium sibiricum) was genetically engineered to form a 9-deoxy variant with diminished cardiotoxicity, a testament to the potential of future engineering efforts.[12] Sibiromycin appears to be synthesized from an anthranilate unit and a dihydropyrrole unit that are joined together by NRPSs SibE and SibD, respectively (Scheme 1). SibE has already been well described[13] but little information is available on SibD. Potential substrates for activation and conjugation have now been examined and, most importantly, a new type of ADA has been identified, as reported below.
Scheme 1.

Sibiromycin assembly by SibE and SibD.
Sequence analysis of SibD suggests that it is a termination module since it contains highly conserved sequence motifs of C and R domains as well as A and T domains.[14] The boundary between each domain was predicted using the Udwary-Merski algorithm (UMA)[15] (Figure S1, Supporting Information) and guided cloning of a gene fragment encoding the putative A and T didomain SibDAT. Studies began with the didomain, rather than the entire module, to avoid solubility problems frequently encountered with heterologous expression of large and intact NRPSs. The gene for SibDAT was cloned into a pSMT3 vector. SibDAT was fused to an N-terminal His6-SUMO tag during its expression in BAP-1 Escherichia coli[16] to aid protein purification. This cell line was selected for its ability to coexpress a phosphopantetheinyl transferase that installs the phosphopantetheine tether on the T domain that is essential for substrate loading. SibDAT was purified via Ni2+ affinity chromatography, digested by Ulp1 to remove the SUMO tag, and isolated after size exclusion chromatography (Figure S2). Tandem MS analysis of SibDAT digested with trypsin confirmed phosphopantetheinylation of a serine that is conserved in T domains of SibD and other PBD synthesizing homologs (Figure S3).
The A domain in SibDAT is expected to adenylate (first-half reaction) and subsequently transfer an amino acid onto the phosphopantetheinylated T domain (second half-reaction, Scheme 2). A standard partial reaction involving substrate dependent exchange of [32P]pyrophosphate into ATP was used to confirm that the A domain retained its adenylation activity in the absence of the C and R domains.[17] Since the native substrate for SibDAT remains unknown, candidates from related pathways were tested. HrmP, an NRPS that incorporates a pyrrolidine moiety into hormaomycin,[18] also accepts a number of compounds including L-tyr, L-thr, and L-pro as substrates for adenylation.[8h] In analogy, L-tyr was tested for its ability to stimulate the partial reaction of SibDAT. A reaction mixture containing SibDAT and L-tyr confirmed this exchange by detection of [32P]ATP at low levels that were consistently above background (Figure 1A). A high background is typical of this assay and does not reflect spontaneous exchange since this process requires participation of an amino acid to generate the required aminoacyl adenylate intermediate. The modest stimulation in the presence of L-tyr was not surprising since a dihydropyrrole rather than its precursor L-tyr was expected to be loaded onto SibD during sibiromycin biosynthesis.[10] Replacing L-tyr with L-thr also stimulated exchange and the likely formation of its aminoacyl adenylate as well (Figure 1A). The similarly low activity induced by a possible substrate or a close analog L-tyr and a non-specific substrate L-thr implied a missing component that could affect catalysis. The identity of this component was subsequently examined using L-tyr as a substrate for SibDAT since this seemed most relevant to the dihydropyrrole biosynthesis of sibiromycin.[10]
Scheme 2.

Adenylation and conjugation to a phosphopantetheinylated T domain of the substrate constitute the two-step reaction catalyzed by the A domain and often assayed indirectly by exchange of [32P]pyrophosphate into ATP.
Figure 1.

Detection of amino acid adenylation by SibDAT using a standard partial reaction that measures [32P]ATP production resulting from back exchange of [32P]pyrophosphate into ATP.[17] The [32P]ATP levels are represented as counts per minute (cpm). Error bars represent one standard deviation of uncertainty. Additional experimental details are provided as supporting information. SibDAT (0.25 μM) was reacted with A) each amino acid substrate (0.8 mM) at 37 °C for 50 min in the presence of ATP (3.7 mM), TCEP (1.0 mM), MgCl2 (7.5 mM), glycerol (7.5% v/v), sodium pyrophosphate (1.0 mM), and [32P]pyrophosphate (1 μCi) in HEPES (35 mM pH 7.5). B) Activation of L-tyr (4 mM) adenylation was assessed under equivalent conditions with the added presence of SibD domain(s) (0.25 μM).
The SibDAT fragment had the potential to adopt a more active and selective conformation in the presence of the remaining domains of SibD through interdomain interactions similar to those observed previously between the C and A domains of another NRPS SrfA-C.[1c] To examine this, the gene fragments coding for the putative C and R domains of SibD were cloned into a pACYCDuet-1 vector and used along with the vector containing the gene for SibDAT to transform BAP-1 E. coli for co-expression of all SibD domains. Limitations in vector design resulted in co-expression of His6-SUMO fusions of SibDC and SibDAT and untagged SibDR. Only SibDC and SibDAT were detected after co-purification of all domains. If SibDR had expressed under these conditions, it was not observed. SibDR could have formed inclusion bodies or may have lacked sufficient affinity to SibDAT or SibDC for its co-purification. As an alternative, SibDR was cloned independently as a His6-SUMO fusion. This yielded soluble protein that was purified similarly to SibDAT. Reaction mixtures containing SibDAT and L-tyr were supplemented with one molar equivalent of SibDC, SibDR, or both domains in an attempt to reconstitute maximal activity of the A domain. All reaction mixtures generated levels of [32P]ATP similar to that lacking an added domain. Consequently, the C and R domains prepared as discrete polypeptides under these conditions did not stimulate the exchange activity (Figure 1B) and were not included in further assays used to identify activators of SibDAT.
The prevalence of MLPs used by nature to activate A domains prompted a search for such a protein that would affect the activity of SibDAT. No candidate was initially apparent in the sibiromycin gene cluster since no coding was evident for a PF03621 domain expected in MLPs.[5] The full genome of S. sibiricum is not yet available, and hence the presence of an MLP-encoding gene elsewhere in the genome is not known. In some examples, MLPs from certain hosts can replace the function of MLPs from native sources.[3,6b,7,8c,8g] For instance, the MLPs CloY and SimY coded by gene clusters from different Streptomyces species responsible for the biosynthesis of chlorobiocin and simocyclinone D8, respectively can activate both tyrosine adenylases that initiate assembly of a common aminocoumarin moiety.[8g] Thus, a search for an MLP for SibDAT was extended to species with homologous gene clusters involved in synthesis of related dihydropyrrole moieties. For example, a gene (hrmR) in the hormaomycin producer codes for HrmR, an MLP that activates HrmP in pyrrolidine biosynthesis.[8h] Additionally, two MLP-encoding genes (mlp1 and mlp2) were identified in Nocardia brasiliensis outside a putative PBD biosynthetic gene cluster discovered using the antibiotics and secondary metabolite analysis shell[19] (antiSMASH) prediction software.
The MLP-encoding genes hrmR, mlp1, and mlp2 were each subcloned into a pTYB12 vector, and the corresponding MLPs were heterologously expressed with an N-terminal chitin binding domain-intein tag. Each MLP was purified via chitin binding affinity chromatography and subjected to DTT-induced tag cleavage based on a protocol reported previously (Figure S4).[8i] To assay the effect of MLPs on SibDAT activity, reaction mixtures containing SibDAT and L-tyr were supplemented with four molar equivalents of the MLPs to ensure sufficient stoichiometries that were previously thought to range from 1:1[3,8c,8g–i] to 1.7:1.0.[4a] All reaction mixtures generated equivalent levels of [32P]ATP regardless of these MLPs (Figure 2A). Thus, the MLPs described in this section did not promote the A domain activity of SibDAT. Consequently, the search for an ADA was extended beyond the traditional candidates.
Figure 2.

Effectors of amino acid adenylation by SibDAT as detected by a standard partial reaction that measures [32P]ATP production resulting from back exchange of [32P]pyrophosphate into ATP.[17] The [32P]ATP levels are represented as cpm. Error bars represent one standard deviation of uncertainty. Conditions are equivalent to those described in Figure 1A except for the added presence of either A) proteins containing MLP signature sequences (1 μM) or B) SibB (1 μM).
Attention returned to the most obvious source, the sibiromycin gene cluster of 26 ORFs. Functional roles had been assigned to 24 of these ORFs primarily from bioinformatics analysis.[10] No roles had yet been ascribed to the two ORF products, SibB and SibR, although subsequent examination found that SibR is related by 41% sequence identity to TylU, a transcription regulator.[20] The remaining ORF, SibB, shared no sequence homology with MLPs but was predicted to share a similar size (93 amino acids). SibB also exhibits a trivial level of sequence identity with the two ADAs known to lack MLP signature sequences (6% with the intermediate domain in RubC1 and 3% with the N-terminal domain in NPS3, Figure S5A).[6] Thus, the size of SibB alone compelled investigation of this as a new type of ADA (Figure S5A). To determine if SibB promotes adenylation by SibDAT, the SibB-encoding gene was cloned into the pSMT3 vector to generate a His6-SUMO fusion for expression and purification of SibB in analogy to that applied to SibDAT (Figure S6). When four equivalents of SibB were added to a standard reaction mixture containing SibDAT and L-tyr, [32P]ATP levels above background increased five-fold (Figure 2B). Analogous results were also observed when L-thr was tested in place of L-tyr (Figure 2B). These results provide the first evidence that SibB represents a new class of ADAs.
This role for SibB could not have been predicted from the information available on MLPs since SibB lacks their signature sequences including a tryptophan dyad that forms a hydrophobic cleft for binding to A domains.[7] SibB also lacks the universally conserved sequences surrounding the tryptophans (SxWP and PxGW) in MLPs (Figure S5A),[5] but three other tryptophans (residues 59, 73, and 92) may form a similar cleft. Additionally, the A domain in SibD and its homologs exhibit a residue (proline or alanine, Figure S7) thought to recognize the hydrophobic cleft of ADAs as evident from all MLP-dependent A domains.[7] SibB apparently represents a new class of ADAs that may act through a common mechanism for activation of A domains but does not exhibit standard sequence motifs of the MLPs.
Successful activation of SibDAT also provided a platform to test which biosynthetic intermediate derived from L-tyr might be loaded onto the NRPS. L-DOPA, the first intermediate of this process, was available commercially and a later intermediate 1 was prepared enzymatically as described previously (Scheme 3).[21] A final intermediate in dihydropyrrole biosynthesis 2b was assembled by aid of a key allyl coupling step (Scheme S1). Briefly, an enol triflate of N-Boc-4-oxo-L-proline methyl ester (5) was prepared, cross-coupled with an allyl-tin reagent, demethylated, and deprotected to form 2b. A related derivative 2a was generated analogously with the use of a vinyl-tin reagent. Alternative addition of these compounds (L-DOPA, 1, 2a, 2b) to a standard exchange assay with SibDAT and SibB generated only background levels of [32P]ATP (Figure S8). Thus, none of these species undergo reversible adenylation. Alternative activation and loading of 3 seems unlikely since its tautomer 2b was inactive as well. The remaining intermediate 4 was evaluated indirectly through use of the structural analogues L-trans-4-methylproline 9, L-cis-4-hydroxyproline 10, and L-3,4-dehydroproline 11. These too were incapable of stimulating [32P]ATP formation in equivalent exchange reactions (Figure S8).
Scheme 3.

Proposed biosynthesis of the dihydropyrrole moiety in sibiromycin originating from L-tyr.[10]
Casual analysis of these trends might suggest that the A domain simply exhibits a very high level of selectivity. However, this would not be consistent with its ability to adenylate L-thr that is not involved in the dihydropyrrole synthesis (Figure S8). Many of the purported intermediates in sibiromycin biosynthesis remain speculative and may require revisions similar to those recently published on the formation of a dihydropyrrole intermediate in lincomycin A.[22] Results from gene deletion in this related pathway indicate that methylation appears to target 1 rather than 2a. In addition, the SibY and SibS homologs in lincomycin A biosynthesis have been reassigned to a hydrolase and isomerase, respectively[22] and challenge the previous suggestion of possible endocyclic and exocyclic unsaturation in 3 and 4.
Identification of SibB as a new ADA will certainly help screen future candidates for loading onto SibD but speculation based on the lack of ATP/[32P]pyrophosphate exchange should be considered carefully when the T domain is present. Successful formation of [32P]ATP requires reversible dissociation of the pyrophosphate formed by aminoacyl adenylation and regeneration of ATP (k−1) that is either fast and/or independent of the subsequent thiolation step (k2, Scheme 2). Only direct detection of the last process offers an unambiguous method for evaluating possible NRPS loading.
The amino acids L-tyr and L-thr that were previously shown to activate [32P]ATP exchange provided the best opportunity to observe loading onto the T domain. These amino acids were individually incubated with SibDAT, SibB, and ATP, and the proteins were then denatured and digested with trypsin. Acylation of the phosphopantetheinylated peptide was monitored by LC-MS/MS. Only L-tyr appeared to load onto SibB•SibDAT (SibB•SibDAT-S-tyr) as detected by its phosphopantetheinylated peptide conjugate with an experimental mass of 3639.784 Da (m/z = 910.954 Da, z=4) that deviates by only 13 ppm from its theoretical mass of 3639.830 Da (Scheme 4 and Table S1) (tret 30.2 min, Figure S9). Several daughter b and y ions characteristic of the parent peptide further support this assignment. For example, the acylated fragment peptides ions (y17, y23, and y24) were observed within a mass deviation of only 29, 18, and 15 ppm, respectively. Concurrent detection of the phosphopantetheinylated peptide lacking the tyrosyl conjugation (tret 31.6 min, Figures S9 and S10) suggests a mixture of SibB•SibDAT and SibB•SibDAT-S-tyr was likely generated during incubation. In contrast, no acylation of the phosphopantetheinylated peptide was detected after incubation with L-thr (Figures S11 and S12) despite the ability of this amino acid to stimulate ATP/[32P]pyrophosphate exchange. Either the corresponding SibB•SibDAT-S-thr is less stable than its L-tyr derivative, or transfer of the activated L-threonyl derivative to the T domain is less efficient. The later alternative suggests the potential for substrate discrimination at both the activation and transfer steps of loading in a strategy reminiscent of aminoacyl tRNA formation catalyzed by aminoacyl tRNA synthetases.23
Scheme 4.

UPLC-MS detection of a phosphopantetheinylated peptide conjugated with tyrosine (tret 30.2 min). This peptide was generated via partial tryptic digestion of SibDAT after its incubation with SibB and L-tyr under substrate loading conditions. Fragmentation was observed at the illustrated amide bonds extending from the amino (b ions) and carboxyl (y ions) termini (see Table S1).
Loading studies with 2a and 2b indicated that these derivatives were also unable to acylate the phosphopantetheinylate moiety of SibB•SibDAT (Figures S13 and S14). Thus, the inability of 2a and 2b to stimulate [32P]ATP formation was due to a lack of initial adenylation and not the result of fast acyl transfer and slow [32P]pyrophosphate exchange into ATP. From the data available to date, loading by the SibB•SibD protein complex may start as early as the initial L-tyr that initiates the dihydropyrrole biosynthetic pathway or involve an intermediate that has not yet been identified.[11b]
The ability of L-tyr to stimulate ATP/[32P]pyrophosphate exchange with SibDAT reveals an important function of SibB in sibiromycin biosynthesis whether or not L-tyr is ultimately confirmed to be a substrate for SibD in vivo. SibB represents a new type of ADA and significantly expands their known diversity.[2] The preponderance of reported ADAs are MLPs, and only the examples acting in cis from rubradirin and lysine biosynthesis deviate from this pattern of protein activators. SibB is the first ADA without the signature sequences of MLPs to activate an A domain in trans. Unlike the ADA in rubradirin biosynthesis, SibB cannot be replaced by MLPs from related biosynthetic pathways. Protein BLAST analysis of SibB reveals uncharacterized homologs in other actinobacteria including an antitumor producer Streptomyces globsiporus, a pathogen Mycobacterium abscessus, and Streptacidiphilus rugosus (44%, 40%, and 30% sequence identity, respectively) (Figure S5B). Future searches for small effector proteins controlling NRPS catalysis should therefore not be restricted to MLPs.
Supplementary Material
Acknowledgments
We thank Dr. Barbara Gerratana for leading initial efforts on sibiromycin biosynthesis, Dr. Chris Lima for the pSMT3 plasmid and Ulp1 expression vector, Dr. Chaitan Khosla for the BAP-1 strain of E. coli, Dr. I. Phil Mortimer for assistance with LC-MS/MS experiments, Dr. Gary Posner for advice on preparation of the vinyl and allyl compounds, and Drs. Jeanne Davidsen and Craig Townsend for our introduction to MLPs. This research was supported in part by the National Institutes of Health (GM084473).
Footnotes
Experimental Section
All experimental details including the construction of expression vectors, production and purification of proteins, synthesis and characterization of all compounds, and assays for radioisotope exchange and NRPS acylation are described in supporting information.
References
- 1.a) Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; b) Strieker M, Tanović A, Marahiel MA. Curr Opin Struct Biol. 2010;20:234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]; c) Tanovic A, Samel SA, Essen LO, Marahiel MA. Science. 2008;321:659–663. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- 2.a) Baltz RH. J Ind Microbiol Biotechnol. 2011;38:1747–1760. doi: 10.1007/s10295-011-1022-8. [DOI] [PubMed] [Google Scholar]; b) Baltz RH. J Ind Microbiol Biotechnol. 2014;41:357–369. doi: 10.1007/s10295-013-1360-9. [DOI] [PubMed] [Google Scholar]
- 3.Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, Thomas MG. Biochemistry. 2010;49:8815–8817. doi: 10.1021/bi1012854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. Chem Biol. 2010;17:1077–1083. doi: 10.1016/j.chembiol.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zolova OE, Garneau-Tsodikova S. Med Chem Commun. 2012;3:950–955. [Google Scholar]; c) Al-Mestarihi AH, Garzan A, Kim JM, Garneau-Tsodikova S. ChemBioChem. 2015;16:1307–1313. doi: 10.1002/cbic.201402711. [DOI] [PubMed] [Google Scholar]; d) McMahon MD, Rush JS, Thomas MG. J Bacteriol. 2012;194:2809–2818. doi: 10.1128/JB.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hiratsuka T, Koketsu K, Minami A, Kaneko S, Yamazaki C, Watanabe K, Oguri H, Oikawa H. Chem Biol. 2013;20:1523–1535. doi: 10.1016/j.chembiol.2013.10.011. [DOI] [PubMed] [Google Scholar]; f) Zolova OE, Garneau-Tsodikova S. J Antibiot. 2014;67:59–64. doi: 10.1038/ja.2013.98. [DOI] [PubMed] [Google Scholar]
- 5.Drake EJ, Cao J, Qu J, Shah MB, Straubinger RM, Gulick AM. J Biol Chem. 2007;282:20425–20434. doi: 10.1074/jbc.M611833200. [DOI] [PubMed] [Google Scholar]
- 6.a) Kalb D, Lackner G, Rappe M, Hoffmeister D. ChemBioChem. 2015;16:1426–1430. doi: 10.1002/cbic.201500190. [DOI] [PubMed] [Google Scholar]; b) Boll B, Heide L. ChemBioChem. 2013;14:43–44. doi: 10.1002/cbic.201200633. [DOI] [PubMed] [Google Scholar]
- 7.Herbst DA, Boll B, Zocher G, Stehle T, Heide L. J Biol Chem. 2013;288:1991–2003. doi: 10.1074/jbc.M112.420182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Wolpert M, Gust B, Kammerer B, Heide L. Microbiology. 2007;153:1413–1423. doi: 10.1099/mic.0.2006/002998-0. [DOI] [PubMed] [Google Scholar]; b) Lautru S, Oves-Costales D, Pernodet JL, Challis GL. Microbiology. 2007;153:1405–1412. doi: 10.1099/mic.0.2006/003145-0. [DOI] [PubMed] [Google Scholar]; c) Zhang W, Heemstra JR, Walsh CT, Imker HJ. Biochemistry. 2010;49:9946–9947. doi: 10.1021/bi101539b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tatham E, Chavadi SS, Mohandas P, Edupuganti U, Angala S, Chatterjee D, Quadri LE. BMC Microbiol. 2012;12:118. doi: 10.1186/1471-2180-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang C, Kong L, Liu Q, Lei X, Zhu T, Yin J, Lin B, Deng Z, You D. PLoS One. 2013;8:e56772. doi: 10.1371/journal.pone.0056772. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Al-Mestarihi AH, Villamizar G, Fernández J, Zolova OE, Lombó F, Garneau-Tsodikova S. J Am Chem Soc. 2014;136:17350–17354. doi: 10.1021/ja510489j. [DOI] [PubMed] [Google Scholar]; g) Boll B, Taubitz T, Heide L. J Biol Chem. 2011;286:36281–36290. doi: 10.1074/jbc.M111.288092. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Crüsemann M, Kohlhaas C, Piel J. Chem Sci. 2013;4:1041–1045. [Google Scholar]; i) Davidsen JM, Bartley DM, Townsend CA. J Am Chem Soc. 2013;135:1749–1759. doi: 10.1021/ja307710d. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Kittilä T, Schoppet M, Cryle MJ. ChemBioChem. 2016;17:576–584. doi: 10.1002/cbic.201500555. [DOI] [PubMed] [Google Scholar]
- 9.Bosello M, Zeyadi M, Kraas FI, Linne U, Xie X, Marahiel MA. J Nat Prod. 2013;76:2282–2290. doi: 10.1021/np4006579. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Khullar A, Chou S, Sacramo A, Gerratana B. Appl Environ Microbiol. 2009;75:2869–2878. doi: 10.1128/AEM.02326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Thurston DE, Bose DS, Howard PW, Jenkins TC, Leoni A, Baraldi PG, Guiotto A, Cacciari B, Kelland LR, Foloppe MP, Rault S. J Med Chem. 1999;42:1951–1964. doi: 10.1021/jm981117p. [DOI] [PubMed] [Google Scholar]; b) Gerratana B. Med Res Rev. 2012;32:254–293. doi: 10.1002/med.20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yonemoto IT, Li W, Khullar A, Reixach N, Gerratana B. ACS Chem Biol. 2012;7:973–977. doi: 10.1021/cb200544u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giessen TW, Kraas FI, Marahiel MA. Biochemistry. 2011;50:5680–5692. doi: 10.1021/bi2006114. [DOI] [PubMed] [Google Scholar]
- 14.Schwarzer D, Finking R, Marahiel MA. Nat Prod Rep. 2003;20:275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 15.Udwary DW, Merski M, Townsend CA. J Mol Biol. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeifer BA, Admiraal SJ, Gramajo H, Cane D, Khosla CE. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 17.Linne U, Marahiel MA. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 18.Hofer I, Crusemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J. Chem Biol. 2011;18:381–391. doi: 10.1016/j.chembiol.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 19.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bate N, Bignell DRD, Cundliffe E. Mol Microbiol. 2006;62:148–156. doi: 10.1111/j.1365-2958.2006.05338.x. [DOI] [PubMed] [Google Scholar]
- 21.Saha S, Li W, Gerratana B, Rokita SE. Bioorg Med Chem. 2015;23:449–454. doi: 10.1016/j.bmc.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiraskova P, Gazak R, Kamenik Z, Steiningerova L, Najmanova L, Kadlcik S, Novotna J, Kuzma M, Janata J. Front Microbiol. 2016;7:276. doi: 10.3389/fmicb.2016.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ibba M, Söll D. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.