

Abstract
The use of Polymerase Chain Reaction (PCR) in infectious disease diagnosis, has resulted in an ability to diagnose early and treat appropriately diseases due to fastidious pathogens, determine the antimicrobial susceptibility of slow growing organisms, and ascertain the quantum of infection. This article outlines the PCR, some of its modifications and their application in infectious disease diagnosis.
KEY WORDS: Microbial diagnosis, PCR
Introduction
The study of genetic structure and function requires a sufficient amount of DNA, which was synthesised using a difficult process involving bacteria. A surprisingly simple method for making unlimited copies of DNA fragments was conceived by Kary Mullis in 1983 during a moonlit drive through the mountains of Northern California's redwood country. The polymerase chain reaction (PCR) is a technique for copying a piece of DNA in the laboratory with readily available reagents. With each step of the reaction the number of DNA molecules increases exponentially and in a few hours of running the reaction more than 100 billion copies can be made which can be easily detected. The PCR has progressed to become an important tool in the rapid diagnosis of infectious disease [1].
Watson and Crick in 1953 suggested the double stranded DNA helical structure, the base adenine (A) binding to thymine (T) and guanine (G) to cytosine (C). On raising the temperature the strands separate (melt), and lower temperatures cause them to rejoin (anneal), with the nucleotides realigning to regain the double helical structure. The unique sequences of DNA molecules coding for the pathogen are detected by PCR. This is an easier and more rapid technique than traditional exacting methods for cultivation and identification of fastidious pathogens.
Polymerase chain reaction
To detect a pathogen by the PCR reaction it is essential to know the sequence of nucleotides flanking an unique region on its DNA. Short strands of single stranded DNA called primers are then chemically synthesised. The primers are complimentary to the sequences flanking the unique region. The concentration of the primer is several million folds than the target DNA. Two primers are used, one designed to anneal to the sense strand, the other designed to anneal to the anti sense strand. The primers are mixed with a buffered solution of template DNA, nucleotides (dNTPs), magnesium chloride and DNA polymerase [2]
Earlier DNA polymerases were denatured by heat and required replenishment after every denaturing cycle. A thermostable DNA polymerase called Taq polymerase is derived from the bacillus Thermus acquaticus (found in hot spring). These thermostable DNA polymerases have revolutionised PCR technology and enable the reaction to be carried out by a single addition of Taq polymerase [3].
The double stranded template DNA is denatured by heating to a temperature above its melting (98°C) and the strands of DNA separate. The temperature is lowered so as to allow annealing of the primer to the template DNA. The Taq DNA polymerase extends the primer if it has bound to the target template strand of DNA by adding appropriate complementary nucleotides to the three prime [3’] end of the bound primer (Fig 1). High primer concentration ensures that primer annealing and extension are preferred over template strand reannealing. The reaction is stopped by raising the temperature and strand denaturation occurs. The temperature is then lowered to permit primer annealing, and the cycle is repeated. The PCR reaction is represented diagrammatically in Fig 1.
Fig. 1.
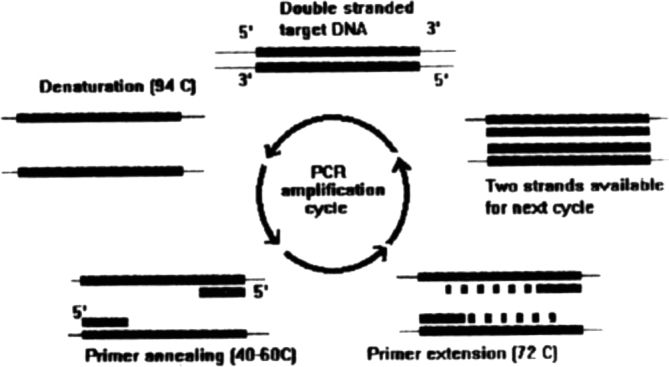
Shows the amplification of target DNA by PCR using two DNA primers. Taq polymerase and nucleotides. The repetitive cycle of deanturation, annealing, extention results in two copies of the target DNA, which is now available for the next cycle of amplification, resulting in a logarithmic increase in DNA copies after each cycle.
After the first three cycles the majority of the DNA strands would be those which are bound by the primers at both ends. Further amplification cycles result in exponential increase of the amplified DNA and thus would be of a defined length. The reaction is carried out as a 50 ul volume reaction mixture. The product is electrophoresed on an agarose gel. On staining with ethidium bromide and visualisation in UV light the DNA molecules fluoresce. Molecular weight measurement is carried out by comparing migration with markers of known molecular weight [2].
Specific methods for nucleic acid amplification reactions
Some important modifications of the PCR are:
Multiplex PCR In multiplex PCR simultaneous amplification of more than one genetic locus, using more than one set of primers is carried out in the same reaction. This can be used to differentiate between etiological agents responsible for a lesion. Depending on the molecular weight of the fragment amplified the etiologic agent can be determined [4].
Randomly amplified polymorphic DNA (RAPD) typing: This is used for epidemiological identification of bacterial isolates. One or more short primers of variable length are arbitrarily selected, and allowed to anneal to the template DNA at a low stringency. PCR amplification is carried out and the products resolved electrophoretically to yield DNA fingerprints which differs according to the degree of relatedness of the strains under investigation [5].
Reverse Transcriptase (RT) PCR: This technique uses RNA as a starting template. RNA is converted to cDNA by a retroviral reverse transcriptase. The cDNA is amplified and the amplified band detected [6]. A recombinant modified form of Thermus thermophilius DNA polymerase gene expressed in E.coli, the enzyme rTth, has an efficient reverse transcriptase activity in the presence of manganese, and DNA polymerase activity in presence of magnesium. Use of this enzyme has enabled the determination of cellular mRNA expression by a single step procedure [7]. The importance of RT PCR lies in its ability to detect RNA Viruses (e.g. HIV), study intracellular signals (e.g. intracellular interleukin expression) and therapeutic to quantify viral loads (e.g. semi quantitative RT PCR for HIV loads to monitor response to anti retroviral therapy).
Nucleic acid sequence based amplification (NASBA) : This is an ingenious method which is isothermal and does not require thermal cycling. RNA is converted to cDNA which carries a T7 RNA polymerase sequence at one end in the initiation phase and the adherent RNA is removed by RNase H. The Reverse transcriptase (RT) then synthesises a complementary DNA molecule to acquire a double stranded structure. The T7 RNA polymerase then synthesises large number of RNA copies. The cycle is repeated to produce antisense RNA as the final amplified product [8].
Ligase chain reaction(LCR) : This uses four primers so designed that on annealing the primers bind such that they are immediately adjacent and completely cover the target sequence. The DNA ligase present joins both the fragments, which can then be detected [9].
Applications of the PCR in Infectious disease diagnosis
Tuberculosis : With increasing incidence of both HIV infection and multi drug resistant strains of M. tuberculosis, early detection is vital for diagnosis and therapy. Classical techniques for detection of infection have a drawback in that the organism is fastidious and grows slowly. Many molecular strategies for detection of mycobacteria have been developed. Important ones are PCR, transcription mediated amplification, nucleic acid sequence based amplification and ligase chain reaction [10]. Mycobacterial speciation which is time consuming and exacting by conventional techniques, can be differentiated relatively easily by Multiplex PCR [4]. Molecular susceptibility testing for first line drugs INH and rifampicin is based on the fact that there is a mutation in the cat gene and the rpoB gene. This can be detected by a PCR amplification of the gene fragment followed by a simple electrophoresis in denaturing gels for single stranded conformational polymorphism (SSCP) analysis. This technique is sensitive and is able to detect even point mutations [11, 12] The above procedures lead to early diagnosis of infections by M tuberculosis and initiation of appropriate therapy.
Streptococcal pharyngitis: In developed countries PCR diagnosis of Gp A haemolytic streptococci by PCR (Gp A Streptococcus direct test or GASD) is gaining importance since its definitive exclusion could curtail empirical treatment of pharyngitis, curtail use of beta lactam antibiotics, and reduce overall costs of therapy [12].
Atypical pneumonia: A multiplex PCR based strategy wherein rapid diagnosis of infections caused by Chlymydia pneumoniae, Mycoplasma pneumonia and legionellae could help reshape the management of atypical pneumonias [14].
Persistent illness: Persistent infections by HSV, CMV, EBV, VZV, HHV, JC virus, measles, hepatitis, rarely respect organ boundaries, are difficult to diagnose and treat. In the immune competent host these diseases present usually as an acute illness and recovery is followed by a strong protective immunity. However in the immunocompromised host these infections may be reactivated. They cause not only a diagnostic dilemma but also the detection of their nucleic acid by amplification procedures may not be of clinical relevance. Quantitative assays for their presence are important and have been developed [14].
Acute febrile illness like falciparum malaria, salmonellosis, babesiosis, have been identified using PCR. Especially with falciparum infections use of a single PCR reaction and hybridisation assays with various probes is used in species identification [15].
Ulcerative urogenital infections: A multiplex PCR for differentiating etiological causes of ulcerative urogenital infections due to Haemophilus ducreyi, T. pallidum, HSV has been reported [16].
Conclusions: The advent of nucleic acid detection techniques in medical microbiology has revolutionised disease diagnosis and prognostication. Difficult organisms can now be detected with greater sensitivity and precision. Drug resistance monitoring of mycobacteria using nucleic acid based methods is going to provide an important technique for early markers for drug resistance enabling start of early appropriate therapy. PCR is thus a valuable tool in disease diagnosis. Incorporation of nucleic acid based testing into laboratory services will result in a challenging and exciting time for the clinical microbiologist.
REFERENCES
- 1.Mullis MB. The unusual origin of the polymerase chain reaction. Scientific American. 1990:59–65. doi: 10.1038/scientificamerican0490-56. [DOI] [PubMed] [Google Scholar]
- 2.Erlich HA. Basic Methodology. In: Erlich HA, editor. PCR Technology. 1st edition. Stockton press; New York: 1989. pp. 1–5. [Google Scholar]
- 3.Erlich HA, Gelfand D, Sninsky JJ. Recent advances in Polymerase chain reaction. Science 1991;251:1643-50 [DOI] [PubMed]
- 4.Cormican M, Glenon M. Ni Riain U, Flynn J. Multiplex PCR for identifying mycobacterial isolates, J Clin Pathol. 1995;48:203–205. doi: 10.1136/jcp.48.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Power EGM. RAPD typing in microbiology-a technical review. J Hosp Inf 1996;34:247-65 [DOI] [PubMed]
- 6.Meyers TW, Sigua CL. Amplification of RNA: High temperature reverse transcription and DNA amplification with Thermus thermophilius DNA polymerase. In: Innis MA, Gelfand DH, Sninsky JJ, editors. PCR Strategies. 1st edition. Academic press; New York: 1995. pp. 58–68z. [Google Scholar]
- 7.Myers TW, Gelfland DH. Reverse transcription and DNA amplification by a Thermus Thermophilus. DNA polymerase Biochemistry. 1991;30:7661–7667. doi: 10.1021/bi00245a001. [DOI] [PubMed] [Google Scholar]
- 8.Guatelli JC, Whitfied KM, Kwoh DY. Isothermal in vitro amplification of nucleic acids by multienzyme reaction modelled after retroviral replication. PCNAS (USA) 1990;87:1874–1888. doi: 10.1073/pnas.87.5.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landegren U, Kaiser R, Sanders J. A ligase mediated gene detection technique. Science 1988;241:1077-80 [DOI] [PubMed]
- 10.Shaw RJ. Polymerase chain reaction. In: Davis PDO, editor. Clinical Tuberculosis. 1st edn. Chapman and Hall Medical; London: 1994. pp. 381–391. [Google Scholar]
- 11.Zhang Y, Heym B, Alen B. The catalase peroxidase gene and INH resistance of [DOI] [PubMed]
- 12.Musser JM. Antimicrobial agents resistance to mycobacteria: Molecular genetic insights. Clin Microbial Review. 1995;8:496–514. doi: 10.1128/cmr.8.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whelen AC, Persing DH. The role of nucleic acid amplification and detection in the clinical microbiology. Annu Rev Microbiol. 1996;50:349–373. doi: 10.1146/annurev.micro.50.1.349. [DOI] [PubMed] [Google Scholar]
- 15.Kimura M, Miyake H, Kim HS, Tanabe M, Arai M. Species specific PCR detection of malaria parasite by microtiter plate hybridisation: clinical study with malaria patients. J Clin Microbiol. 1995;33:2342–2346. doi: 10.1128/jcm.33.9.2342-2346.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orle KA, Gates CA, Martin DH, Body BA, Weiss JB. Simultaneous PCR Detection of Hducreyi, T. pallidum and Herpes simplex virus from genital ulcers. J Clin Microbiol. 1996;34:49–54. doi: 10.1128/jcm.34.1.49-54.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uncited reference
- 13.Heiter BJ, Bourbeau PP. Comparison of the Genprobe GpA Streptococcus Direct Test with culture and rapid Streptococcal antigen detection assay for diagnosis of streptococcal pharyngitis. J Clin Microbial 1993;31:2070-3 [DOI] [PMC free article] [PubMed]