

Abstract
Malignant gliomas (MG), tumors of glial origin, are the most commonly diagnosed primary intracranial malignancies in adults. Currently available treatments have provided only modest improvements in overall survival and remain limited by inevitable local recurrence, necessitating exploration of novel therapies. Among approaches being investigated, one of the leading contenders is immunotherapy, which aims to modulate immune pathways to stimulate the selective destruction of malignant cells. Dendritic cells (DCs) are potent initiators of adaptive immune responses and therefore crucial players in the development and success of immunotherapy. Clinical trials of various DC-based vaccinations have demonstrated the induction of anti-tumor immune responses and prolonged survival in the setting of many cancers. In this review, we summarize current literature regarding DCs and their role in the tumor microenvironment, their application and current clinical use in immunotherapy, current challenges limiting their efficacy in anti-cancer therapy, and future avenues for developing successful anti-tumor DC-based vaccines.
Keywords: Malignant glioma, Glioblastoma, Immunotherapy, Dendritic cell based vaccine
Introduction
It has been established that natural antitumor immune responses develop in a variety of cancer types, primarily mediated by CD8+ cytotoxic lymphocytes (CTLs) and CD4+ T helper-1 (Th1) cells [1–3]. However, these responses are often insufficient to completely eliminate tumor presence and may be overcome by tumor-induced immunosuppression [3, 4]. Based on these observations, cancer immunotherapy aims to modulate immune pathways to initiate or augment otherwise inadequate anti-tumor immune responses. One promising approach is through therapeutic vaccination, whereby immune cells such as dendritic cells (DCs) are sensitized to tumor-associated antigens (TAAs) and stimulated to mobilize effector responses resulting in selective destruction of malignant cells.
DCs are bone marrow-derived lymphoid cells uniquely capable of activating primary immune responses through the presentation of antigens to naïve CD4+ and CD8+ T cells [1, 5]. Through the release of interleukin-15 (IL-15), DCs stimulate the development of memory T cells and lasting protective immunity. DCs are also potent activators of B cells, natural killer (NK), and natural killer T (NKT) cells [5]. In the steady-state, DCs reside in peripheral tissues, internalizing antigens from their surrounding environment. These antigens are degraded into short peptide fragments, loaded onto major histocompatibility complex (MHC) molecules to form peptide-MHC (p-MHC) complexes, and displayed on the DC’s surface. Following exposure to pathogenic or inflammatory molecules, DCs mature and migrate to secondary lymphoid organs to activate T cells [1]. Antigen presentation by immature DCs induces antigen-specific tolerance, a pathway crucial in preventing autoimmunity and often exploited by tumors to avoid elimination by the immune system [6, 7]. As crucial modulators of innate and adaptive immune responses, DCs provide a promising foundation for immunotherapy.
Malignant gliomas (MGs), tumors of glial origin, are the most frequently diagnosed primary intracranial malignancies in adults. The most common, and most aggressive, subtype of MG is glioblastoma (GBM). The prognosis for patients with GBM is extremely poor, with median life expectancy of 14.6 months and 5-year survival rates less than 10% [8, 9]. The current standard treatment for MG is maximal surgical resection followed by radiotherapy and temozolamide (TMZ) [9]. However, these nonspecific therapies are limited by systemic toxicities and damage to healthy surrounding tissues and ultimately fail to result in complete, sustained tumor eradication. There is therefore a great need to explore novel approaches, such as immunotherapy, to improve outcomes for these patients.
Immunotherapy in the context of MG is a highly evolving field that has shown significant promise in pre-clinical and early clinical testing. With a better understanding of cancer antigens on a basic genomic level and growing knowledge of immune checkpoint pathways, the last several years have seen great advancements in the fields of cancer immunology and immunotherapy. Understanding these recent developments is critical in designing the next generation of successful anti-glioma DC-based vaccines.
Dendritic cell biology
DC subtypes and DC trafficking
DCs are a morphologically and functionally heterogeneous group of cells derived from CD34+ hematopoietic progenitor cells in the bone marrow. There are two distinct subtypes of human DCs: a larger subset of CD11c+ “myeloid” dendritic cells (mDCs), and a smaller subset of CD11c-“plasmacytoid” dendritic cells (pDCs). Both mDCs and pDCs are antigen-presenting cells (APCs) and can activate T cells, though they differ in tissue distribution, surface molecule expression, and cytokine release [1].
mDCs are found in most lymphoid and nonlymphoid tissues and tend to be potent stimulators of cell-mediated immunity. Activated mDCs release IL-12, which induces IFN-γ secretion and CD4+ Th1 differentiation, promoting Th1-mediated antitumor responses [10, 11]. Present in secondary lymphoid organs, pDCs have the unique ability to produce large amounts of interferon-α (IFN-α) in response to viral infection. They express Toll-like receptor-7 (TLR-7) and TLR-9 to recognize viral nucleic acids [5, 12]. Type I IFNs, like IFN-α, are potent activators of antiviral and antitumor responses.
Following development in the bone marrow, immature mDCs and pDCs disperse throughout the body, guided by chemokines such as MIP-1α, MIP-1β, RANTES, MCP-3, and MIP-5. Detection of pathogenic or inflammatory molecules stimulates DC trafficking to sites of infection or tissue damage [1].
Molecules expressed by DCs
DCs express surface molecules specialized for T cell interactions including antigen presentation (CD1 and MHC class I and II), costimulatory (CD80/B7.1 and CD86/B7.2), and adhesion (CD11, CD50, CD54, CD58) molecules [2]. Present on most cells, MHC class I molecules display internally derived antigens, including self and viral peptides, to CD8+ T cells. Unique to APCs, MHC class II molecules present exogenous peptides to CD4+ T cells. Antigens captured by DCs are typically loaded onto MHC class II molecules, but may be targeted to MHC class I molecules and presented to CD8+ T cells in a process termed “cross-presentation.”[1, 13] CD1 molecules present endogenous and exogenous lipid antigens [1]. DCs also express a variety of intracellular and extracellular receptors through which they sense antigens, chemokines, and activating stimuli in their environment. Among these are C-type lectin receptors (DEC-205, DC-SIGN), Fcγρεχεπτορσ (CD64, CD32), integrins, TLRs, TNF-family receptors, cytokine and chemokine receptors, and scavenger receptors [1, 13].
Mature versus immature DCs
Immature DCs are specialized for antigen sampling and express high concentrations of receptors mediating antigen recognition and uptake. They reside in peripheral tissues, continuously internalizing antigens from their environment. However, in this immature state they express low levels of surface MHC and costimulatory molecules and therefore are inefficient antigen presenting cells and poor initiators of T cell activation [7]. Detection of pathogenic or inflammatory molecules, such as LPS or TNF-α, initiates DC maturation, leading to an enhanced ability to activate T cells. Maturing DCs downregulate receptors specialized for antigen uptake, upregulate surface expression of MHC and costimulatory molecules, and undergo cytoskeletal changes to improve motility and maximize surface area for T cell interactions. Maturation also induces DCs to secrete cytokines, chemokines, and growth factors to attract other immune cells and promote the activation, proliferation, and differentiation of effector cells. Pathogenic molecules and other tissue factors present during maturation influence the specific cytokine release profile of mature DCs [7]. Activated DCs upregulate CCR7 expression and migrate to the paracortex of draining lymph nodes, attracted by chemokines MIP-3β and SLC [1, 7]. Here, DCs receive the final stimulus for their maturation: ligation of surface CD40 molecules by T cell CD40 ligand. CD40/CD40L interactions further increase DC costimulatory molecule expression and cytokine secretion, strengthening DC-T cell interactions and effector responses [1, 2].
Immunogenic versus tolerogenic DCs
Antigen presentation to naïve T cells has the potential to result in antigen-specific immunity or antigen-specific tolerance [7]. Although both mature and immature DCs are capable of antigen presentation, only mature DCs possess the high levels of surface MHC and costimulatory molecules necessary to activate naïve T cells. Antigen presentation by immature or incompletely matured DCs leads to antigen-specific tolerance by inducing T cell anergy, apoptosis, or differentiation into immunosuppressive regulatory T (Treg) cells [14].
DC-mediated generation of antigen-specific tolerance is fundamental in preventing autoimmune destruction of self-proteins. During T cell development in the thymus, DCs induce clonal deletion of strongly self-reactive double-positive thymocytes in a process termed “negative selection.”[6] However, not all self-antigens are present in the thymus and some are only expressed later in life. The identification and removal of T cells with receptors specific for these antigens is mediated peripherally by immature DCs. Early development of self-tolerance is especially important given that in sites of inflammation, DCs are exposed to both self and non-self antigens in an activating environment. A tolerogenic state also occurs in a variety of tumor types, allowing malignant cells to evade detection and elimination by the immune system [3, 4].
DC-T cell interaction (Fig. 1)
Fig. 1.
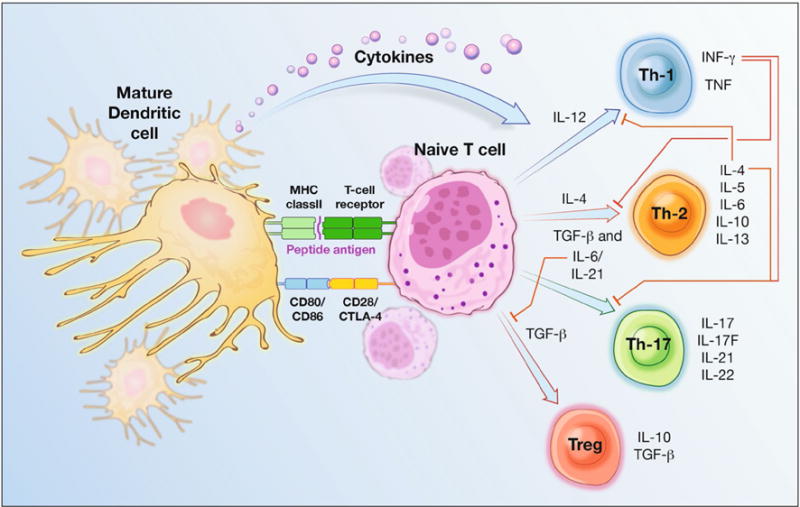
Demonstrating DC-T cell interaction. DCs provide three key signals to activate naïve T cells and initiate adaptive immune responses. First, DC surface p-MHC complexes are recognized by antigen-specific T cell receptors (TCRs). A second costimulatory interaction occurs between DC CD80/CD86 molecules and T cell CD28 molecules. T cells may also express CTLA-4 molecules, which interact with DC CD80/CD86 and transmit signals inhibitory to T cell activation. Depending on the presence of local cytokines, activated T cells may terminally differentiate along one of several specialized subtypes. Among these are Th1, Th2, Th17, and Treg lineages. Th1, Th2, and Th17 comprise the “activating” arms of T cell responses, and Treg cells form the “suppressive” arm. IL-12 stimulates the differentiation of Th1 cells, which produce IFN-g, an important activator of innate and adaptive immune responses. Their differentiation is inhibited by IL-4. IL-4 promotes the differentiation of Th2 cells, which secrete IL-4, IL-5, IL-6, IL-10, and IL-13, activating eosinophils, mast cells, and B cells to support humoral immunity and parasite resistance. Th2 differentiation is inhibited by IFN-g. Th17 cells develop in the presence of TGF-b and IL-6 or IL-21. They secrete IL-17, IL-17F, IL-21, and IL-22 and play key roles in the development of autoimmune tissue inflammation and resistance to infection with extracellular bacteria. The presence of TGF-b promotes the development of regulatory T cells. Suboptimal p-MHC-TCR or costimulatory interactions, which may occur through antigen presentation by immature DCs, and delivery of inhibitory signals via DC co-inhibitory molecules or CTLA-4 activation may also induce Treg cell differentiation. Treg cell development is inhibited by the IL-6. Tregs secrete IL-10 and TGF-b to suppress immune responses
Within secondary lymphoid tissues, DCs present antigens to naïve T cells. This encounter is characterized by several key interactions, illustrated in Fig. 1. The strength and nature of the elicited response is determined by the state of DC maturation, concentration of p-MHC complexes, affinity of T cell receptors for p-MHC complexes, type and strength of B7 interactions, and presence of local cytokines. Of particular interest to cancer immunotherapy is the polarization of T cell differentiation. Th1 cells secrete pro-inflammatory cytokines to activate downstream effector responses and support CD8+ CTL maturation, forming the basis for antitumor immunity, whereas Th2 and Treg responses are not cytotoxic, impair Th1-mediated tumor destruction, and allow tumor persistence [10]. Gradual dysfunction of Th1-mediated cellular immunity and the development of Th2 and Treg responses are associated with cancer progression and a poor prognosis in many malignancies [15, 16].
DC in tumor microenvironment
Role of DCs in various cancers
DCs are naturally exposed to a variety of antigens within the tumor microenvironment. These tumor-associated antigens (TAAs) may be uniquely tumor-specific (MAGE, BAGE, GAGE), viral (HPV, CMV), tissue-specific differentiation antigens (MART-1, gp100, tyrosinase), or mutated and/or overexpressed self-proteins (HER-2/neu, EGFRvIII) [17, 18]. Chemotherapy and radiation, standard components of anticancer treatment, trigger widespread tumor cell death further promoting TAA release [19]. These antigens can be captured by tumor-infiltrating DCs and presented to naïve T cells to initiate antigen-specific antitumor responses.
However, many tumors employ mechanisms to avoid immune-mediated rejection. Malignant cells may develop reduced immunogenicity through mutations of TAAs, defects in antigen processing or presentation, or downregulation of MHC class I expression [4]. Tumors also actively promote the development of a tolerogenic environment, suppressing the effectiveness of antitumor immune responses through the release of immunosuppressive cytokines, modulation of immune checkpoint pathways, and attraction of immunosuppressive leukocytes. Many tumors overexpress the STAT3 protein, which prevents tumor cell apoptosis, downregulates MHC and costimulatory molecule expression, promotes immunosuppressive cytokine release, inhibits IL-12 and IFN-γ expression, and suppresses Th1 immune responses [20]. Tumors also contain an abundance of compounds known to inhibit DC maturation and function, including IL-10, TGF-β, VEGF, IL-6, and PGE2 [21]. DCs in tumor-bearing patients often express an immature or incompletely matured phenotype and have a reduced capacity to activate antitumor immune responses, instead promoting Treg cell development and antigen-specific tolerance [21].
Role of DC in glioma
A prominent feature of MG is profound suppression of cell-mediated immunity [22]. Gliomas often contain high proportions of apoptotic and dysfunctional T lymphocytes [22, 23]. T cells obtained from patients with MG demonstrate defective signaling, impaired cytokine release, and a diminished ability to proliferate and carry out effector responses [22]. Gliomas actively suppress T cell function by modulating inhibitory checkpoint pathways and releasing immunosuppressive cytokines like TGF-β and IL-10 [22]. TGF-β inhibits the activation, proliferation, and differentiation of effector T cells [4] and is independently associated with a poor prognosis in MG [24]. Often overexpressed by malignant cells and tumor-infiltrating DCs, programmed-death-ligand-1 (PD-L1) binds to PD-1 molecules on activated T cells to induce their anergy and apoptosis. T cells can also be stimulated to express CTLA-4, which competes with CD28 molecules for binding to DC CD80/CD86 and sends inhibitory signals to T cells, preventing their activation [25]. CTLA-4 interactions with DCs can induce DC expression of indoleamine-2,3-deoxygenase (IDO), an enzyme involved in tryptophan catabolism, which arrests T cell proliferation, induces T cell apoptosis, and promotes Treg cell generation [13, 25]. IDO overexpression is observed in aggressively growing tumors and associated with reduced effector T cell infiltrations [25].
The dysfunction exhibited by T cells of glioma patients occurs in APCs as well. This concept can be illustrated by exploring the role of pDCs in glioma development and progression. Both animal models and human clinical trials have demonstrated that pDCs can induce antitumor immune responses through T cell activation and IFN-α production [12, 13]. However, other studies have associated pDC tumor infiltration with disease progression and a poor prognosis [26, 27]. These findings can be attributed to the systemic dysfunction of pDCs often observed in the setting of cancer. Tumor-associated pDCs exhibit a reduced capacity to secrete IFN-α, likely due to downregulation of TLR-9, and impaired antigen presentation. These pDCs are poor initiators of antitumor immunity and promote Treg cell development [12, 26, 27]. Our group has shown that in a murine model of glioma, selective pDC depletion in the early stages of tumor formation reduces Treg cell presence and prolongs survival [26]. Patients with glioma possess greater proportions of Treg cells, the accumulation of which are associated with suppression of antitumor responses and a poor prognosis [16, 27–29]. Selective Treg cell depletion has been shown to restore effector cell functions, augment antitumor immune responses, and improve survival outcomes in many cancers [30, 31].
DC based theraputic vaccine
As crucial regulators of innate and adaptive immune responses, DCs provide a solid foundation for the development of immunotherapies. DC-based immunotherapies manipulate DCs to initiate immune responses against TAAs. Numerous preclinical studies have established the ability of various DC-based vaccinations to induce robust and highly specific antitumor T cell responses, leading to prolonged survival and protective antitumor immunity in animal models [32–34]. Initially evaluated in human clinical trials in the 1990s, DC-based vaccines were shown to be beneficial in the treatment of patients with B-cell lymphoma [35], melanoma [36], and prostate cancer [37]. In the years since, DC-based vaccines have been adapted and studied in a variety of other malignancies, listed in Table 1 [35–48]. The first patient with a primary intracranial tumor treated with DC-based immunotherapy is described in a case report published in 2000 by Liau et al. Following surgical resection, this patient received three immunizations of autologous DCs pulsed with allogeneic MHC class I glioblastoma peptides as treatment for recurrent brainstem GBM. The vaccine was well tolerated and the patient developed measurable cellular immune responses against vaccine antigens, however an objective clinical response was not evident [49]. Results of early clinical trials, listed in Table 2, have since confirmed the safety, feasibility, and immune-stimulating activity of DC-based immunotherapy in patients with MG [24, 28, 29, 49–64]. Given its reliance on final effector functions of the immune system, a concern with immunotherapy is its efficacy in conjunction with lymphocyte-depleting treatments, like chemotherapy. In subsequent studies of DC-based immunotherapy in combination with standard treatment for MG, vaccinated patients receiving concurrent TMZ demonstrated enhanced humoral and cellular antitumor responses correlating with prolonged survival, confirming the efficacy of immunotherapy in a lymphopenic environment [57]. There are two well-established avenues for introducing antigens to DCs: direct targeting of antigens to DCs in vivo and ex vivo generation of antigen-loaded DCs [5, 13].
Table 1.
Clinical trials of DC based vaccine for cancer
| Malignancy | Reference | Phase | N | Antigen | Vaccine construct |
|---|---|---|---|---|---|
| B-cell lymphoma | Hsu et al. [35] | Pilot | 4 | Idiotype protein | DCs + idiotype protein |
| Melanoma | Nestle et al. [36] | Pilot | 16 | HLA-restricted peptides or autologous tumor lysate | GM-CSF IL-4 DCs + tumor peptides or autologous tumor lysates |
| Prostate cancer | Murphy et al. [37] | II | 25 | PSM-P1 and -P2 | GM-CSF IL-4 DCs + tumor peptides |
| Melanoma | Thurner et al. [38] | I | 11 | Mage-3A1 | GM-CSF IL-4 DCs (activated with TNF-α) + Mage-3A1 tumor peptide |
| Melanoma | Mackensen et al. [39] | I | 14 | MAGE-1, MAGE-3, melan-A, gp100, tyrosinase | CD34 + progenitor derived DCs (activated with TNFα) + tumor peptides |
| Colorectal, NSCLC | Fong et al. [40] | I | 12 | 610D: altered carcinoembryonic antigen (CEA) peptide | FLT3 ligand-expanded DCs + 610D peptide |
| Melanoma | Banchereau et al. [41] | I | 18 | MART-1, tyrosinase, MAGE-3, gp100 | CD34 + progenitor derived DCs (activated with TNF-α) + tumor peptides |
| Melanoma | Schuler-Thurner et al. [42] | I | 16 | Tumor peptides | GM-CSF IL-4 cryopreserved DCs (activated with TNF-α, IL-1β, IL-6, PGE2) + tumor peptides |
| Lymphoma | Timmerman et al. [43] | Pilot | 35 | Idiotype protein | DCs (spontaneously matured) + idiotype protein; idiotype and KLH protein vaccination |
| Cervical | Ferrara et al. [44] | Pilot | 15 | Recombinant HPV16 E7 or HPV18 E7 oncoprotein | GM-CSF IL-4 DCs (matured with IL-1β, IL-6, TNF-α, and PGE2) + recombinant tumor protein |
| Melanoma | Salcedo et al. [45] | I/II | 15 | Allogeneic tumor lysate | GM-CSF IL-13 DCs + allogeneic tumor lysate; DC + hepatitis B surface protein (HBs) and/or tetanus toxoid |
| Melanoma | Palucka et al. [46] | I | 20 | Allogeneic tumor lysate (Colo829 melanoma cells) | GM-CSF IL-4 DCs (activated with TNF and CD40L) + allogeneic tumor lysate |
| Renal cell carcinoma | Wierecky et al. [47] | I/II | 20 | M1.1 and M1.2 | GM-CSF IL-4 DCs (matured with TNF-α) + tumor peptides; !L-2 vaccination |
| Prostate cancer | Kantoff et al. [48] | III | 341 | PA2024: PAP-GM-CSF recombinant fusion protein | DCs + PA2024 recombinant fusion protein |
Table 2.
Clinical trials of DC based vaccine for glioma
| Reference | Phase | N | Malignancy | Antigen | Vaccine construct | Median TTP (months) | Median PFS (months) | Median OS (months) |
|---|---|---|---|---|---|---|---|---|
| Liau et al. [49] | Case report | 1 | Recurrent brainstem glioma | Allogeneic tumor peptides | DCs + allogeneic tumor peptides | |||
| Yu et al. [50] | I | 9 | Newly diagnosed AA (2), GBM (7) | TRP-1, MAGE-1, and gp100 | GM-CSF IL-4 DCs + autologous tumor peptides | 15 | ||
| Kikuchi et al. [51] | I | 8 | Recurrent malignant glioma | DC-tumor cell fusion | GM-CSF IL-4 TNFα DC-tumor cell fusion | |||
| Yamanaka et al. [52] | I/II | 10 | Recurrent AA (3), GBM (7) | Autologous tumor lysate | GM-CSF IL-4 DCs + autologous tumor lysate + KLH | |||
| Yu et al. [53] | I | 14 | AA (3/4 recurrent), GBM (9/10 recurrent) | Autologous tumor lysate | GM-CSF IL-4 DCs + tumor lysate | 30.7 | ||
| Rutkowski et al. [54] | I | 12 | Recurrent GBM (10), malignant pleomorphic xanthoastrocytoma (1) | Autologous tumor lysate | GM-CSF IL-4 DCs (matured with TNF-α, IL-1β, and PGE2) + autologous tumor lysate | 3 | 10.5 | |
| Kikuchi et al. [55] | I/II | 15 | Recurrent AA (7), GBM (6), AOA (2) | DC-tumor cell fusion | GM-CSF IL-4 TNF-α DC-tumor cell fusion; injections of rIL-12 | |||
| Yamanaka et al. [56] | I/II | 24 | Recurrent AA (2), GBM (18), glioma (1), anaplastic mixed glioma (1), AOA (1), AO (1) | Autologous tumor lysate | GM-CSF IL-4 DCs (± matured with OK-432) + autologous tumor lysate + KLH | 15.8 | ||
| Liau et al. [24] | I | 12 | GBM (5/12 recurrent) | Autologous tumor peptides | GM-CSF IL-4 DCs + autologous tumor peptides | 15.5 | 23.4 | |
| Wheeler et al. [57] | II | 32 | GBM (21/32 recurrent) | Autologous tumor lysate | GM-CSF IL-4 DCs + autologous tumor lysate | 10.1 (5.5 in non-responders) | 21.4 (14.1 in non-responders) | |
| Walker et al. [58] | I | 9 | AA (3/4 recurrent), GBM (2/9 recurrent) | Autologous tumor cells | GM-CSF IL-4 DCs + irradiated autologous tumor cells | |||
| Ardon et al. [59] | Pilot | 8 | Newly diagnosed GBM | Autologous tumor lysate | DCs (matured with TNF-α, IL-1β, and PGE2) + tumor lysate | 18 | 24 | |
| Fadul et al. [60] | II | 10 | Newly diagnosed GBM | Autologous tumor lysate | DCs (matured with TNF-α and PGE2) + autologous tumor lysate | 9.5 | 28 | |
| Okada et al. [61] | I/II | 22 | Recurrent AA(5), GBM (13), AO (3), AOA (1) | EphA2, interleukin (IL)-13 receptor-2, YKL-40, and gp100 | Type 1 polarized DC (matured with IL-1β, TNF-α, IFN-α, IFN-γ, and poly-I:C) + synthetic tumor peptides; poly-ICLC injection | 4 | ||
| Chang et al. [62] | I/II | 17 | Recurrent AA (1), GBM (6/14 recurrent), malignant oligodendroglioma (1/2 recurrent) | Irradiated autologous tumor cells | GM-CSF IL-4 DCs + autologous tumor cells | 17.1 | ||
| Prins et al. [28] | I | 23 | GBM (8/23 recurrent) | Autologous tumor lysate | GM-CSF IL-4 DCs + autologous tumor lysate; imiquimod cream or poly ICLC injection | 15.9 | 31.4 | |
| Ardon et al. [63] | I/II | 77 | Newly diagnosed GBM | Autologous tumor lysate | DCs (matured with TNF-α, IL-1β, and PGE2) + autologous tumor lysate | 10.4 | 18.3 | |
| Phuphanich et al. [64] | I | 21 | GBM (3/19 recurrent), brainstem glioma (1) | HER2, TRP-2, gp100, MAGE-1, IL13Ra2, and AIM-2 | GM-CSF IL-4 DCs (matured with TNF-α) + synthetic tumor peptides | 16.9 | 38.4 | |
| Prins et al. [29] | I | 6 | GBM (2/4 recurrent), anaplastic glioma (2) | TRP-2, gp100, her-2/neu, survivin | GM-CSF IL-4 DCs (matured with TNF-α, IL-6, IL-1β, PGE2) + synthetic glioma-associated antigen (GAA) peptides | 14.5 | ||
| NCT02366728 | II | Newly diagnosed GBM | CMV pp65 peptide | DCs + CMV pp65 mRNA ± tetanus diphtheria toxoid (Td) | Ongoing | |||
| NCT02465268 | II | Newly diagnosed GBM | CMV pp65 peptide | DCs + CMV pp65 mRNA + GM-CSF + Td | Ongoing | |||
| NCT00045968; NCT02146066 | III | Newly diagnosed GBM/Recurrent GBM | Autologous tumor lysate | DCs + autologous tumor lysate | Ongoing | |||
| NCT01567202 | II | Newly diagnosed or recurrent GBM | Autogeneic glioma stem-like cells (A2B5+) | DCs + stem-like cell antigens from irradiated GBM | Ongoing |
TTP time to progression, PFS progression free survival, OS overall survival
In vivo DC targeting
The targeted delivery of antigens to DCs in vivo can be achieved by coupling antigens to antibodies specific to DC surface molecules like C-type lectins, Fcγ, MHC class II, or CD40 [65, 66]. As different DC receptors have differential influences on the capacity of mature DCs to polarize T cell differentiation, this method allows targeting of specific receptors known to stimulate DC-mediated induction of Th1 responses [27]. However, a major limitation of this approach is the possibility of antigen uptake by immature DCs, leading to the induction of tolerogenic responses [65, 66]. This risk is especially relevant for cancer patients, who may suffer from immune system dysfunction induced by tumor-secreted factors or chemotherapy or radiation treatments. Concurrent administration of DC maturation activators, such as TLR ligands or CD40 agonists, can minimize this risk [66].
Ex-vivo generated DCs
Autologous DCs can be generated and expanded ex vivo, charged with TAAs, exposed to maturation stimuli, and then reintroduced into the patient as a vaccine. DCs may be directly isolated from the peripheral blood or differentiated in vitro from monocytes or CD34+ hematopoietic progenitor cells. The most widely used method involves the in vitro differentiation of peripheral blood monocytes in the presence of GM-CSF and IL-4 [67]. DCs may be exposed to antigens in the form of peptides or proteins, whole killed tumor cells or lysates, tumor stem cells, mRNA or cDNA encoding TAAs, or through direct fusion with tumor cells, and subsequently cultured with molecules to induce maturation. Among those commonly used are TNF-α, IL-1β, IL-6, PGE2, LPS, and IFN-γ [21, 68, 69].
Antigen selection introduces unique benefits and limitations to vaccine generation and efficacy. Commonly used in clinical trials, peptide antigens can be generated for key sequences of tumor-specific proteins, modified to enhance immunogenicity, and targeted directly to DC surface MHC molecules in culture [5]. Their known structure facilitates monitoring of antigen-specific immune responses, however limits their use to patients with HLA subtypes possessing inherent affinity for these sequences; in a recent clinical trial of DC-based vaccination with glioma-associated antigens (GAAs), HLA subtype restrictions only allowed for treatment of 40% of initially enrolled patients [29]. Larger proteins and tumor mRNA molecules must be internalized and processed by DCs, allowing the selection and presentation of a variety of epitopes compatible with the patient’s own HLA type, though the sequences selected may not be strongly immunogenic [2]. Whole tumor lysates, tumor stem cells, and tumor cell-DC fusion vaccines expose DCs to a tumor’s unique, complete antigen profile without the need for individual antigen identification, which may be useful in highly heterogeneous tumors and those in which specific TAAs are unknown. However, this type of vaccine depends on the availability of autologous tumor material, which may be limited in patients having undergone previous treatments.
Ex vivo DC generation is expensive, labor-intensive, and must be personalized for individual patients. Optimization of in vitro conditions to yield high-quality DCs capable of inducing strong cytotoxic responses in vivo remains a topic of continued research [21, 68, 70, 71]. Despite these challenges, ex vivo DC generation offers greater control over the phenotype and quality of DCs and their encounter with antigen. Expanded and matured away from tumor-induced immunosuppression, these DCs are poised to activate tumor-specific immunity rather than tolerance and may be particularly useful in patients with weakened immune systems that cannot respond to in vivo delivered stimuli [5, 21].
Limitations of DC based vaccine
The field of immunotherapy has seen significant advancements over the past decade. DC-based vaccination is well tolerated and induces systemic antitumor responses and prolonged survival in a subset of vaccinated patients with a variety of tumor types. However, objective clinical response rates remain low, leaving much room for improvement. Furthermore, comparing results between clinical trials has been limited by variability in vaccine composition and preparation and lack of established criteria for objective evaluation of immunological and clinical responses.
Selection of optimal conditions for vaccine development to generate high-quality DCs capable of inducing robust antitumor immune responses remains a significant area of research. Recent reports suggest that DCs matured ex vivo are less effective than their natural counterparts in activating T cells and inducing effective antitumor immunity [68, 71]. Also fundamental to the development of DC-based vaccines is the selection of immunogenic antigens with which to prime the immune system. This is limited by the heterogeneous antigen profile of MG, both between patients and within individual tumors [5]. Vaccination also introduces selective pressures for tumor antigenic mutation and the development of antigen-loss variants, particularly with single-antigen vaccines and those targeting antigens non-essential for cell survival [13]. Emergence of antigen-negative metastases following DC-based vaccination has been documented in several studies [38, 64]. Another complication associated with immune system manipulation is the unintentional activation of effector responses against self-proteins. Although uncommon, autoimmune responses have occurred following immunotherapy. Most notably, the development of vitiligo has been observed in several patients receiving DC-based vaccination for metastatic melanoma [39, 41, 46].
Tumor-induced immunosuppression remains one of the greatest challenges currently facing immunotherapy. Through elaboration of cytokines like IL-10 and TGF-β, induction of negative checkpoint regulators such as CTLA-4 and PD-L1, recruitment of immunosuppressive leukocytes, and downregulation of tumor cell immunogenicity, tumors evade immune detection, suppress DC and effector cell function, and limit the efficacy of DC-based vaccination [4, 5].
Ways to overcome limitations and future direction
The effectiveness of DC-based immunotherapy hinges on an ability to stimulate robust antigen-specific CD4+ and CD8+ effector responses that are not overcome by tumor-induced immunosuppression. Our group has shown that in a murine model of glioma, mDCs and pDCs behave differently in DC-based anti-glioma vaccines, significantly impacting the resulting antitumor immune response. These results demonstrate the importance of selecting an optimal DC subtype during vaccination development, and specifically that using natural mDCs and selectively depleting pDCs can enhance the efficacy of DC-based immunotherapy [26].
The ideal vaccine antigen is unique to malignant cells, commonly expressed between patients and within individual tumors, crucial to tumor survival, and not restricted to certain HLA subtypes. Although the molecular heterogeneity of MGs has complicated the identification of tumor-specific antigens, clinical trials targeting antigens like TRP-2, gp100, MAGE-1, HER2, and CMV pp65 have demonstrated success and continue to be investigated (NCT02366728, NCT02465268) [29, 50, 61, 64]. Peptide antigens can also be modified to increase affinity for MHC molecules, promote immune cell activation, or enhance immunogenicity, particularly for overexpressed self-antigens to which the immune system has developed tolerance [40]. Sipuleucel-T, a DC-based vaccine utilizing a recombinant antigen-GM-CSF fusion protein, has significantly advanced the treatment of metastatic prostate cancer, extending overall survival by 4.1 months [48]. Other vaccine trials focus on whole tumor materials, which allow DCs of any HLA subtype to be loaded with multiple antigens expressed within an individual tumor [28, 51–60, 62, 63]. Targeting multiple antigens or epitopes reduces tumor selection for antigen-loss variants [13]. Phase III clinical trials of DC-based vaccination utilizing autologous tumor lysate in patients with MG are currently ongoing (NCT00045968, NCT02146066).
Targeting multiple immune pathways through combination therapy has potential to induce a multi-faceted response that is more effective than DC-based vaccination alone. Combination treatment with radiotherapy and chemotherapy has been shown to enhance the efficacy of DC-based vaccination in preclinical studies and human clinical trials [57, 63, 72]. In addition to direct killing of tumor cells, these therapies stimulate immune responses that complement the antitumor effects of DC-based immunotherapy [73]. Both modalities increase tumor cell immunogenicity and susceptibility to immune-mediated destruction by upregulating the expression of MHC, costimulatory, and adhesion molecules, stress ligands, and death receptors. Through induction of DNA damage and ER stress, they stimulate a particularly inflammatory form of cell death, releasing TAAs, cytokines, chemokines, and other immune-stimulating danger signals that attract DCs, leading to the activation of adaptive immune responses [73]. Combination with other immune-modulating treatments like oncolytic virotherapy or adoptive T cell transfer has potential to further enhance immune responses to DC-based immunotherapy. Vaccine efficacy may also be improved with the addition of immunogenic adjuvants or stimulators of APC function. Imiquimod, a TLR-7 agonist, and poly-ICLC, a TLR-3 agonist, enhance DC survival and trafficking to lymphoid tissues [28, 61, 74]. IL-12, fundamental in the generation of antitumor immunity, has also been shown to augment DC-based vaccine effectiveness [55, 75]. IL-2, a potent stimulator of T cell proliferation, has shown some immunotherapeutic promise, however its effectiveness is limited by a propensity to promote Treg cell development [27, 47, 70].
Mitigating tumor-induced immunosuppression will be fundamental in the development of the next generation of DC-based vaccines. This can be accomplished through neutralization of immunosuppressive cytokines, blockade of negative regulators of T cell function, or depletion of immunosuppressive cells. Antibody-mediated inhibition of CTLA-4 has augmented antitumor responses in animal models and human clinical trials and, in combination with intratumoral IL-12, dramatically reduced Treg cell presence and increased the proportion of functional effector T cells in patients with MG [74]. However, its use is limited by systemic toxicity and life-threatening autoimmune responses secondary to unrestricted T cell activation [76]. Human clinical trials of MDX-1106, an anti-PD-1 monoclonal antibody, demonstrated evidence of clinical efficacy in the treatment of advanced metastatic cancers and exhibited a more favorable side effect profile than CTLA-4 inhibitors [77]. PD-1 inhibition continues to be investigated in clinical trials for MG (NCT02423343, NCT02529072). A recent phase I trial of simultaneous PD-1 and CTLA-4 inhibition in patients with advanced melanoma has shown significant promise as a combination therapy, with 65% of patients showing evidence of antitumor responses to vaccination and 53% of patients experiencing significant tumor regression of 80% or more [78]. A phase II clinical trial investigating simultaneous CTLA-4 and PD-1 inhibition in patients with MG is currently ongoing (NCT02794883).
Results of multiple clinical trials have established the ability of DC-based immunotherapy to induce strong antigen-specific antitumor immune responses and prolong survival in a variety of malignancies, including MG. However, these benefits are still not realized in the majority of vaccinated patients. While the induction of antitumor effector responses is an important endpoint of vaccination, fully addressing complex elements such tumor-induced immunosuppression remains a significant challenge in the development of effective immunotherapies. In addition, effect of epigenetics on immune system has to be taken into consideration to predict vaccine mediated immune activation at a personal level. Moving forward, future of DC cancer vaccination will include rewiring DC molecular pathways and targeting natural DCs both in vivo and ex vivo to generate mature activated DCs that are refractory to tumor induced immunosuppression. Ultimately, the best outcomes will likely be seen in the setting of combination therapies that generate a multi-faceted approach to tumor destruction in terms of activating the effector arm and suppressing the regulatory arm of the immune system.
Acknowledgments
This work is supported by NIH NRCDP-K12 and NINDS K08 NS092895 Grant (MD). Authors would like to thank Christopher Brown MS, for his assistance with the image illustrations.
Footnotes
Compliance with ethical standards
Conflict of interest Authors declare no conflict of interest.
References
- 1.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YG, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Timmerman JM, Levy R. Dendritic cell vaccines for cancer immunotherapy. Annu Rev Med. 1999;5:507–529. doi: 10.1146/annurev.med.50.1.507. [DOI] [PubMed] [Google Scholar]
- 3.Zou W. Immunosuppressive networks in the tumor environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near future. Int J Cancer. 2001;94(4):459–473. doi: 10.1002/ijc.1503. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 7.Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23(9):445–449. doi: 10.1016/s1471-4906(02)02281-0. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 9.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 10.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol. 1996;26(3):659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 11.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123(3):326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candolfi M, King GD, Yagiz K, Curtin JF, Mineharu Y, Muhammad AKMG, Foulad D, Kroeger KM, Barnett N, Josien R, et al. Plasmacytoid dendritic cells in the tumor microenvironment: immune targets for glioma therapeutics. Neoplasia. 2012;14(8):757–770. doi: 10.1593/neo.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neill DW, Adams S, Bhardwaj N. Manipulating dendritic cell biology for the active immunotherapy of cancer. Blood. 2004;104(8):2235–2246. doi: 10.1182/blood-2003-12-4392. [DOI] [PubMed] [Google Scholar]
- 14.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh P, Komschlies KL, Cippitelli M, Longo DL, Subleski J, Ye JP, Sica A, Young HA, Wiltrout RH, Ochoa AC. Gradual loss of T-helper 1 populations in spleen of mice during progressive tumor growth. J Natl Cancer Inst. 1995;87(19):1478–1483. doi: 10.1093/jnci/87.19.1478. [DOI] [PubMed] [Google Scholar]
- 16.Grauer OM, Nierkens S, Bennink E, Toonen LWJ, Boon L, Wesseling P, Sutmuller RPM, Adema GJ. CD4+ FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121(1):95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 17.Boon T, Coulie PG, vandenEynde Tumor antigens recognized by T cells. Immunol Today. 1997;18(6):267–268. doi: 10.1016/s0167-5699(97)80020-5. [DOI] [PubMed] [Google Scholar]
- 18.Stewart B, Wild CP. World cancer report 2014. International agency for research on cancer, WHO; Geneva: 2014. [PubMed] [Google Scholar]
- 19.Melief CJM. Cancer immunotherapy by dendritic cells. Immunity. 2008;29(3):372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalinski P, Okada H. Polarized dendritic cells as cancer vaccines: directing effector-type T cells to tumors. Semin Immunol. 2010;22(3):173–182. doi: 10.1016/j.smim.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100(1–2):216–232. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 23.Walker DG, Chuah T, Rist MJ, Pender MP. T-cell apoptosis in human glioblastoma multiforme: implications for immunotherapy. J Neuroimmunol. 2006;175(1–2):59–68. doi: 10.1016/j.jneuroim.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, Lin JW, Chute DJ, Mischel PS, Cloughesy TF, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 25.Zhai L, Lauing KL, Chang AL, Dey M, Qian J, Cheng Y, Lesniak MS, Wainwright DA. The role of IDO in brain tumor immunotherapy. J Neurooncol. 2015;123(3):395–403. doi: 10.1007/s11060-014-1687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dey M, Chang AL, Miska J, Wainwright DA, Ahmed AU, Balyasnikova IV, Pytel P, Han Y, Tobias A, Zhang LJ, et al. Dendritic cell-based vaccines that utilize myeloid rather than plasmacytoid cells offer a superior survival advantage in malignant glioma. J Immunol. 2015;195(1):367–376. doi: 10.4049/jimmunol.1401607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM. Gene expression profile correlates with T cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prins RM, Wang XY, Soto H, Young E, Lisiero DN, Fong B, Everson R, Yong WH, Lai A, Li G, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother. 2013;36(2):152–157. doi: 10.1097/CJI.0b013e3182811ae4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casares N, Arribillaga L, Sarobe P, Dotor J, de Cerio ALD, Melero I, Prieto J, Borras-Cuesta F, Lasarte JJ. CD4(+)/CD25(+) regulatory cells inhibit activation of tumor-primed CD4(+) T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J Immunol. 2003;171(11):5931–5939. doi: 10.4049/jimmunol.171.11.5931. [DOI] [PubMed] [Google Scholar]
- 31.Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174:99–106. doi: 10.1111/j.1749-6632.2009.04939.x. [DOI] [PubMed] [Google Scholar]
- 32.Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186(7):1177–1182. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liau LM, Black KL, Prins RM, Sykes SN, DiPatre PL, Cloughesy TF, Becker DP, Bronstein JM. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90(6):1115–1124. doi: 10.3171/jns.1999.90.6.1115. [DOI] [PubMed] [Google Scholar]
- 34.Heimberger AB, Archer GE, Crotty LE, McLendon RE, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery. 2002;50(1):158–164. doi: 10.1097/00006123-200201000-00024. [DOI] [PubMed] [Google Scholar]
- 35.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2(1):52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 36.Nestle FO, Alijagic S, Gilliet M, Sun YS, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 37.Murphy GP, Tjoa BA, Simmons SJ, Jarisch J, Bowers VA, Ragde H, Rogers M, Elgamal A, Kenny GM, Cobb OE, et al. Infusion of dendritic cells pulsed with HLA-A2-specific prostate-specific membrane antigen peptides: a phase II prostate cancer vaccine trial involving patients with hormone-refractory metastatic disease. Prostate. 1999;38(1):73–78. doi: 10.1002/(sici)1097-0045(19990101)38:1<73::aid-pros9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 38.Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H, Bender A, Maczek C, Schreiner D, von den Dresch P, et al. Vaccination with Mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190(11):1669–1678. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackensen A, Herbst B, Chen JL, Kohler G, Noppen C, Herr W, Spagnoli GC, Cerundolo V, Lindemann A. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int J Cancer. 2000;86(3):385–392. doi: 10.1002/(sici)1097-0215(20000501)86:3<385::aid-ijc13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 40.Fong L, Hou YF, Rivas A, Benike C, Yuen A, Fisher GA, Davis MM, Engleman EG. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. 2001;98(15):8809–8814. doi: 10.1073/pnas.141226398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61(17):6451–6458. [PubMed] [Google Scholar]
- 42.Schuler-Thurner B, Schultz ES, Berger TG, Weinlich G, Ebner S, Woerl P, Bender A, Feuerstein B, Fritsch PO, Romani N, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med. 2002;195(10):1279–1288. doi: 10.1084/jem.20012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99(5):1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 44.Ferrara A, Nonn M, Sehr P, Schreckenberger C, Pawlita M, Durst M, Schneider A, Kaufmann AM, et al. Dendritic cell-based tumor vaccine for cervical cancer II: results of a clinical pilot study in 15 individual patients. J Cancer Res Clin Oncol. 2003;129(9):521–530. doi: 10.1007/s00432-003-0463-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salcedo M, Bercovici, Taylor R, Vereecken P, Massicard S, Duriau D, Vernel-Pauillac F, Boyer A, Baron-Bodo V, Mallard E, et al. Vaccination of melanoma patients using dendritic cells loaded with an allogeneic tumor cell lysate. Cancer Immunol Immunother. 2006;55(7):819–829. doi: 10.1007/s00262-005-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palucka AK, Ueno H, Connolly J, Kerneis-Norvell F, Blanck JP, Johnston DA, Fay J, Banchereau J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8(+) T-cell immunity. J Immunother. 2006;29(5):545–557. doi: 10.1097/01.cji.0000211309.90621.8b. [DOI] [PubMed] [Google Scholar]
- 47.Wierecky J, Muller MR, Wirths S, Halder-Oehler E, Dorfel D, Schmidt SM, Hantschel M, Brugger W, Schroder S, Horder MS, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66(11):5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 48.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 49.Liau LM, Black KL, Martin NA, Sykes SN, Bronstein JM, Jouben-Steele L, Mischel PS, Belldegrun A, Cloughesy TF. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case report. Neurosurg Focus. 2000;9(6):e8. doi: 10.3171/foc.2000.9.6.9. [DOI] [PubMed] [Google Scholar]
- 50.Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, Yong WH, Incardona F, Thompson RC, Riedinger MS, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61(3):842–847. [PubMed] [Google Scholar]
- 51.Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50(7):337–344. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamanaka R, Abe T, Yajima N, Tsuchiya N, Tsuchiya N, Homma J, Kobayashi T, Narita M, Takahashi M, Tanaka R. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89(7):1172–1179. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64(14):4973–4979. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 54.Rutkowski S, DeVleeschouwer S, Kaempgen E, Wolff JEA, Kuhl J, Demaerel P, Warmuth-Metz M, Flamen P, Van Calenbergh F, Plets C, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91(9):1656–1662. doi: 10.1038/sj.bjc.6602195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, Ryan JL, Kufe DW, Ohno T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27(6):452–459. doi: 10.1097/00002371-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 56.Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, Yoshida S, Abe T, Narita M, Takahashi M, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11(11):4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 57.Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, Goldfinger D, Ng HS, Irvin D, Yu JS. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–5964. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 58.Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15(2):114–121. doi: 10.1016/j.jocn.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 59.Ardon H, Van Gool S, Lopes IS, Maes W, Sciot R, Wilms G, Demaerel P, Bitjttebier P, Claes L, Goffin J, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010;99(2):261–272. doi: 10.1007/s11060-010-0131-y. [DOI] [PubMed] [Google Scholar]
- 60.Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, Szczepiorkowski ZM, Tosteson TD, Rhodes CH, Wishart HA, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34(4):382–389. doi: 10.1097/CJI.0b013e318215e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, Mintz AH, Engh JA, Bartlett DL, Brown CK, et al. Induction of CD8(+) T-Cell responses against novel glioma–associated antigen peptides and clinical activity by vaccinations with alpha-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, Yang WK. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18(8):1048–1054. doi: 10.1016/j.jocn.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 63.Ardon H, Van Gool SW, Verschuere T, Maes W, Fieuws S, Sciot R, Wilms G, Demaerel P, Goffin J, Van Calenbergh F, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother. 2012;61(11):2033–2044. doi: 10.1007/s00262-012-1261-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang HQ, Nuno MA, Richardson JE, Fan XM, Ji JF, Chu RM, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–135. doi: 10.1007/s00262-012-1319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8(+) T cell tolerance. J Exp Med. 2002;196(12):1627–16338. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kastenmuller W, Kastenmuller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines—hope or hype? Nat Rev Immunol. 2014;14(10):705–711. doi: 10.1038/nri3727. [DOI] [PubMed] [Google Scholar]
- 67.Thurner B, Roder C, Dieckmann D, Heuer H, Kruse M, Glaser A, Keikavoussi P, Kampgen E, Bender A, Schuler G. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J Immunol Methods. 1999;223(1):1–15. doi: 10.1016/S0022-1759(98)00208-7. [DOI] [PubMed] [Google Scholar]
- 68.Schreibelt G, Benitez-Ribas D, Schuurhuis D, Lambeck AJA, van Hout-Kuijer M, Schaft N, Punt CJ, Figdor CG, Adema GJ, de Vries IJM. Commonly used prophylactic vaccines as an alternative for synthetically produced TLR ligands to mature monocyte-derived dendritic cells. Blood. 2010;116(4):564–574. doi: 10.1182/blood-2009-11-251884. [DOI] [PubMed] [Google Scholar]
- 69.Kalinski P, Muthuswamy R, Urban J. Dendritic cells in cancer immunotherapy: vaccines and combination immunotherapies. Expert Rev Vaccines. 2013;12(3):285–295. doi: 10.1586/ERV.13.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Engell-Noerregaard L, Hansen TH, Andersen MH, Straten PT, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother. 2009;58(1):1–14. doi: 10.1007/s00262-008-0568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJM. Paradigm shift in dendritic cell-based immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front Immunol. 2014 doi: 10.3389/fimmu.2014.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Derer A, Frey B, Fietkau R, Gaipl U. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol Immunother. 2016;65(7):779–786. doi: 10.1007/s00262-015-1771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nikitina EY, Gabrilovich DI. Combination of γ-irradiation and dendritic cell administration induces a potent antitumor response in tumor-bearing mice: approach to treatment of advanced stage cancer. Int J Cancer. 2001;94(6):825–833. doi: 10.1002/1097-0215(20011215)94:6<825::aid-ijc1545>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 74.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, Miller JK, Liau LM. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176(1):157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 75.Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A, Weller M, Becher B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell–mediated glioma rejection. J Exp Med. 2013;210(13):2803–2811. doi: 10.1084/jem.20130678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]