

Abstract
Traumatic brain injury (TBI) is one of the most disabling clinical conditions that could lead to neurocognitive disorders in survivors. Our group and others previously reported that prophylactic enrichment of dietary omega-3 polyunsaturated fatty acids (n-3 PUFAs) markedly ameliorate cognitive deficits after TBI. However, it remains unclear whether a clinically relevant therapeutic regimen with n-3 PUFAs administered after TBI would still offer significant improvement of long-term cognitive recovery. In the present study, we employed the decline of spatial cognitive function as a main outcome after TBI to investigate the therapeutic efficacy of post-TBI n-3 PUFA treatment and the underlying mechanisms. Mice were subjected to sham operation or controlled cortical impact, followed by random assignment to receive the following four treatments: (1) vehicle control; (2) daily intraperitoneal injections of n-3 PUFAs for 2 weeks, beginning 2 h after TBI; (3) fish oil dietary supplementation throughout the study, beginning 1 day after TBI; or (4) combination of treatments (2) and (3). Spatial cognitive deficits and chronic brain tissue loss, as well as endogenous brain repair processes such as neurogenesis, angiogenesis, and oligodendrogenesis, were evaluated up to 35 days after TBI. The results revealed prominent spatial cognitive deficits and massive tissue loss caused by TBI. Among all mice receiving post-TBI n-3 PUFA treatments, the combined treatment of fish oil dietary supplement and n-3 PUFA injections demonstrated a reproducible beneficial effect in attenuating cognitive deficits although without reducing gross tissue loss. Mechanistically, the combined treatment promoted post-TBI restorative processes in the brain, including generation of immature neurons, microvessels, and oligodendrocytes, each of which was significantly correlated with the improved cognitive recovery. These results indicated that repetitive and prolonged n-3 PUFA treatments after TBI are capable of enhancing brain remodeling and could be developed as a potential therapy to treat TBI victims in the clinic.
Keywords: Omega-3 polyunsaturated fatty acid (n-3 PUFA), Water maze, Hippocampus, Angiogenesis, Oligodendrogenesis
Introduction
Traumatic brain injury (TBI) is the leading cause of mortality and disability in children and young adults and a major socioeconomic burden in the US. Currently, strategies to treat severe TBI are largely limited to supportive management, for example, stabilizing the function of vital organs, whereas no direct therapies are available that can effectively decelerate brain injury progression or promote brain repair1. Several approaches of TBI treatment, such as anti-inflammatory and antioxidative strategies2, hypothermia3, and cell-based therapies4, demonstrate efficacy in preclinical studies; however, none of them have been successfully translated to clinical use5,6. TBI induces rapid and sustained pathophysiological changes in the brain, characterized by initial primary injuries resulting from mechanical forces, as well as prolonged secondary injuries through various complex cellular processes, which eventually leads to neuronal death, axonal injury, and neurological deficits7,8. Simultaneously, active tissue remodeling including neurogenesis9, angiogenesis10, and white matter repair11 occurs in the postinjury brain, which could positively influence post-TBI functional recovery. It is imperative to develop novel TBI therapies to battle brain injury development and/or boost brain repair, thereby achieving the improvement of long-term patient outcome.
Numerous preclinical studies by our and other groups have suggested omega-3 polyunsaturated fatty acid (n-3 PUFA) as an emerging candidate for TBI therapy12,15. The most important n-3 PUFAs for human health, that is, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), must be acquired through dietary intake, with the primary sources being fish and fish oil16,17. n-3 PUFAs exist abundantly in the brain and play a crucial role in essential neuronal functions, such as axonal guidance, synapse and dendrite formation, neurotransmission, etc.16,18. Following experimental TBI, n-3 PUFAs exert potent protective effects through multifaceted actions, for example, amelioration of oxidative stress19, mitigation of endoplasmic reticulum stress20, modulation of microglial activation21, and improvement of white matter integrity15. The use of n-3 PUFAs to treat TBI has not hitherto been translated to the clinic, although there are sporadic case studies using n-3 PUFAs acutely after human TBI22,23. A large portion of the preclinical studies on n-3 PUFAs employed a pre-TBI treatment paradigm, achieved through prophylactic dietary supplement or genetic engineering12,15,19, which provided limited information when considering translating this treatment from bench to bedside. While some studies delivered invaluable mechanistic insights of post-TBI n-3 PUFA treatment14,20, concerns still remain over the short delivery time window or the lack of long-term functional evaluation. To facilitate future investigations on n-3 PUFAs toward patient use, we aim to develop a clinically feasible treatment regimen using manageable delivery routes and time window, hoping to validate the use of n-3 PUFAs post-TBI and potentially benefit the TBI victims.
In this study, we designed the post-TBI therapeutic regimens of n-3 PUFAs, in which the first intervention (daily injections) did not start until 2 h after TBI. We evaluated the efficacy of this treatment, together with fish oil dietary supplementation that started at 1 day after injury, on TBI outcome in a mouse model. Furthermore, we examined the underlying mechanisms of post-TBI n-3 PUFA treatments, with a focus on postinjury tissue remodeling processes.
Materials and Methods
Animals
C57BL/6J mice (male, 10-12 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and used in our experiments. Mice were housed in a temperature- and humidity-controlled animal facility with a 12-h light-dark cycle. Food and water were available ad libitum. All animal procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Efforts were made to minimize animal suffering and the number of animals used.
Traumatic Brain Injury
All mice were randomly assigned into experimental groups with the use of a lottery-drawing box. TBI was induced in mice by a controlled cortical impact (CCI) as described previously24. Briefly, mice were anesthetized with 3% isoflurane (Butler Schein Animal Health, Dublin, OH, USA) vaporized in 67% N2O/30% O2 mixture until they were unresponsive to the tail pinch test. Anesthesia was maintained with 1.5% isoflurane in 67% N2O/30% O2 by fixing the mouse to a nose cone with consistent blowing of gas. Following a craniotomy (diameter: 3.0 cm), the CCI was performed (2.0 mm lateral to midline and 0.5 mm anterior to bregma) with a pneumatically driven CCI device (Precision Systems and Instrumentation, Fairfax, VA, USA) using a 3-mm flat-tipped impounder (velocity: 3.50 m/s; duration: 150 ms; depth: 1.5 mm). The bone flap was then replaced and sealed. Rectal temperature was maintained at 37 ± 0.5°C during surgery and up to 30 min after TBI using a heating pad. Sham animals were subjected to all aspects of the surgery and handling (including the craniotomy) except for the CCI. Animals that died during or immediately after surgery (less than 5%) were excluded from the studies. All outcome measurements were carried out by investigators blinded to the experimental group assignment.
Administration of n-3 PUFAs After TBI
Mice were randomly assigned to receive the following treatments of n-3 PUFAs after TBI: (1) vehicle control: mice were fed a regular laboratory rodent diet (Prolab Isopro RMH 3000 5P76; LabDiet, St. Louis, MO, USA), in which the n-3 PUFA content was low (0.36%), and received intraperitoneal (IP) injections of 0.9% NaCl (300 μl per injection, 2 h after TBI and then daily for 14 days); (2) injections of mixed n-3 PUFAs (“N3”): mice were fed a regular diet and received IP injections of EPA and DHA (7 mg of EPA and 3 mg of DHA/kg body weight, diluted in 300 μl of 0.9% NaCl per injection, 2 h after TBI and then daily for 14 days); (3) fish oil dietary supplementation (FO): mice were fed a diet supplemented with n-3 PUFAs (DHA and EPA, triple-strength n-3 fish oil; Puritan's Pride, Oakdale, NY, USA) to reach a 4% final n-3 PUFA concentration beginning 1 day after TBI and for up to 35 days and received IP injections of 0.9% NaCl (300 μl per injection, 2 h after TBI and then daily for 14 days); (4) combination of treatments (2) and (3) (“N3 + FO”): mice were fed a diet supplemented with fish oil and received injections of DHA and EPA as described above. The EPA/DHA ratio (70%/30%) in the injectable N3 mixture was dictated by their respective contents in the triple-strength n-3 fish oil (624 mg of EPA and 244 mg of DHA in 1,360 mg of fish oil per capsule).
Morris Water Maze
The Morris water maze test was performed at 29-34 days after TBI to evaluate long-term spatial cognitive deficits as previously described25. Briefly, a pool (diameter: 109 cm) with opaque water was divided into four quadrants by phantom lines. A square platform (11 cm × 11 cm) was submerged 1 cm under the water in one of the four quadrants. Three trials were performed on each testing day. For each trial, the mouse was placed into the pool from each of the other three quadrants without the platform and allowed 60 s to swim and locate the hidden platform (learning phase of the test). The time at which the mouse found the platform (escape latency) was recorded for each trial. At the end of each trial, the mouse was placed on the platform or allowed to stay on the platform for 10 s with prominent spatial cues displayed around to memorize the position of the platform. Mice were pretrained for 3 consecutive days before TBI (three trials on each day) (Fig. 1A). After TBI, three trials were performed daily for 5 consecutive days from days 29 to 33, and the average latency to escape was calculated. At day 34 after TBI, a single, 60-s probe trial was performed in which the platform was removed. The time the mouse spent in the target quadrant where the platform was previously located was recorded (memory phase of the test). Swim speed was recorded on each testing day from days 29 to 34 after TBI as a parameter to assess the gross loco-motor function of the mouse.
Figure 1.
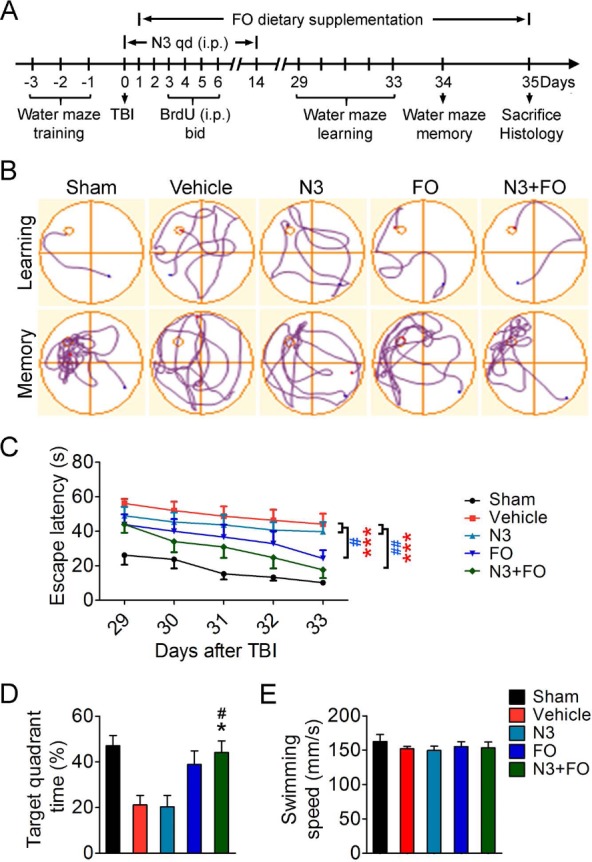
Combined treatment of n-3 PUFAs after TBI elicits long-term improvement of spatial cognitive function. (A) Experimental timelines. Mice were pretrained for the Morris water maze test for 3 days before traumatic brain injury (TBI). Mixed docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) (together called N3) were injected intraperitoneally (IP) 2 h after TBI and then daily for 14 days. Fish oil (FO) dietary supplements started 1 day after TBI. qd, once a day; bid, twice a day. Water maze learning tests were performed at days 29 to 33, and memory tests were performed at day 34 after TBI. Mice were sacrificed at 35 days after TBI for histological examinations. (B) Representative images showing mice swim paths from each group at the learning or memory phase of the Morris water maze. (C) The latency for mice to escape to the submerged platform in the learning test at 29-33 days after TBI. (D) Spatial memory was measured at 34 days after TBI by the time spent in the target quadrant where the platform was previously located. (E) Average swimming speed at 29-34 days after TBI indicates no significant difference of gross motor function among all groups. n = 6 mice for sham group; n = 8 mice for vehicle, N3, and FO groups; n = 7 mice for N3 + FO group. *p ≤ 0.05, ***p ≤ 0.001, FO or N3 + FO versus vehicle; #p ≤ 0.05, ##p ≤ 0.01, FO or N3 + FO versus N3 by two-way analysis of variance (ANOVA) (C) or oneway ANOVA (D).
Immunohistochemistry and Image Analysis
At day 35 after TBI, mice were deeply anesthetized and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Brains were collected and cryoprotected in 30% sucrose in PBS. Frozen serial coronal brain sections (25 μm thick) were cut by a cryostat (Microm HM450; Thermo Fisher Scientific, Florence, K Y, USA). Immunohistochemistry was performed on free-floating sections. Briefly, sections were blocked with 5% donkey serum in PBS for 1 h, followed by overnight incubation (4°C) with the following primary antibodies: rabbit anti-NeuN (1:500; EMD Millipore, Billerica, MA, USA), goat anti-doublecortin (DCX; 1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rat anti-CD31 (1:200; BD Biosciences, San Jose, CA, USA), and mouse anti-adenomatous polyposis coli (APC; 1:400; EMD Millipore). After a series of washing, sections were incubated for 1 h at 37°C with the appropriate donkey secondary antibodies conjugated with Alexa Fluor 488 or Cy3 (1:1,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Alternate sections from each experimental condition were incubated in all solutions except the primary antibodies to assess nonspecific staining. Sections were then mounted and coverslipped with Fluoromount-G containing 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, Pittsburgh, PA, USA). Fluorescence images were captured with an Olympus FluoView FV1000 confocal microscope using FV10-ASW 2.0 software (Olympus America, Center Valley, PA, USA). Alternatively, whole-brain images of NeuN fluorescence were acquired with an inverted Nikon Diaphot 300 fluorescence microscope equipped with a SPOT RT slider camera and Meta Series Software 5.0 (Molecular Devices, Sunnyvale, CA, USA).
Brain tissue loss was analyzed with ImageJ as described previously15. Briefly, six equally spaced NeuN-stained coronal brain sections encompassing the CCI territory (from bregma 1.10 mm to bregma −1.34 mm) were selected. The tissue loss in each section was calculated by subtracting the NeuN immunosignal-positive area in the contralateral hemisphere from that in the ipsilateral hemisphere. The volume of tissue loss was calculated by multiplying the mean area of tissue loss in each section by the thickness of the tissue evaluated.
Examination of Recently Proliferated Cells
Recently proliferated cells were labeled with the S-phase marker 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich, St. Louis, MO, USA) as previously described26. Briefly, BrdU was injected IP twice a day at a dose of 50 mg/kg body weight at 3-6 days after TBI. At 35 days after TBI, mice were sacrificed, and coronal brain sections were prepared as described above. Sections were treated with 2 N HCl for 1 h at 37°C followed by 0.1 M boric acid (pH 8.5) for 10 min at room temperature. Sections were then blocked with a mouse on mouse (MOM) detection kit (Vector Laboratories, Burlingame, CA, USA) for 1 h, followed by incubation with purified mouse anti-BrdU antibody (1:1,000; BD Biosciences) for 1 h at room temperature and then overnight at 4°C. After a series of washes, sections were incubated with the Alexa Fluor 488-conjugated AffiniPure donkey anti-mouse IgG (1:1,000; Jackson ImmunoResearch) for 1 h at room temperature. Fluorescence images were captured as described above. BrdU-immunopositive cells were counted using ImageJ (NIH, Bethesda, MD, USA) and expressed as the number of cells per mm2 or per microscopic field (0.16 mm2 at 40× magnification). Post-TBI neurogenesis, angiogenesis, and oligodendrogenesis in the perilesion areas (within 300 μm to the lesion edge) were evaluated on BrdU/NeuN, BrdU/DCX, BrdU/CD31, and BrdU/APC double-stained sections. Two microscopic fields from the cerebral cortex and striatum, and one microscopic field in each of the subventricular zone (SVZ), hippocampal CA1, CA3, and dentate gyrus (DG) areas were randomly sampled in every section.
Statistical Analyses
All data are presented as mean ± standard error of the mean (SEM). The differences between means of multiple groups were assessed by one- or two-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test. The Pearson product linear regression analysis was used to correlate the mouse's spatial learning performance (at 33 days after TBI) and memory performance in the Morris water maze with various histological parameters. A value of p < 0.05 was considered to be statistically significant.
Results
Post-TBI Treatment with n-3 PUFAs was Effective in Improving Long-Term Cognitive Recovery
Cognitive impairments are common complications caused by TBI and are closely related to daily functioning of the TBI victims7,27. We previously reported that n-3 PUFA-enriched diets can substantially elevate brain n-3 PUFA content when applied to mice on a long-term basis26. This prophylactic treatment before the onset of TBI, in combination with the continuous n-3 PUFA supplement after injury, was able to ameliorate long-term spatial learning and memory deficits in a mouse CCI model15. To test whether n-3 PUFAs hold potential to be used on TBI victims, we designed an n-3 PUFA interventional regimen starting 2 h after TBI via IP injections of an EPA and DHA mixture (Fig. 1A). The EPA/DHA ratio (7:3) of the injection mixture was dictated by their respective compositions in the triple-strength n-3 fish oil. This acute treatment, together with long-term FO dietary supplement that started 1 day after TBI, features a potentially manageable therapeutic window and delivery route in clinical settings. To determine the efficacy of n-3 PUFA treatment against TBI-induced cognitive decline, we assessed the long-term spatial learning and memory performance by mice in the Morris water maze at 29-34 days after TBI (Fig. 1B). Spatial learning was observed in all groups during the 5-day testing period, as demonstrated by the gradually decreased latency for the mice to find the submerged platform at 29-33 days after TBI (Fig. 1C). Compared to sham-operated mice, TBI induced prominent spatial learning deficits in the vehicle group (p < 0.001 vehicle vs. sham by two-way ANOVA) (Fig. 1C). Post-TBI N3 injections did not reduce the escape latency compared to the vehicle group. In contrast, FO dietary supplementation alone or combined with N3 injections significantly facilitated the recovery of spatial learning, compared to either the vehicle- or N3-injected group (Fig. 1C). Consistently, N3 + FO-treated mice, but not either treatment alone, also improved the spatial memory 34 days after TBI compared to the vehicle- or N3-injected group (Fig. 1D). All five groups showed comparable swimming speed during the 6-day testing (Fig. 1E). Therefore, the observed difference in the mice's learning and memory performance was not a consequence of varied gross locomotor function.
We next examined whether the improved cognitive function elicited by n-3 PUFA treatment was associated with less neuronal tissue loss after TBI, as determined on NeuN-immunostained coronal sections (Supplementary Fig. 1A; supplementary material available at https://pitt.app.box.com/v/PuCellTransplantSuppleFigs). However, the volumes of the tissue loss in all four groups of TBI mice were comparable (Supplementary Fig. 1B). In summary, these data indicate that long-term FO dietary supplementation after TBI, especially when combined with acute N3 injections, significantly improves cognitive recovery. Mechanisms other than the reduction of gross neuronal tissue lost might underlie this improved cognitive function.
Post-TBI n-3 PUFA Treatment Does Not Alter Histological Hippocampal Integrity
We further investigated the underlying mechanisms accounting for the improved cognitive functions observed in mice receiving post-TBI n-3 PUFA treatments. Since spatial learning and memory are believed to largely rely on the histological and functional integrity of the hippocampus25,28, we measured the number of viable neurons (NeuN+ cells) in the ipsilesional hippocampal CA1, CA3, and DG at 35 days after TBI (Fig. 2A). The results (Fig. 2B) revealed that TBI caused significant neuronal loss in the ipsilateral CA1 (vehicle 686.59 ± 70.49 cells/mm2 vs. sham 1,065.88 ± 53.13 cells/mm2, p < 0.05) and CA3 (vehicle 563.29 ± 68.24 cells/mm2 vs. sham 946.83 ± 92.64 cells/mm2, p < 0.05), but not in the DG (vehicle 1,489.22 ± 80.30 cells/mm2 vs. sham 1,434.42 ± 63.64 cells/mm2, p > 0.05). Post-TBI N3 injections, FO supplements, or combined N3 and FO treatment did not elicit significant protection on hippocampal integrity in CA1 or CA3 when examined at 35 days after TBI (Fig. 2B). Furthermore, neurogenesis was barely observed in the entire hippocampus, shown by very few BrdU+ cells or BrdU+/NeuN+ cells in CA1, CA3, and DG (Fig. 2A and C). Pearson product linear regression analysis demonstrated moderate but yet statistically significant correlations between the numbers of CA3 viable neurons, but not CA1 viable neurons, and the mice's performance in the water maze learning and memory test (Fig. 2D and E). In summary, these data suggest that the post-TBI n-3 PUFA treatment did not significantly preserve hippocampal neuronal integrity, although the CA3 viable neurons are partially linked to improved spatial cognitive functions. Thus, other mechanisms must contribute to the cognitive improvement in mice receiving the N3 + FO treatment.
Figure 2.
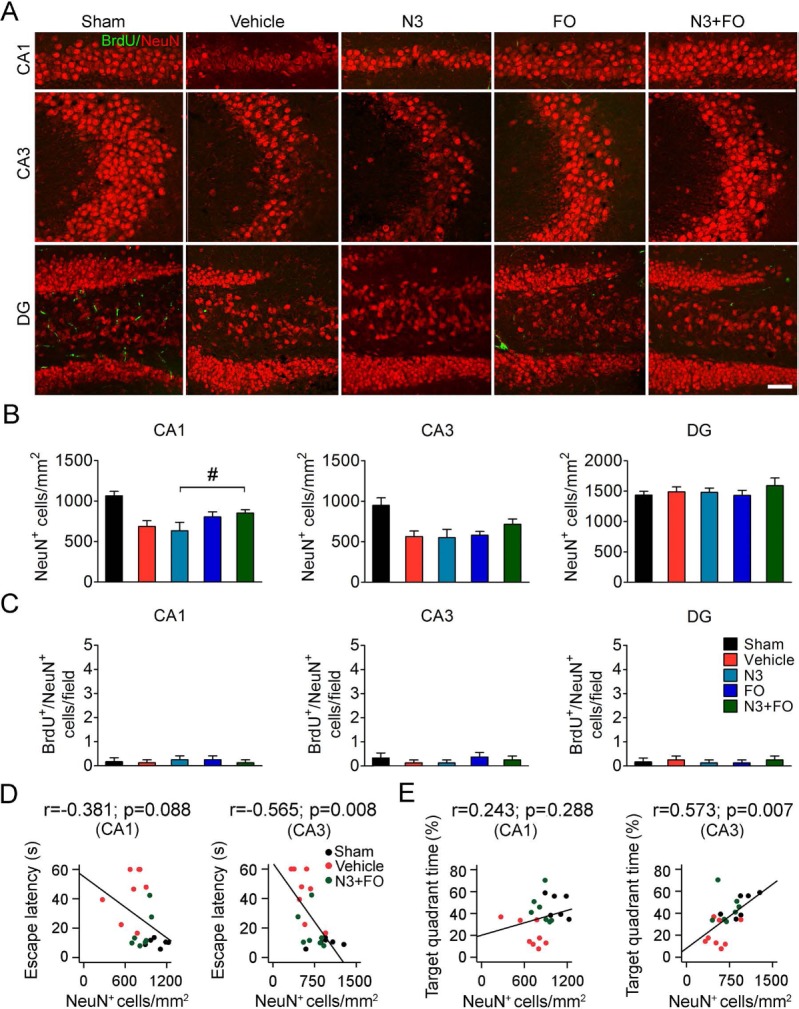
Post-TBI n-3 PUFA treatment does not improve hippocampal integrity. (A) Representative images showing double-label immunofluorescence of 5-bromo-2′-deoxyuridine (BrdU) and NeuN in the CA1, CA3, and dentate gyrus (DG) of the ipsilesional hippocampus at 35 days after traumatic brain injury (TBI). Scale bar: 50 μm. (B) Quantification of total surviving neurons (NeuN+ cells) in the ipsilesional CA1, CA3, and DG. (C) Quantification of newly generated mature neurons (BrdU+/NeuN+ cells) in the ipsilesional CA1, CA3, and DG. Barely any newly generated mature neurons were present in all regions of the hippocampus in all groups. #p ≤ 0.05, N3 + FO (fatty acid injection and fish oil dietary supplementation) versus N3 by one-way analysis of variance (ANOVA). (D, E) Pearson correlation between the mice's spatial learning (D) and memory (E) performance in the Morris water maze and the numbers of total viable neurons in the ipsilesional CA1 and CA3. n = 6 mice for sham group; n = 8 mice for vehicle, N3, and FO groups; n = 7 mice for N3 + FO group.
Delayed n-3 PUFA Treatment Enhances the Survival of Cortical and Striatal Neurons
Despite the absence of direct protection of hippocampal neurons, post-TBI n-3 PUFA treatment elicited dramatic improvement of cognitive recovery, suggesting the participation of nonhippocampal mechanisms. It is a generally accepted concept that a functionally integrated neural network is required to properly navigate the Morris water maze, which is dependent not only on the hippocampus but also on other brain regions such as the cerebral cortex and striatum29,31. Since the present CCI model produced substantial lesion in both the cortex and striatum, we examined the neuronal survival in these regions at 35 days after TBI (Fig. 3A and B). Post-TBI FO treatment alone resulted in a significant increase in viable neurons in the perilesion (within 300 μm from the injury border) striatal areas compared to mice receiving vehicle treatment (Fig. 3C and D). Combined N3 + FO treatment further enhanced the protection and significantly increased viable neurons in both the perilesion cortex and striatum (Fig. 3C and D). In comparison, N3 injections alone did not produce a significant effect on neuronal loss (N3 943.60 ± 48.38 cells/mm2 vs. vehicle 938.72 ± 45.79 cells/mm2 in the cortex and N3 1,185.30 ± 47.02 cells/mm2 vs. vehicle 1,087.65 ± 82.65 cells/mm2 in the striatum, p > 0.05) (Fig. 3D). Some BrdU+ cells were observed in the perilesion cortex and striatum, but only a very small portion expressed the mature neuronal marker NeuN (Fig. 3C and E). Furthermore, n-3 PUFA treatment did not alter the number of BrdU+/NeuN+ cells in either the cortex or the striatum (Fig. 3E). These data suggest that increased viable neurons in FO- or N3 + FO-treated mice may be a result of protection against cell death rather than increased neurogenesis. The number of viable neurons in both the cortex and the striatum showed a moderate but statistically significant negative correlation with the escape latency in the water maze learning task (Fig. 3F) and a positive correlation with spatial memory (Fig. 3G). Together, these results suggest that the combined FO dietary supplementation and N3 injections may improve post-TBI cognitive functions partially through the promotion of neuron survival in the cortex and striatum.
Figure 3.

Post-TBI n-3 PUFA treatment attenuates cortical and striatal neuronal death. (A) A representative image of one coronal brain section showing NeuN immunofluorescence at 35 days after traumatic brain injury (TBI). Boxes demonstrate the ipsilateral perilesion areas in the cortex (CTX) and striatum (STR) where images in (C) were taken. Scale bar: 1 mm. (B) A representative image from the perilesion cortex showing typical double-label immunofluorescence of 5-bromo-2′-deoxyuridine (BrdU) and NeuN. Arrow: BrdU+/NeuN+ cell (yellow). Arrowheads: BrdU+/NeuN− cells (green). Scare bar: 50 μm. (C) Representative images of BrdU (green) and NeuN (red) immunofluorescence in the perilesion cortex and striatum at 35 days after TBI. Scale bar: 50 μm. (D, E) Quantification of total viable neurons (D) and newly generated mature neurons (E) in the perilesion cortex and striatum. *p ≤ 0.05, ***p ≤ 0.001 versus vehicle; ##p ≤ 0.01, ###p ≤ 0.001 versus N3 (fatty acid injection) by one-way analysis of variance (ANOVA). (F, G) Pearson correlation between the mice's spatial learning (F) and memory (G) performance in the Morris water maze and the numbers of total viable neurons in the cortex and striatum. n = 6 mice for sham group; n = 8 mice for vehicle, N3, and fish oil dietary supplementation (FO) groups; n = 7 mice for N3 + FO group.
Combined N3 and FO Treatment After TBI Stimulates the Generation of Immature Neurons
TBI has been shown to accelerate neurogenesis in the hippocampus and SVZ, which may contribute to the functional recovery of the brain9,32,33. In the present study, very few BrdU+/NeuN+ cells were detected throughout the brain after TBI (Figs. 2 and 3). To determine whether this was due to a deficiency of neural stem/progenitor cell (NSC/NPC) generation per se, we examined the expression of DCX, a marker for neuronal precursor cells and immature neurons, at 35 days after TBI (Fig. 4A). TBI stimulated endogenous neurogenesis, as shown by increased BrdU+/DCX+ cells in the perilesion cortex (vehicle 1.94 ± 0.20 cells/field vs. sham 0 cells/field, p = 0.009) (Fig. 4B). Among all four treatments, FO dietary supplementation alone or combined with N3 injections increased the BrdU+/DCX+ newly generated immature neurons in both the perilesion cortex and striatum (Fig. 4B). Some DCX+/BrdU− cells were also present, indicating that the antibody used exhibited some nonspecificity. Nevertheless, there was no difference in BrdU+/DCX+ cell numbers among all groups in the SVZ at 35 days after TBI (Supplementary Fig. 2), indicating that the newborn immature neurons in the SVZ largely migrated to the cortex and striatum at earlier stages after TBI. Importantly, the increase in newborn immature neurons in the striatum significantly correlated with the mice's spatial learning and memory performance in the Morris water maze (Fig. 4C and D). These results suggest that post-TBI n-3 PUFA supplementation was capable of stimulating the generation of immature neurons and their migration toward the perilesion brain regions. Although not fully differentiated into mature neurons, these immature cells might play a role in the improvement of post-TBI cognitive recovery.
Figure 4.

Post-TBI n-3 PUFA treatment stimulates the generation of neural progenitor cells. (A) Representative images of 5-bromo-2′-deoxyuridine (BrdU) and doublecortin (DCX) immunofluorescence in the ipsilateral perilesion cortex and striatum at 35 days after traumatic brain injury (TBI). Boxes illustrate the regions enlarged in high-power images (the sixth column). Arrows: BrdU+/DCX+ cells (yellow). Arrowheads: BrdU+/DCX− cells (green). Scale bars: 50 μm. (B) Quantification of BrdU+/DCX+ cells in the perilesion cortex and striatum. $$p ≤ 0.01 versus sham; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 versus vehicle; #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001 versus fatty acid injection (N3) by one-way analysis of variance (ANOVA). (C, D) Pearson correlation between the mice's spatial learning (C) and memory (D) performance in the Morris water maze and the numbers of BrdU+/DCX+ cells in the cortex and striatum. n = 6 mice for sham group; n = 8 mice for vehicle, N3, and fish oil dietary supplementation (FO) groups; n = 7 mice for N3 + FO group.
Delayed n-3 PUFA Treatments Promote Angiogenesis After TBI
Angiogenesis is an essential restorative mechanism in the brain after TBI and supplies a “vascular niche” that is necessary for the survival of surrounding cells34,35. We examined the impact of delayed n-3 PUFA treatment on angiogenesis in the perilesion regions in the cortex and striatum 35 days after TBI by double-label immunostaining of BrdU and the endothelial marker CD31 (Fig. 5A). TBI stimulated endogenous angiogenesis in the perilesion cortex (vehicle 56.01 ± 7.92 cells/mm2 vs. sham 13.97 ± 2.59 cells/mm2, p = 0.011) (Fig. 5B). FO dietary supplementation alone or combined with N3 injections significantly increased the numbers of newly generated microvessels in both the cortex and striatum (Fig. 5B), whereas N3 injections alone did not alter angiogenesis compared to vehicle treatment (N3 53.59 ± 4.35 cells/mm2 vs. vehicle 56.01 ± 7.92 cells/mm2 in the cortex; N3 59.63 ± 4.39 cells/mm2 vs. vehicle 47.55 ± 6.27 cells/mm2 in the striatum; p > 0.05) (Fig. 5B). Meanwhile, the spatial learning and memory abilities in the water maze showed a significant correlation with angiogenesis (Fig. 5C and D). Together, these results further demonstrated that the beneficial effect of n-3 PUFA treatments on post-TBI cognitive function was partially achieved through promoting angiogenesis in the surviving brain tissues.
Figure 5.

Delayed n-3 PUFA treatment promotes post-TBI angiogenesis. (A) Representative images of 5-bromo-2′-deoxyuridine (BrdU) and CD31 immunofluorescence in the ipsilateral perilesion cortex (CTX) and striatum (STR) at 35 days after traumatic brain injury (TBI). Boxes indicate areas that were enlarged in high-power images (the sixth column). Arrows: BrdU+ cells on vessels (yellow) Arrowheads: BrdU+/CD31− cells (green). Scale bars: 50 μm. (B) Quantification of newly generated endothelial cells (BrdU+/CD31+ cells) in the perilesion cortex and striatum. $p ≤ 0.05 versus sham; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 versus vehicle; #p ≤ 0.05, ##p ≤ 0.01 versus N3 (fatty acid injection) by one-way analysis of variance (ANOVA). (C, D) Pearson correlation between the mice's spatial learning (C) and memory (D) performance in the Morris water maze and the numbers of BrdU+/CD31+ cells in cortex and striatum. n = 6 mice for sham group; n = 8 mice for vehicle, N3, and fish oil dietary supplementation (FO) groups; n = 7 mice for N3 + FO group.
Post-TBI n-3 PUFA Treatments Boost Oligodendrogenesis
TBI-induced demyelination and axonal injury are important contributors to cognitive decline7,11. Oligodendrogenesis, including the proliferation of oligodendrocyte progenitor cells (OPCs) and their differentiation into myelinating oligodendrocytes, facilitates remyelination, white matter repair, and neurological recovery after TBI11. We therefore examined post-TBI oligodendrogenesis in the perilesion cortex and striatum 35 days after TBI by double-label immunostaining of BrdU and APC, a marker for mature oligodendrocytes36. In vehicle-treated mice, few BrdU+/APC+ cells were observed in the perilesion cortex or striatum (p > 0.05 vehicle vs. sham) (Fig. 6A and B), indicating negligible post-TBI oligodendrogenesis. While N3 injections did not cause an increase in BrdU+/APC+ cell numbers compared to the vehicle group, long-term FO dietary supplement led to a significant increase in the newly generated mature oligodendrocytes in both the cortex and striatum (FO 82.20 ± 10.10 cells/mm2 vs. vehicle 28.20 ± 4.38 cells/mm2 in the cortex; FO 74.94 ± 8.95 cells/mm2 vs. vehicle 29.01 ± 4.87 cells/mm2 in the striatum; p < 0.001 and p < 0.05, respectively) (Fig. 6A and B). Notably, when N3 injections were combined with FO supplementation, oligodendrogenesis was further enhanced (151.04 ± 18.33 cells/mm2 in the cortex and 144.59 ± 18.33 cells/mm2 in the striatum; p < 0.001 vs. vehicle and N3). It is likely that the enhanced oligodendrogenesis associated with the N3 + FO treatment contributed to the improved spatial cognitive function, as the mice's performance in the learning tests of the Morris water maze showed moderate but statistically significant correlations, whereas the memory ability exhibited strong correlations with the extent of oligodendrogenesis in both the cortex and striatum (Fig. 6C and D). These results suggest that improved oligodendrogenesis might be another underlying mechanism in improving cognitive function by n-3 PUFA treatments.
Figure 6.

Post-TBI n-3 PUFA treatment enhances oligodendrogenesis. (A) Representative images of 5-bromo-2′-deoxyuridine (BrdU) and adenomatous polyposis coli (APC) immunofluorescence in the ipsilateral perilesion cortex and striatum at 35 days after traumatic brain injury TBI. Boxes indicate areas that were enlarged in high-power images (the sixth column). Arrows: BrdU+/APC+ cells (yellow). Arrowheads: BrdU+/APC− cells (green). Scale bars: 50 μm. (B) Quantification of newly generated mature oligoden-drocytes (BrdU+/APC+ cells) in the perilesion cortex (CTX) and striatum (STR). *p ≤ 0.01, ***p ≤ 0.001 versus vehicle; ##p ≤ 0.01, ###p ≤ 0.001 versus N3 by one-way analysis of variance (ANOVA). (C, D) Pearson correlation between the mice's spatial learning (C) and memory (D) performance in the Morris water maze and the total number of BrdU+/APC+ cells in the cortex and striatum. n = 6 mice for sham group; n = 8 mice for vehicle, fatty acid injection (N3), and fish oil dietary supplementation (FO) groups; n = 7 mice for N3 + FO group.
Discussion
The present study characterized the therapeutic efficacy of n-3 PUFAs against TBI-induced cognitive deficits using an administration paradigm that combines repeated post-TBI DHA/EPA injections with long-term FO dietary supplementation. The results demonstrated that the combined N3 and FO treatment significantly improved spatial cognitive recovery even without affecting the tissue contusion after TBI. Further mechanistic explorations revealed that combined N3 and FO treatment not only protected neurons against TBI-induced cell death but also promoted multiple endogenous restorative mechanisms, such as generation of immature neurons, microvessels, and oligodendrocytes, all of which could contribute to the improved cognition in TBI mice.
The extent of TBI-induced neuronal death strongly correlates with the development of cognitive deficits37,38. TBI leads to rapid neuronal cell death resulting from direct contusion, followed by progressive secondary cell death in surrounding tissues8. n-3 PUFAs have been shown to exert potent protection against oxidative stress, glutamate-induced excitotoxicity, apoptotic cell death, and inflammation16,39,40. In mice receiving 2 months of prophylactic n-3 PUFA dietary supplementation, TBI caused a less extensive loss of CA3 neurons compared to mice on a regular diet15. In the present study, we confirmed that delayed n-3 PUFA treatment after TBI also provided significant protection against neuronal loss in the cerebral cortex and striatum. Interestingly, long-term dietary supplementation with FO (for up to 35 days after TBI) effectively reduced neuronal loss (Fig. 3). It is worth noting that although N3 injections alone were not sufficient to be neuroprotective in the long run, they appeared to boost the beneficial effect of FO supplementation (Fig. 3). Future research is needed to further determine how the delayed n-3 PUFA treatments alter the spatial and temporal profile of neuronal cell death after TBI and the underlying signaling mechanisms.
Importantly, the number of viable neurons in the CA3 region, cortex, and striatum was in significant correlation with post-TBI cognitive functions. We speculate that the increased viable neurons might largely result from neuroprotection because it is seldom that any BrdU+/NeuN+ cells are present in TBI brains with or without FO treatment. TBI was shown to stimulate neurogenesis in the granule cell layer of the DG, and the new cells were thought to be able to mature into neurons and contribute to the restoration of hippocampal functions32,41. The absence of robust neurogenesis in the DG after TBI in the present study may be partially explained by the differences in injury sites of the CCI model between our study and the previous studies by others. Compared to the model used in the literature32,42, our CCI model produces a lesion at more anterior coronal levels, which may trigger fewer neurorestorative responses in the DG (Fig. 2C). Nevertheless, the lack of BrdU+/NeuN+ cells throughout various brain regions in our study suggests two possibilities, that is, there was a lack of NSC/NPC generation or the NSCs/NPCs did not fully differentiate into mature neurons.
We therefore examined the BrdU+/DCX+ cells and found that n-3 PUFA treatments did promote the proliferation of NSCs in the post-TBI cortex and striatum. However, these immature neurons apparently failed to differentiate into mature neurons during the 35 days post-TBI period. Interestingly, the number of BrdU+/DCX+ immature neurons was nevertheless significantly correlated with post-TBI cognitive recovery, suggesting that the induced neurogenesis could contribute to functional recovery through mechanisms independent of neuronal replacement. For instance, immature neurons generated via neurogenesis can produce and release a plethora of trophic factors43, thereby supporting the survival of surrounding neurons and nourishing a favorable microenvironment for tissue repair.
The brain has some plasticity for spontaneous functional recovery after injury, through multiple endogenous restorative processes such as angiogenesis, axonal sprouting, and white matter repair44,47. In addition to the increase in immature neurons discussed above, we observed enhanced angiogenesis and oligodendrogenesis in the perilesion brain tissue upon post-TBI n-3 PUFA treatment (Figs. 5 and 6). The generation of new microvessels helps to form the neurovascular niches, facilitates tissue remodeling such as neurogenesis and synaptogenesis, and supports the survival and migration of NSCs/NPCs, all of which may lead to functional recovery44,48. Another important restorative mechanism is oligodendrogenesis, which is essential for remyelination and white matter repair after TBI49. Following ischemic brain injury, delayed n-3 PUFA administration is capable of promoting angiogenesis and oligodendrogenesis, which are associated with improved functional recovery50,51. In the present study, we noticed an enhancement of post-TBI angiogenesis and oligodendrogenesis upon n-3 PUFA treatment that lasted up to 35 days after injury. Interestingly, our analysis shows that although all of these restorative responses, for example, generation of immature neurons, vessels, and oligodendrocytes, correlated with cognitive recovery, the correlations were all just moderate. Such an observation may imply that any single component of post-TBI restoration may be necessary but not sufficient enough to lead to an overall functional improvement. To this extent, n-3 PUFAs target multiple aspects of tissue remodeling simultaneously and for an extended period of time after injury, which could suggest therapeutic potential for clinical use. We found that for multiple histological parameters, including neuroprotection, angiogenesis, and oligodendrogenesis, FO supplements demonstrated more significant effects than N3 injections alone, and the combined N3 and FO treatment has the most consistent beneficial effect among all treatment paradigms. Together, these results suggest that combined acute injections of n-3 PUFAs, together with chronic supplementation, may be a promising strategy to treat TBI victims.
Promoting long-term neurological recovery has been a major focus in rehabilitation for TBI victims. Cognitive deficits are core characteristics as a result of TBI and can persist for a long period of time. To date, the development of effective neurotherapeutic interventions against TBI has been largely fruitless despite the growing public awareness and research endeavors. Preclinical studies should take into consideration using clinically manageable methods to eventually translate the therapy to TBI clinics. Fish oil is a safe and economical prophylactic supplement that has been shown to protect against brain disorders in many animal models. In our experiments, we began repetitive EPA/DHA injections at 2 h after TBI; however, EPA/DHA injections alone did not improve cognitive functions. Compared to previous studies by other groups showing DHA-mediated neuroprotection when delivered 5 min after TBI20, we might have missed the therapeutic time window for n-3 PUFAs to exert acute protection. Nevertheless, when FO dietary supplementation was given to mice even as late as 1 day after TBI, most outcome parameters examined (e.g., spatial learning and memory, neuronal viability in the cortex and striatum, regeneration of immature neurons, angiogenesis, and oligodendrogenesis) were improved. When FO dietary supplementation was combined with acute N3 injections, substantial therapeutic effects were achieved. These data suggest the importance of a long-term supply of n-3 PUFAs to promote tissue repair and functional recovery even if the treatment starts as late as 1 day after TBI. With long-term brain tissue restoration as the objective, the n-3 PUFA treatment regimen described here has a clinically relevant time window of efficacy, making it a legitimate candidate for testing in TBI clinical trials. However, some caveats exist in our study. For example, some of the neurovascular restorative processes that could be affected by n-3 PUFAs were not examined in the present study, for example, the effect of n-3 PUFAs on microglial activation, inflammatory responses, and white matter integrity52,55. Another limitation is the use of the CCI model. Despite being one of the most widely used TBI models, the CCI model replicates some, but not all, aspects of human TBI pathology. The diffusive effects of CCI are not as obvious as in several other models (e.g., closed head injury, blast, fluid percussion injury)56. Therefore, future preclinical research is warranted to test the therapeutic efficacy of post-TBI n-3 PUFA delivery using other neurotrauma models in order to facilitate the clinical translation of this promising therapeutic intervention for TBI.
Acknowledgments
This project was supported by the US National Institutes of Health grants NS045048, NS095671, and NS095029 (to J.C.), and the National Natural Science Foundation of China grants 30970939 and 81272804 (to L.C.), 81529002 (to J.C.), and 81100978 (to W.Z.). J.C. is also supported by the US Department of Veterans Affairs Senior Research Career Scientist Award. The authors are indebted to Pat Strickler for the excellent administrative support. J.C., L.C., and H.P. designed the research. H.P., X.J., Z.W., S.H., W.Z., J.L., and H.M. performed the research. X.J. and Y.S. analyzed the data. H.P., X.J., Y.S., D.H., and J.C. wrote the manuscript. All authors reviewed and approved the manuscript. The authors declare no conflicts of interest.
References
- 1.Brain Trauma F., American Association of Neurological Surgeons, Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma 2007; 24(suppl 1): S1–106. [DOI] [PubMed] [Google Scholar]
- 2.Hellewell S., Semple B.D., Morganti-Kossmann M.C. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016; 1640(Pt A): 36–56. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich W.D., Bramlett H.M. Therapeutic hypothermia and targeted temperature management in traumatic brain injury: Clinical challenges for successful translation. Brain Res. 2016; 1640(Pt A): 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel K., Sun D. Strategies targeting endogenous neurogenic cell response to improve recovery following traumatic brain injury. Brain Res. 2016; 1640(Pt A): 104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline A.E., Leary J.B., Radabaugh H.L., Cheng J.P., Bondi C.O. Combination therapies for neurobehavioral and cognitive recovery after experimental traumatic brain injury: Is more better? Prog Neurobiol. 2016; 142: 45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein D.G. Embracing failure: What the phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015; 29(11): 1259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeKosky S.T., Ikonomovic M.D., Gandy S. Traumatic brain injury—Football, warfare, and long-term effects. N Engl J Med. 2010; 363(14): 1293–6. [DOI] [PubMed] [Google Scholar]
- 8.Stoica B.A., Faden A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010; 7(1): 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson R.M., Singh A., Sun D., Fillmore H.L., Dietrich D.W. 3rd, Bullock M.R. Stem cell biology in traumatic brain injury: Effects of injury and strategies for repair. J Neurosurg. 2010; 112(5): 1125–38. [DOI] [PubMed] [Google Scholar]
- 10.Prakash R., Carmichael S.T. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. 2015; 28(6): 556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi H., Hu X., Leak R.K., Shi Y., An C., Suenaga J., Chen J., Gao Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp Neurol. 2015; 272: 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu A., Ying Z., Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma 2004; 21(10): 1457–67. [DOI] [PubMed] [Google Scholar]
- 13.Mills J.D., Bailes J.E., Sedney C.L., Hutchins H., Sears B. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg. 2011; 114(1): 77–84. [DOI] [PubMed] [Google Scholar]
- 14.Shin S.S., Dixon C.E. Oral fish oil restores striatal dopamine release after traumatic brain injury. Neurosci Lett. 2011; 496(3): 168–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pu H., Guo Y., Zhang W., Huang L., Wang G., Liou A.K., Zhang J., Zhang P., Leak R.K., Wang Y., Chen J., Gao Y. Omega-3 polyunsaturated fatty acid supplementation improves neurologic recovery and attenuates white matter injury after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2013; 33(9): 1474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasadsri L., Wang B.H., Lee J.V., Erdman J.W., Llano D.A., Barbey A.K., Wszalek T., Sharrock M.F., Wang H.J. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J Neurotrauma 2013; 30(11): 897–906. [DOI] [PubMed] [Google Scholar]
- 17.Kris-Etherton P.M., Taylor D.S., Yu-Poth S., Huth P., Moriarty K., Fishell V., Hargrove R.L., Zhao G., Etherton T.D. Polyunsaturated fatty acids in the food chain in the United States. Am J Clin Nutr. 2000; 71(1 Suppl): 179S–88S. [DOI] [PubMed] [Google Scholar]
- 18.Hering H., Lin C.C., Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci. 2003; 23(8): 3262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren H., Yang Z., Luo C., Zeng H., Li P., Kang J.X., Wan J.B., He C., Su H. Enriched endogenous omega-3 fatty acids in mice ameliorate parenchymal cell death after traumatic brain injury. Mol Neurobiol. 2016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Begum G., Yan H.Q., Li L., Singh A., Dixon C.E., Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci. 2014; 34(10): 3743–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harvey L.D., Yin Y., Attarwala I.Y., Begum G., Deng J., Yan H.Q., Dixon C.E., Sun D. Administration of DHA reduces endoplasmic reticulum stress-associated inflammation and alters microglial or macrophage activation in traumatic brain injury. ASN Neuro. 2015; 7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis M., Ghassemi P., Hibbeln J. Therapeutic use of omega-3 fatty acids in severe head trauma. Am J Emerg Med. 2013; 31(1): 273 e5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts L., Bailes J., Dedhia H., Zikos A., Singh A., McDowell D., Failinger C., Biundo R., Petrick J., Carpenter J. Surviving a mine explosion. J Am Coll Surg. 2008; 207(2): 276–83. [DOI] [PubMed] [Google Scholar]
- 24.Wang G., Shi Y., Jiang X., Leak R.K., Hu X., Wu Y., Pu H., Li W.W., Tang B., Wang Y., Gao Y., Zheng P., Bennett M.V., Chen J. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci USA 2015; 112(9): 2853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang G., Jiang X., Pu H., Zhang W., An C., Hu X., Liou A.K., Leak R.K., Gao Y., Chen J. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: Scriptaid protects against TBI via AKT. Neurotherapeutics 2013; 10(1): 124–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W., Wang H., Zhang H., Leak R.K., Shi Y., Hu X., Gao Y., Chen J. Dietary supplementation with omega-3 polyunsaturated fatty acids robustly promotes neurovascular restorative dynamics and improves neurological functions after stroke. Exp Neurol. 2015; 272: 170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverberg N.D., Crane P.K., Dams-O'Connor K., Holdnack J., Ivins B.J., Lange R.T., Manley G.T., McCrea M., Iverson G.L. Developing a cognition endpoint for traumatic brain injury clinical trials. J Neurotrauma 2017; 34(2): 363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson C.L., Bath P.M., Murphy S.P. G-CSF reduces infarct volume and improves functional outcome after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005; 25(4): 431–9. [DOI] [PubMed] [Google Scholar]
- 29.Okada M., Nakanishi H., Tamura A., Urae A., Mine K., Yamamoto K., Fujiwara M. Long-term spatial cognitive impairment after middle cerebral artery occlusion in rats: No involvement of the hippocampus. J Cereb Blood Flow Metab. 1995; 15(6): 1012–21. [DOI] [PubMed] [Google Scholar]
- 30.Yonemori F., Yamaguchi T., Yamada H., Tamura A. Spatial cognitive performance after chronic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1999; 19(5): 483–94. [DOI] [PubMed] [Google Scholar]
- 31.Block F., Kunkel M., Schwarz M. Quinolinic acid lesion of the striatum induces impairment in spatial learning and motor performance in rats. Neurosci Lett. 1993; 149(2): 126–8. [DOI] [PubMed] [Google Scholar]
- 32.Dash P.K., Mach S.A., Moore A.N. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001; 63(4): 313–9. [DOI] [PubMed] [Google Scholar]
- 33.Gao X., Chen J. Moderate traumatic brain injury promotes neural precursor proliferation without increasing neurogenesis in the adult hippocampus. Exp Neurol. 2013; 239: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S.H., Wang J.J., Chen C.H., Chang H.K., Lin M.T., Chang F.M., Chio C.C. Umbilical cord blood-derived CD34(+) cells improve outcomes of traumatic brain injury in rats by stimulating angiogenesis and neurogenesis. Cell Transplant. 2014; 23(8): 959–79. [DOI] [PubMed] [Google Scholar]
- 35.Xiong Y., Mahmood A., Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs 2010; 11(3): 298–308. [PMC free article] [PubMed] [Google Scholar]
- 36.Fancy S.P., Harrington E.P., Baranzini S.E., Silbereis J.C., Shiow L.R., Yuen T.J., Huang E.J., Lomvardas S., Rowitch D.H. Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci. 2014; 17(4): 506–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker K.R., Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front Aging Neurosci. 2013; 5: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuloaga K.L., Zhang W., Yeiser L.A., Stewart B., Kukino A., Nie X., Roese N.E., Grafe M.R., Pike M.M., Raber J., Alkayed N.J. Neurobehavioral and imaging correlates of hippocampal atrophy in a mouse model of vascular cognitive impairment. Transl Stroke Res. 2015; 6(5): 390–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bazan N.G. Cell survival matters: Docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006; 29(5): 263–71. [DOI] [PubMed] [Google Scholar]
- 40.Wang X., Zhao X., Mao Z.Y., Wang X.M., Liu Z.L. Neuroprotective effect of docosahexaenoic acid on glutamate-induced cytotoxicity in rat hippocampal cultures. Neuro report 2003; 14(18): 2457–61. [DOI] [PubMed] [Google Scholar]
- 41.Lazarov O., Hollands C. Hippocampal neurogenesis: Learning to remember. Prog Neurobiol. 2016; 138–140: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X., Gao X., Michalski S., Zhao S., Chen J. Traumatic brain injury severity affects neurogenesis in adult mouse hippocampus. J Neurotrauma 2016; 33(8): 721–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu P., Jones L.L., Snyder E.Y., Tuszynski M.H. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003; 181(2): 115–29. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y., Mahmood A., Chopp M. Neurorestorative treatments for traumatic brain injury. Discov Med. 2010; 10(54): 434–42. [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J., Venkat P., Zacharek A., Chopp M. Neurorestorative therapy for stroke. Front Hum Neurosci. 2014; 8: 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X., Ye R., Yan T., Yu S.P., Wei L., Xu G., Fan X., Jiang Y., Stetler R.A., Liu G., Chen J. Cell based therapies for ischemic stroke: From basic science to bedside. Prog Neurobiol. 2014; 115: 92–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao X., Enikolopov G., Chen J. Moderate traumatic brain injury promotes proliferation of quiescent neural progenitors in the adult hippocampus. Exp Neurol. 2009; 219(2): 516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z.G., Chopp M. Neurorestorative therapies for stroke: Underlying mechanisms and translation to the clinic. Lancet Neurol. 2009; 8(5): 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Waly B., Macchi M., Cayre M., Durbec P. Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci. 2014; 8: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pu H., Jiang X., Hu X., Xia J., Hong D., Zhang W., Gao Y., Chen J., Shi Y. Delayed docosahexaenoic acid treatment combined with dietary supplementation of omega-3 fatty acids promotes long-term neurovascular restoration after ischemic stroke. Transl Stroke Res. 2016; 7(6): 521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang X., Pu H., Hu X., Wei Z., Hong D., Zhang W., Gao Y., Chen J., Shi Y. A post-stroke therapeutic regimen with omega-3 polyunsaturated fatty acids that promotes white matter integrity and beneficial microglial responses after cerebral ischemia. Transl Stroke Res. 2016; 7(6): 548–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossi D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015; 130: 86–120. [DOI] [PubMed] [Google Scholar]
- 53.Franco R., Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015; 131: 65–86. [DOI] [PubMed] [Google Scholar]
- 54.Verkhratsky A., Steardo L., Parpura V., Montana V. Translational potential of astrocytes in brain disorders. Prog Neurobiol. 2016; 144: 188–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dallerac G., Rouach N. Astrocytes as new targets to improve cognitive functions. Prog Neurobiol. 2016; 144: 48–67. [DOI] [PubMed] [Google Scholar]
- 56.Osier N.D., Dixon C.E. The controlled cortical impact model: Applications, considerations for researchers, and future directions. Front Neurol. 2016; 7: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]