

Abstract
Background
The availability of complete genome sequences enables all the members of a gene family to be identified without limitations imposed by temporal, spatial or quantitative aspects of mRNA expression. Using the nearly completed human genome sequence, we combined in silico and experimental approaches to define the complete human nuclear receptor (NR) set. This information was used to carry out a comparative genomic study of the NR superfamily.
Results
Our analysis of the human genome identified two novel NR sequences. Both these contained stop codons within the coding regions, indicating that both are pseudogenes. One (HNF4 γ-related) contained no introns and expressed no detectable mRNA, whereas the other (FXR-related) produced mRNA at relatively high levels in testis. If translated, the latter is predicted to encode a short, non-functional protein. Our analysis indicates that there are fewer than 50 functional human NRs, dramatically fewer than in Caenorhabditis elegans and about twice as many as in Drosophila. Using the complete human NR set we made comparisons with the NR sets of C. elegans and Drosophila. Searches for the >200 NRs unique to C. elegans revealed no human homologs. The comparative analysis also revealed a Drosophila member of NR subfamily NR3, confirming an ancient metazoan origin for this subfamily.
Conclusions
This work provides the basis for new insights into the evolution and functional relationships of NR superfamily members.
Background
The complete genomic sequences of four eukaryotic organisms have been reported (Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae and Arabidopsis thaliana). These genome sequences have been used to demonstrate the utility of comparative genomics in discerning evolutionary as well as possible functional relationships within protein superfamilies [1, 2]. With the recent addition of the complete human genome sequence available within the human genome project database (>94% sequence available as of February 2001), it is now possible to begin comparative genomic studies using human superfamily members.
We are particularly interested in the nuclear receptor (NR) class of proteins. NRs appear to be restricted to metazoans, in which they have key roles in integrating the complexities of homeostasis and development [3]. In general, NRs function as transcriptional regulators, working in concert with coactivators and corepressors to activate and suppress target gene expression [4]. The transcriptional activities of about half of the NRs studied in humans are regulated by small lipophilic ligands whereas the other, so-called orphan, receptors await ligand identification. Because of their potential for modulation by exogenous compounds and their central roles in metabolism, these receptors are extremely important targets in human disease. Additionally, NRs represent possible new targets for the control of invertebrate pests in agriculture. The latter approach would be especially promising if certain classes of NRs are shown to be specific to selected invertebrate subgroups. Comparison of complete sets of NRs from evolutionarily divergent organisms should tell us more about the feasibility of these approaches as well as shed light on general phylogenetic relationships among NRs and among species.
For the past year, known NR sets have presented an interesting puzzle. When the C. elegans genome sequence was reported, over 220 NR members were found [5], and subsequent sequence releases have brought the number of predicted C. elegans NR genes to 270 (A.S., unpublished data). This dramatic increase over the number of currently published human NRs (48) led to speculation that the human NR set could also expand dramatically [6]. Surprisingly, only 21 total NRs were found in the recently reported Drosophila genome sequence [7]. An intriguing question now is whether the total set of human NRs will reflect the diversity seen in C. elegans or instead will parallel that found in Drosophila. We employed a combined bioinformatic/molecular biology approach to answer this question.
Results and discussion
We have developed a genomic sequence analysis pipeline utilizing BLAST searches [8] followed by HMMER domain analysis [9] to identify NR sequences within the human genome. Domain analysis was facilitated by the knowledge that the NR superfamily is unified by a common modular structure [9]. One hallmark structure that characterizes the family is a DNA-binding domain (DBD) characterized by two C4-type zinc fingers contained in the amino-terminal half of the proteins. A second characteristic feature, the ligand-binding domain (LBD), is found at the distal carboxyl terminus and contains a highly conserved transcriptional transactivation function (AF2) [10]. The complete known complement of human NRs was used as a query set to identify candidate novel NR sequences from public human genome databases. Identified candidate sequences were followed up with more detailed bioinformatic and, when warranted, molecular biology analysis. Using this approach, we identified two novel NR sequences. The closest homologs of these sequences were represented by FXR (NR1H4) and HNF4γ (NR2A2).
The FXR-related gene sequence (FXR-r) was mapped in silico to chromosomal position 1p13.1-1p13.3, distinct from the chromosomal location of FXR (12q23.1-20). The predicted coding sequence of FXR-r was not contiguous within the genome. A total of seven intronic gaps separated the regions of coding similarity. Interestingly, the positions of the introns within FXR-r were at the same relative positions within the coding sequence as in FXR, suggesting a close evolutionary relationship between these two sequences. The predicted coding sequence of FXR-r displayed similarity to FXR across nearly the entire length (48% sequence identity at the amino acid level) but contained multiple stop codons (Figure 1a). The sequences of multiple stop codons were confirmed by PCR amplification and subsequent sequencing of FXR-r genomic DNA fragments (see Materials and methods). Sequence analysis thus indicated that this gene does not code for a functional NR and is likely to be a pseudogene. Surprisingly, real-time quantitative PCR (RTQ-PCR) detected relatively high levels of expression of FXR-r mRNA in testis (data not shown) indicating that this gene is a transcribed pseudogene.
Figure 1.

Amino acid alignments of the novel NR sequences FXR-r and HNF4γ-r. (a) FXR-r amino acid alignment with FXR (NR1H4). The nucleotide sequences from complete and ordered clone AL358372 contained eight fragments with amino acid sequence similarity with FXR (from 5'-3' relative to FXR-r, nucleotide positions 13144-13480; 15053-15199; 21541-21672; 21781-21879; 22015-22115; 23681-23795; 24486-23795; 27041-27262). The positions of the intron gaps within FXR-r and FXR are boxed. Positions of termination codons within FXR-r are indicated by an asterisk. (b) HNF4γ-r amino acid alignment with HNF4γ (NR2A2). The complete and ordered clones AL449103 and AL138997 contained contiguous HNF4γ-r sequence. Seven frameshifts (indicated by asterisks) were required to preserve the reading frame of HNF4γ-r relative to HNF4γ. Intron locations in the HNF4γ gene are boxed.
The second novel NR gene (HNF4γ-r) was mapped in silico to chromosome position 13q14.11 - 13q14.3, unlinked to the known HNF4γ gene at position 12q12. The HNF4γ-r sequence showed sequence similarity across nearly the entire length of the coding region of HNF4γ (71.4% sequence identity at the amino acid level). Like FXR-r, HNF4γ-r coding sequence contained multiple stop codons (Figure 1b) and thus also appears to represent a pseudogene. Nine frame-shifts were necessary to maintain the amino acid reading frame relative to HNF4γ. The predicted HNF4γ-r sequence was confirmed by sequence analysis of human genomic DNA (see Materials and methods). The predicted coding sequence of HNF4γ-r was contiguous within the genome, consistent with possible retrotransposition into the genome [11]. Unlike FXR-r, no expression of HNF4γ-r mRNA was detected in any of the tissues examined (data not shown).
Only one other NR pseudogene has been reported to date, a pseudogene related to the ERRα receptor [12]. The identification of FXR-r and HNF4γ-r brings the total human NR pseudogene number to three. Further evidence that these genes are pseudogenes includes the fact that no homologs of the HNF4γ-r and FXR-r genes could be found in available mouse, rat, Fugu, or Drosophila genome sequences. Pseudogene sequences would not be expected to be conserved between genomes even as closely related as human and mouse. In addition, and in contrast to their closest functional gene homologs HNF4 and FXR, HNF4γ-r or FXR-r did not display conservation of their amino acid sequences relative to their nucleic acid sequences. This result is also consistent with the pseudogene characterization of these sequences.
FXR-r and HNF4γ-r were found using searches that utilized other known human NRs as query sequences. Certain C. elegans receptors contain LBD sequences that differ significantly from mammalian LBDs [5]. It remained possible, then, that NR LED sequences existed in the human genome that represented homologs of C. elegans NRs but were not identified using mammalian NRs as a query set. To address this question, we scanned the human genome with the complete sets of C. elegans and Drosophila NRs. Extensive analysis using these receptors as query sequences did not reveal a single novel mammalian homolog of these sequences (no BLAST hits with p value < 10). From our analysis, we conclude that the human NR set will not be expanded by orthologs resembling the large number of NRs found in C. elegans.
Phylogenetic analysis of vertebrate NRs has defined six ancestral NR subfamilies [13, 14, 15]. Five of the subfamilies are also represented among both the C. elegans and Drosophila NRs (Figure 2), consistent with the proposed ancient metazoan origin of these subfamilies [14]. The earlier analysis identified no arthropod or nematode members of the NR3 subfamily, suggesting that this subfamily might be more recently derived and specific to the deuterostome lineage [14]. More recently, however, the Drosophila genome sequence [1] has revealed a previously unknown NR sequence (CG7407) that falls into the NR3 subfamily (Figures 2,3). The observation that NR3 is represented in both protostomes and deuterostomes indicates that NR3, like the other major NR subfamilies, is of an ancient metazoan origin. Notably, the C. elegans genome does not encode a member of the NR3 subfamily. It will be of interest to learn if NR3 is represented in any nematode species, as absence of the NR3 subfamily from nematodes in general would suggest that arthropods and vertebrates may share an evolutionary history that occurred after separation of the nematode lineage. Such an early divergence of the nematode evolutionary lineage would be in disagreement with a recent hypothesis placing nematodes and arthropods in a common evolutionary clade of molting invertebrates [16] and would be more consistent with the traditional placement of nematodes in a lineage that diverged from other metazoans before separation of the major protostome and deuterostome lineages [17].
Figure 2.
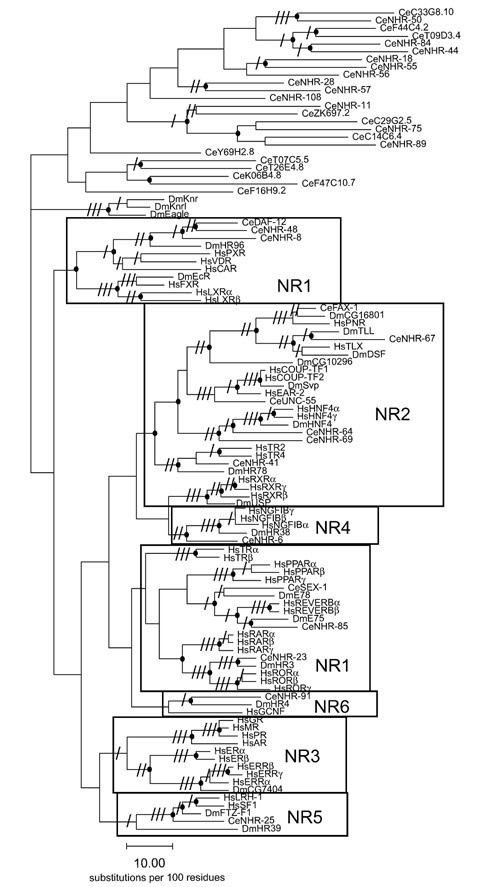
Relationships within the completed sets of NR superfamily members from humans, C. elegans and Drosophila. A neighbor-joining tree of NR DNA-binding domain sequences was generated using the paupsearch feature of the GCG 10.1 program package (1,000 bootstrap replicates, midpoint rooting); analysis methods are described in detail in Sluder et al. [5]. Significant bootstrap support values are indicated by slashes on the appropriate branches: (/) 50-79%; (//) 80-94%; (///) ≥ 95%; branches also supported by parsimony analysis are marked by dots; subfamilies are boxed. All human (Hs) and Drosophila (Dm) NRs are included, except for the two human NRs that lack a canonical DBD (DAX-1 (NR0B1) and SHP (NR0B2)). The C. elegans (Ce) sequences include all members of the six major metazoan NR subfamilies as well as selected representatives of the major groupings of divergent C. elegans NRs evident in a larger tree containing all the nematode sequences.
Figure 3.

Amino acid alignment of Drosophila CG7404 with the sequences of human ERRα, ERRβ, and ERRγ. DmCG7404 exhibits similarity to the human ERRs in both DNA- and ligand-binding domain sequences. Block boxes indicate residues identical in at least two of the four sequences; gray boxes indicate similar residues. Expression of CG7407 as mRNA and the splicing pattern has been confirmed by the recovery and sequencing of cDNA (K.K. et al., unpublished data).
Clearly, dramatically divergent evolutionary pathways have shaped the NR sets in separate phylogenetic lineages. Within the six major NR subfamilies, four groups of NRs are currently known only in vertebrates (thyroid hormone receptors (TR), peroxisome proliferator-activated receptor (PPAR), retinoic acid receptors (RAR) and the steroid receptor group containing glucocorticoid receptor (GR), mineralocorticoid receptors (MR), progesterone receptor (PR) and androgen receptors (AR)). In addition, both invertebrate genomes encode NRs that are not clearly placed in one of the six defined NR subfamilies (Figure 2). As previously noted [13], the three Drosophila members of the Knirps group define an unusual class of NRs that lack similarity to the LBDs of the vertebrate NRs. Most of the C. elegans NRs (255 of 270) are diverged from those found in humans and flies (Figure 2). It is unclear whether these divergent nematode NRs represent new subfamilies [18] or are highly diverged members of one or more of the six recognized subfamilies. In contrast to the situation with the insect Knirps group, analyses of potential structures indicates that the majority of the divergent C. elegans receptors are predicted to contain the canonical antiparallel α-helix sandwich structure characteristic of ligand-regulated NRs (A. Bogan, C. Maina, J.-M. Chandonia, F. Cohen, K. Yamamoto, and A. Sluder, unpublished data). Thus, despite the extensive diversity in sequence of many C. elegans LBD sequences, they are unified by a common structural fold, possibly reflecting the requirement for interaction with a core set of NR cofactors [19]. Since the known structures of NR LBDs contain a hydrophobic cleft in which their endogenous hormone ligands are bound, it is likely that most, if not all, orphan receptors will be amenable to modulation by small molecules.
In sum, we have found a striking difference between humans, Drosophila and C. elegans with respect to their NR sets. There is a finite possibility that the last 5% of the human genome sequence could harbor an additional novel NR sequence, but this is unlikely given that this 5% is enriched in repetitive heterochromatic sequence. Such a finding would not change the general conclusion that there are striking differences between the three genomes. Knowledge of all the members of each NR set defines the unique landscape for NR modulation and provides a basis for more detailed phylogenetic studies. Furthermore, such a whole-genome comparison of the types and numbers of genes only reflects one level of NR complexity in an organism. The impact of transcriptional and post-transcriptional processing events on total NR functional diversity in each proteome will be a subject for future studies.
Materials and methods
Computational identification of human NR sequences
Protein homology searches were performed using individual members of the NR family compared against the human genome database (HTG section from GenBank) using TBLASTN [8]. Results of all sequence hits were stored in an in-house Oracle database. Homologous sequences (those with a BLAST expectation value of 0.1 or lower) were then compared with the entire query NR data set using BLASTN [8]. By choosing the criteria on the basis of automated assessment for 95% sequence identity over 200 base pairs (bp) at the nucleotide level, we could map these hits to individual members of the protein family ('bins'). Those sequence hits that could not automatically be placed in bins on the basis of the selected criteria were either mapped to other protein families (if their sequences matched a known gene in GenBank), considered as potentially novel NR sequences (when key domains were present) or deemed irrelevant (no match to anything). We then applied Hidden Markov Models (HMMs) built on the DBD and LBD domains using the HMMER package [9] to these potential novel sequences. The HMM models were used for further substantiation or for ruling out potential novel hits.
Cross-species comparisons were performed using the following analyses. First, the HMM model for NR DBD (zf-C4.hmm from Pfam) domain was used to search the C. elegans protein database (assembled in house from GenBank) using the HMMER package [9], and identified 252 C. elegans sequences. These 252 C. elegans sequences were compared to the human genomic sequence database by the search strategy described above. No novel human NRs were found from these searches. Second, the above process was repeated for the Drosophila genome and again no novel human NR sequences were found. Third, the majority of the C. elegans NRs were grouped into ten subfamilies based on comparative sequence analysis of the DNA-binding domain sequences (A.S., data not shown). These subfamilies were used to build 10 HMMs. These HMMs were then used to search the database of predicted Drosophila proteins (established in-house by extracting from GenBank) with the HMMER package. The significant hits (score < 0.1) were compared with the 21 known Drosophila NRs. No novel Drosophila NR sequences were found.
With each new update of the human genomic database, the system initiated a new round of searching and filtering, capturing all sequences that were homologous and using a master bin list to filter out sequences that had been annotated in a previous cycle. This eliminated the need to separate out all new sequences in the database at each update. The analysis engine was written in Perl. A Java applet was used as the user's main interface to the analysis engine and the underlying Oracle database. It was embedded in an HTML page from which users could view the sequence record by linking to the in-house and public databases as well as all the sequence and domain alignments. As validation of our bioinformatic search tools, 48 of the 49 known NR sequences in the genome (including the ERR pseudogene) were found. The gene representing the CAR NR (NR1I3) is still absent from high-throughput genomic sequence databases.
Genomic DNA analysis of novel human NR sequences
Confirmation of in silico nucleotide sequence was performed by sequencing FXR-r and HNF4γ-r genomic DNA fragments amplified by PCR. PCR primers were designed using GenBank accession numbers AL358372 (FXR-r) and AL138997 (HNF4γ-r). FXR-r: 5'-GTT GAG TGG AAA TGT GAG AG -3' and 5'-GTA GGA ATG TTG GCA GAA TG -3', product size 566 bp, HNF4γ-r; 5'-AGC TTG ATT TGT AGC TGT TC -3' and 5'-TCC TCT CTG GGG CAG AA-3' product size 1,205 bp. These primers were used to amplify 100 ng Clontech genomic DNA using Amplitaq Gold (PE Biosystems) using standard PCR amplification conditions. DNA sequencing was performed by the GlaxoSmithKline core sequencing facility.
mRNA expression analysis
Human tissues were derived from snap frozen surgical specimens (Duke University, Durham, NC). Total RNA was extracted using Qiagen RNeasy kits according to the supplier's protocol. Some total RNAs were obtained from commercial vendors such as Clontech (Palo Alto, CA) and Zen-Bio (Research Triangle Park, NC). To maximize the sensitivity of the quantitation of transcripts, it was critical to ensure that there was no contaminating genomic DNA. The RNA was treated with DNase using Ambion RNase-free DNase. Effectiveness of the DNase treatment was confirmed by running PCR with human β-actin primers in the absence of reverse transcriptase, to ensure no signal was detected. The RNA was then quantitated using the RNA-specific Ribogreen dye (Molecular Probes), and 25 ng aliquoted into a 96-well plate format. To quantitate mRNA expression, RTQ-PCR was performed using an ABI PRISM 7700 Sequence Detection System instrument and software (PE Applied Biosystems) as described [20, 21]. Gene-specific primers were synthesized (Keystone, Camarillo, CA) for the sequences FXR-r (accession number AL358372) and HNF4γ-r (accession number AL138997).
References
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- Sluder AE, Mathews SW, Hough D, Yin VP, Maina CV. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9:103–120. [PubMed] [Google Scholar]
- Enmark E, Gustafsson J. Nematode genome sequence dramatically extends the nuclear receptor superfamily. Trends Pharmacol Sci. 2000;21:85–87. doi: 10.1016/s0165-6147(99)01417-0. [DOI] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Hidden Markov models. Curr Opin Struct Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11:1025–1033. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchenio T, Segal-Bendirdjian E, Heidmann T. Generation of processed pseudogenes in murine cells. EMBO J. 1993;12:1487–1497. doi: 10.1002/j.1460-2075.1993.tb05792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Beatty B, Squire J, Copeland NG, Gilbert DJ, Jenkins NA, Giguere V. Chromosomal mapping of the human and murine orphan receptors ERRalpha (ESRRA) and ERRbeta (ESRRB) and identification of a novel human ERRalpha-related pseudogene. Genomics. 1997;45:320–326. doi: 10.1006/geno.1997.4939. [DOI] [PubMed] [Google Scholar]
- Laudet V, Hanni C, Coll J, Catzeflis F, Stehelin D. Evolution of the nuclear receptor gene superfamily. EMBO J. 1992;11:1003–1013. doi: 10.1002/j.1460-2075.1992.tb05139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudet V. Evolution of the nuclear receptor superfamily: early diversification from an ancestral orphan receptor. J Mol Endocrinol. 1997;19:207–226. doi: 10.1677/jme.0.0190207. [DOI] [PubMed] [Google Scholar]
- Escriva H, Safi R, Hanni C, Langlois MC, Saumitou-Laprade P, Stehelin D, Capron A, Pierce R, Laudet V. Ligand binding was acquired during evolution of nuclear receptors. Proc Natl Acad Sci USA. 1997;94:6803–6308. doi: 10.1073/pnas.94.13.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387:489–493. doi: 10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- Fitch DHA, Thomas WK. Evolution. In C elegans II Edited by Riddle DL, Blumenthal T, Meyer BJ and Priess JR Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Pres 1997, pp. 815–850.
- The Nuclear Receptors Committee A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- Robyr D, Wolffe AP, Wahli W. Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol Endocrinol. 2000;14:329–347. doi: 10.1210/mend.14.3.0411. [DOI] [PubMed] [Google Scholar]
- Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]