

Abstract
The essential biological roles played by glycosidases, coupled to the diverse therapeutic benefits of pharmacologically targeting these enzymes, provide considerable motivation for the development of new inhibitor classes. Cyclophellitol epoxides and aziridines are recently established covalent glycosidase inactivators. Inspired by the application of cyclic sulfates as electrophilic equivalents of epoxides in organic synthesis, we sought to test whether cyclophellitol cyclosulfates would similarly act as irreversible glycosidase inhibitors. Here we present the synthesis, conformational analysis, and application of novel 1,6-cyclophellitol cyclosulfates. We show that 1,6-epi-cyclophellitol cyclosulfate (α-cyclosulfate) is a rapidly reacting α-glucosidase inhibitor whose 4C1 chair conformation matches that adopted by α-glucosidase Michaelis complexes. The 1,6-cyclophellitol cyclosulfate (β-cyclosulfate) reacts more slowly, likely reflecting its conformational restrictions. Selective glycosidase inhibitors are invaluable as mechanistic probes and therapeutic agents, and we propose cyclophellitol cyclosulfates as a valuable new class of carbohydrate mimetics for application in these directions.
Short abstract
1,6-epi-Cyclophellitol cyclosulfate (“α-cyclosulfate”) is a conceptually new, potent and selective irreversible α-glucosidase inhibitor that acts through mimicry of the α-glucosidase Michaelis complex 4C1 chair conformation.
Introduction
The huge diversity of oligosaccharide, polysaccharide, and glycoconjugate structures found in nature reflect their many roles in biological processes. Such diversity is mirrored in the numerous enzymes that have evolved to degrade these biopolymers. Hydrolytic carbohydrate degrading enzymes, glycoside hydrolases or glycosidases, are classified into over 140 distinct sequence (and hence structural) families in the CAZy database.1 This classification provides a powerful framework upon which aspects of conformational analysis and enzyme inhibition can be constructed.
Most retaining glycosidases employ a conserved mechanism, in which the acetals (occasionally ketals) that make up interglycosidic linkages are hydrolyzed using two key carboxylic acid residues (Asp or Glu), which function as catalytic nucleophile and catalytic acid/base.2 Central to glycosidase activity are the considerable conformational distortions undergone by the sugar substrate throughout the catalytic cycle, which are necessary to satisfy the stereoelectronic and orbital overlap requirements for glycoside hydrolysis.3
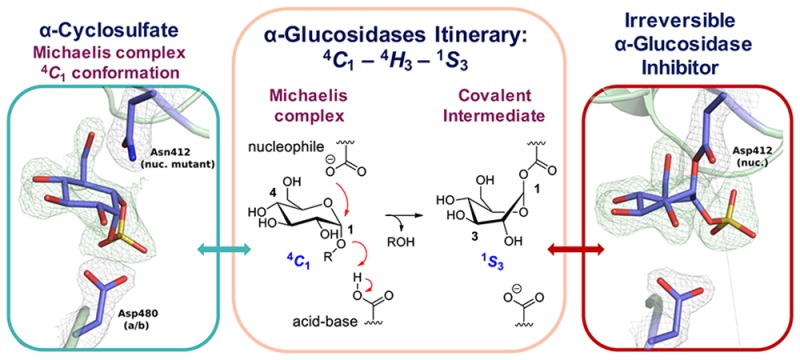
Upon binding to the enzyme, a retaining glycosidase substrate in the Michaelis complex initially adopts a conformation in which the leaving group is axially or pseudoaxially positioned, allowing for favorable in-line attack by the catalytic nucleophile. Nucleophilic attack, with concurrent protonation of the glycosidic oxygen by the catalytic acid/base, leads to a high-energy state (the transition state, TS) with considerable oxocarbenium ion character. This transition state must necessarily adopt a conformation in which C5–O5–C1–C2 are coplanar, to allow for partial oxocarbenium double bond development between O5 and C1. Following this transition state, the substrate forms a covalent intermediate with the glycosidase nucleophile, which is intercepted by water (following the reverse conformational itinerary) to liberate free sugar and enzyme. For retaining α- and β-glucosidases, the typical conformational itineraries for the Michaelis complex → TS → intermediate enzymatic half reactions are 4C1 → 4H3⧧ → 1S3 and 1S3 → 4H3⧧ → 4C1 respectively (Figure 1a), with the 4H3 TS conformation common to both α- and β-glucosidases.4−7
Figure 1.

Conformational itinerary of retaining glucosidase reaction pathways and conformation of covalent inhibitors. (a) Reaction itineraries of retaining β-glucosidases and retaining α-glucosidases, showing conformations of the Michaelis complex, transition state, and covalent intermediates. (b) Structures of cyclophellitol (1), cyclophellitol aziridine (2), 1,6-epi-cyclophellitol (3), 1,6-epi-cyclophellitol aziridine (4), α-cyclosulfate (5), and β-cyclosulfate (6).
Mimicry of transition state features is a powerful strategy for the design of enzyme inhibitors, and thus the majority of competitive glycosidase inhibitors bind by virtue of mimicking either the positive charge or the conformational shape of the transition state.3,8,9 Conformational mimicry can also aid the reaction of irreversible covalent glycosidase inhibitors,10 such as cyclitol epoxides and aziridines.10−12 The natural product, cyclophellitol (1), and its synthetic analogue, cyclophellitol aziridine (2), are glucose configured inhibitors, which mimic the 4H3 conformation adopted by retaining β-glucosidase substrates in their transition state.3,13,14 Upon binding to the enzyme active site, nucleophilic attack by the catalytic nucleophile residue opens the epoxide or aziridine ring, irreversibly inhibiting the enzyme via formation of a covalent enzyme–cyclophellitol (aziridine) adduct.
In general, reactive cyclophellitol analogues, locked into TS-like conformations, are rapid irreversible inactivators of retaining glycosidases.12,15,16 We recently demonstrated that replacement of the cyclophellitol epoxide or aziridine ring by a nonhydrolyzable cyclopropane also yields reversible glycosidase inhibitors with micromolar to nanomolar affinity.9 Thus, conformation, rather than just electrophilic reactivity, may make a substantial contribution to the inhibitory potency of cyclophellitol analogues.
Given the success of reactive sugar epoxides and aziridines as conformationally enhanced retaining glycosidase inactivators,10,12,17 we sought to expand the concept to other electrophilic species. Cyclic sulfate electrophiles are often used in synthetic organic chemistry as equivalents of epoxides in nucleophilic additions, sometimes showing superior reactivity and regioselectivity.18 We therefore sought to test if cyclophellitol configured cyclosulfates could act as a new class of covalent glycosidase inhibitors. Specificity for “α” and “β” enzymes can be conferred simply through choice of the appropriately configured cyclosulfate. We additionally reasoned that substitution of the epoxide in 1 and 3 by a 1,2-cis-cyclic sulfate should yield compounds favoring a 4C1 conformation. “α-Configured” 1,6-epi-cyclophellitol cyclosulfate 5 (hereafter referred to as α-cyclosulfate) should match, conformationally, the 4C1 Michaelis complex conformation of substrates for α-glucosidases and thus be readily poised for in-line nucleophilic attack by the enzyme, yielding a rapid and potent irreversible inhibitor. In contrast, a “β-configured” 1,6-cyclophellitol cyclosulfate 6 (β-cyclosulfate) locked into a 4C1 conformation would likely disfavor reactivity on retaining β-glucosidases, whose Michaelis complex is 1S3.
Here, we present the synthesis of a panel of 1,6-cyclophellitol cyclosulfates which favor 4C1 chair conformations. α-Cyclosulfate 5 irreversibly inhibits retaining α-glucosidases with affinity on a par with 1,6-epi-Cyclophellitol aziridine 4, whereas β-cyclosulfate 6 is a substantially weaker β-glucosidase inhibitor compared to its cognate aziridine 2. 3-D structures of covalent adducts from both “α” and “β” cyclosulfates, bound to representative α- and β-glucosidases, combined with inhibitory kinetic studies and competitive activity based protein profiling (ABPP), demonstrate the high specificity and active center–nucleophile reactivity of these molecules.
The development of selective inhibitors of carbohydrate processing enzymes is of major interest both for the understanding of biological processes involved in glycan processing and in the discovery of new therapeutics.19 Only minor advances have been observed regarding the development of new irreversible glycosidase inhibitors since the discovery of cyclophellitol20 (aziridines)21 or 5-fluoro-22 and 2-deoxy-2-fluoro-23 glycosides, the latter some 30 years ago.24 Cyclophellitol cyclosulfates thus provide a conceptually novel and powerful tool for further study of glycosidases in health and disease.
Results and Discussion
Conformational Free Energy Landscapes of Cyclophellitol Cyclosulfates
Cyclophellitol cyclosulfates 5 and 6 were conceived as putative inhibitors of retaining glucosidases. To determine the likely conformations of these molecules, we first calculated their conformational free energy landscapes (FELs) using ab initio metadynamics (Supporting Information).25 For both 5 and 6, the calculated ground state conformation was centered at 4C1, with a wide energy minimum expanding toward the 2H3–E3–4H3 region (Figure 2a).
Figure 2.
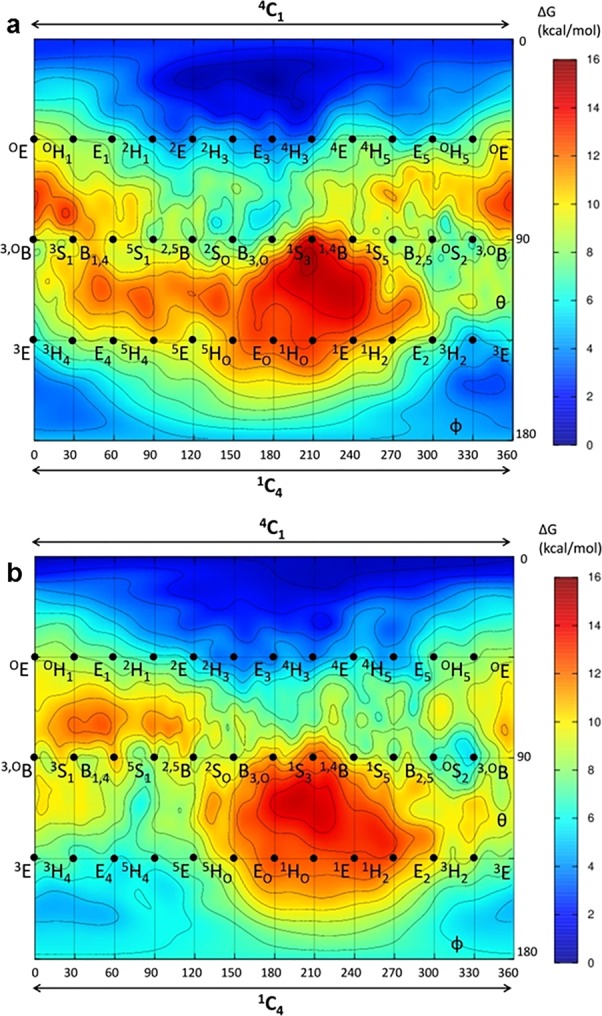
Conformational free energy landscapes of cyclosulfates 5 and 6. Cyclosulfates 5 (a) and 6 (b) adopt 4C1 ground state conformations, with a broad energy minimum extending toward 4H3. The x and y axes of each graph correspond to the φ and θ Cremer–Pople puckering coordinates (in degrees), respectively. Isolines are 1 kcal/mol.
We reasoned that the conformational preference of 5 should render it a potent inactivator of α-glucosidases, as its 4C1 conformation matches that of a typical α-glucosidase Michaelis complex, with the 4H3 TS conformation also energetically accessible. In contrast, 6 should not be able to access the 1S3 β-glucosidase Michaelis complex conformation, although the 4H3 TS conformation could still be accessible. Thus, while 6 may still inhibit β-glucosidases, it should do so with less potency compared to 5 vs α-glucosidases.
Synthesis of Cyclic Sulfates 5 and 6
Key intermediate 7 was synthesized from d-xylose in nine steps as optimized in our group based on the total synthesis of cyclophellitol 1 by Madsen and co-workers (Figure 3a).26,27 Benzylation of the free alcohols in 7 yielded alkene 8, which was oxidized (ruthenium trichloride/sodium periodate) to afford a mixture of cis diols 9 and 10. Compound 10, emulating α-glucopyranosides in configuration, was obtained pure after silica gel column chromatography, whereas β-analogue 9 necessitated recrystallization from methanol and diethyl ether in order to obtain homogeneous material. Generation of the cyclic sulfites by treatment of thionyl chloride and subsequent oxidation gave perbenzylated cyclic sulfates 11 and 12, the benzyl ethers of which were removed by hydrogenolysis using catalytic palladium on carbon to afford compounds 5 and 6. In line with FEL calculations, analysis of experimental J-coupling values for compounds 5 and 6 showed that these analogues indeed adopt a 4C1 conformation in solution (Table S1).
Figure 3.

Synthesis of cyclophellitol cyclosulfates 5 and 6, and inactivation of GAA by compound 5. (a) Compounds 5 and 6 were prepared from cyclohexene 11. Reagents and conditions: (i) BnBr, TBAI, NaH, DMF, rt, 18 h, 70%; (ii) RuCl3, NaIO4, EtOAc, ACN, 0 °C, 2 h (13, 39%; 14, 26%); (iii) (a) SOCl2, Et3N, DCM, 0 °C, (b) RuCl3, NaIO4, CCl4, ACN, 0 °C, 3 h (15, 59%; 16, 62%); (iv) H2, Pd/C, MeOH, rt, 18 h (5, 71%; 6, 72%). (b) Semilogarithmic plots of residual activity of GAA versus time at 9, 8, 7, 6, 5, 4, 3, and 2 μM α-cyclosulfate 5. (c) Plot of pseudo first order rate constants from panel c vs concentration of 5.
In Vitro Inhibition of α- and β-Glucosidases and Kinetic Studies
To establish the activity of cyclosulfates 5 and 6 in relation to their calculated conformations, we assessed their ability to inhibit representative α- and β-glucosidases compared to cyclophellitol epoxides and aziridines 1–4, utilizing both purified glycosidases and human cell/tissue lysates.
We first tested inhibition against human β-glucosidases GBA1 (recombinant Cerezyme protein from Genzyme, classified into the CAZy glycoside hydrolase family GH30) and GBA2 (GBA2 overexpressing HEK293T lysate, family GH116), and α-glucosidases GAA (recombinant Myozyme from Genzyme, family GH31) and GANAB (Pompe disease fibroblast lysates, family GH31; Table 1). Consistent with our previous findings, cyclophellitol 1 and cyclophellitol aziridine 2 were potent inhibitors of GBA1 and GBA2, and 1,6-epi-cyclophellitol 3 and 1,6-epi-cyclophellitol aziridine 4 inhibited GAA and GANAB.10,17,28 In line with our predictions regarding Michaelis complex mimics as strong retaining glycosidase inhibitors, α-cyclosulfate 5 proved to be a potent nanomolar α-glucosidase inhibitor (IC50 82 nM vs GAA; 29 nM vs GANAB) with no reactivity toward β-glucosidases. In contrast, β-cyclosulfate 6 was only a modest inhibitor of β-glucosidases (IC50 119 μM for GBA1 and 58 μM for GBA2). IC50s determined against representative recombinant bacterial glucosidases (β-glucosidases from Thermotoga maritima, TmGH1,3,29 and Thermoanaerobacterium xylanolyticum, TxGH116;30 α-glucosidase from Cellvibrio japonicus, CjAgd31B10,31) showed the same trend as for the human enzymes: strong inhibition by 5, and poor inhibition by 6 (Table S2). To confirm that 5 and 6 were stable under the acidic conditions utilized for these enzymatic assays, the solvolytic stability of these compounds in McIlvaine buffer pH 4.0 was assessed by NMR. We observed no spontaneous hydrolysis after 24 h for either 5 or 6 (Figure S2).
Table 1. Enzyme Inhibition Efficacy of Compounds 1–6a.
| IC50 |
||||
|---|---|---|---|---|
| β-glucosidase |
α-glucosidase |
|||
| compd | GBA1 | GBA2 | GAA | GANAB |
| 1 | 165 ± 4 nM | 139 ± 60 nM | >100 μM | >100 μM |
| 2 | 304 ± 5 nM | 14 ± 1 nM | >100 μM | >100 μM |
| 3 | >100 μM | >100 μM | 15 ± 2 μM | >100 μM |
| 4 | 21 ± 1 μM | >100 μM | 38 ± 3 nM | 1.4 ± 0.1 μM |
| 5 | >100 μM | >100 μM | 82 ± 1 nM | 29 ± 2 nM |
| 6 | 119 ± 9.8 μM | 58 ± 4.5 μM | >100 μM | >100 μM |
Apparent IC50 values for in vitro inhibition of GBA1, GBA2, GAA, and GANAB. Reported values are mean ± standard deviation from 3 technical replicates.
We next determined kinetic parameters of inhibition by 1–6 against recombinant human GBA1 and GAA, as well as bacterial β-glucosidase TmGH1 (Table 2, Figure S3).32 Epoxides 1 and 3 irreversibly inhibited GBA and GAA, with initial binding constants (KI) of 9.2 μM and 1.4 mM, and inactivation rate constants (kinact) of 0.7 and 0.5 min–1 respectively. Consistent with their greater reactivity, aziridines 2 and 4 inhibited GBA and GAA considerably faster than epoxides (full inhibition typically within 30 s at higher concentrations), limiting us to measuring a combined kinact/KI ratio for these molecules.
Table 2. Kinetic Parameters of Compounds 1–6a.
| β-glucosidase
GBA1 |
α-glucosidase
GAA |
|||||
|---|---|---|---|---|---|---|
| compd | kinact (min–1) | KI | kinact/KI (min–1 mM–1) | kinact (min–1) | KI | kinact/KI (min–1 mM–1) |
| 1 | 0.72 | 9.2 μM | 77.7 | >500 μM | ||
| 2 | nd | nd | 19.0 | 9.6 μM* | ||
| 3 | >500 μM | 0.52 | 1.4 mM | 0.37 | ||
| 4 | 6.8 μM* | nd | nd | 58.0 | ||
| 5 | >500 μM | nd | nd | 64.3 | ||
| 6 | 0.015 | 3.6 mM | 4.3 × 10–3 | >500 μM | ||
Inactivation rates and inhibition constants (kinact and KI) for human recombinant β-glucosidase GBA1 and α-glucosidase GAA; nd, not determined due to fast inhibition; *, reversible inhibition observed.
α-Cyclosulfate 5 displayed rapid pseudo first order inhibition kinetics against GAA (kinact/KI = 64 min–1 mM–1), comparable to 1,6-epi-cyclophellitol aziridine 4 (Figure 3b,c, Table 2), illustrating the potency of α-configured cyclosulfates locked in a Michaelis complex conformation favoring nucleophilic interception. Conversely, irreversible inhibition of GBA1 by 6 was several orders of magnitude slower (KI = 3.6 mM; kinact = 0.015 min–1; kinact/KI = 4.33 × 10–3 min–1 mM–1), suggesting a limited ability of this molecule to adopt the 1S3 conformation required for nucleophilic attack and β-glucosidase reactivity. Comparison of KI values for 6 and 1 against GBA1 also showed initial binding of 6 to the enzyme to be substantially weaker compared to 1 (3.5 mM and 9 μM respectively). These values are consistent with the tighter binding affinities often displayed by TS shape analogues compared to molecules locked into other conformations.
Interestingly, while irreversible inhibition by aziridines 2 and 4 was largely specific toward α- or β-glucosidases respectively, these molecules were also observed to be less specific reversible inhibitors for glucosidases of the opposite anomeric specificity (competitive inhibition of GAA by 2, KI = 9.6 μM; competitive inhibition of GBA1 by 4, KI = 6.8 μM; Table 2). We reason that the common 4H3 transition state utilized by both α- and β-glucosidases may allow these enzymes to bind aziridines of either “anomeric” configuration. Thus, the conformational preference of cyclophellitol cyclosulfates presents an advantage in specificity when compared to aziridine type inhibitors, by virtue of mimicking a reactive conformation receptive to nucleophilic attack, not shared between α- or β-glucosidases.
Structural Characterization of Enzyme–Cyclosulfate Interactions
Because of the key role of conformation in the contrasting inhibitory potency of cyclophellitol cyclosulfates against α- vs β-glucosidases, we set out to structurally characterize the conformational itinerary of 5 and 6 when reacted with representative bacterial glucosidases.
Crystal structures of 5 in complex with the GH31 α-glucosidase/tranglycosidase CjAgd31B from Cellvibrio japonicus(10,31) were obtained by soaking crystals of wild type CjAgd31B with 5. We observed density for a covalent adduct bound to the enzyme nucleophile (Asp412) corresponding to ring opened cyclosulfate in a 1S3 conformation (Figure 4a), matching the conformation previously observed for reacted aziridine 4,10 as well as a complex of CjAgd31B reacted with 5-fluoro-α-glucosyl fluoride31 (Figure 4b; PDB codes 5I24 and 4BA0 respectively). Structures of unreacted 5 were also obtained in complex with an inactive D412N nucleophile mutant of CjAgd31B. Density for unreacted 5 in the CjAgd31B active site matched a 4C1 conformation as predicted by FEL calculations and NMR analysis (Figure 4a), demonstrating that 5 indeed binds to α-glucosidases as a conformational Michaelis complex analogue, allowing unimpeded access for in-line attack by the enzyme nucleophile. Difference density in the active site of CjAgd31B D412N after soaking with 4 corresponded to an unreacted cyclophellitol aziridine in 4H3 conformation (Figure 4b), consistent with this molecule acting as a TS shape mimic when unreacted.
Figure 4.
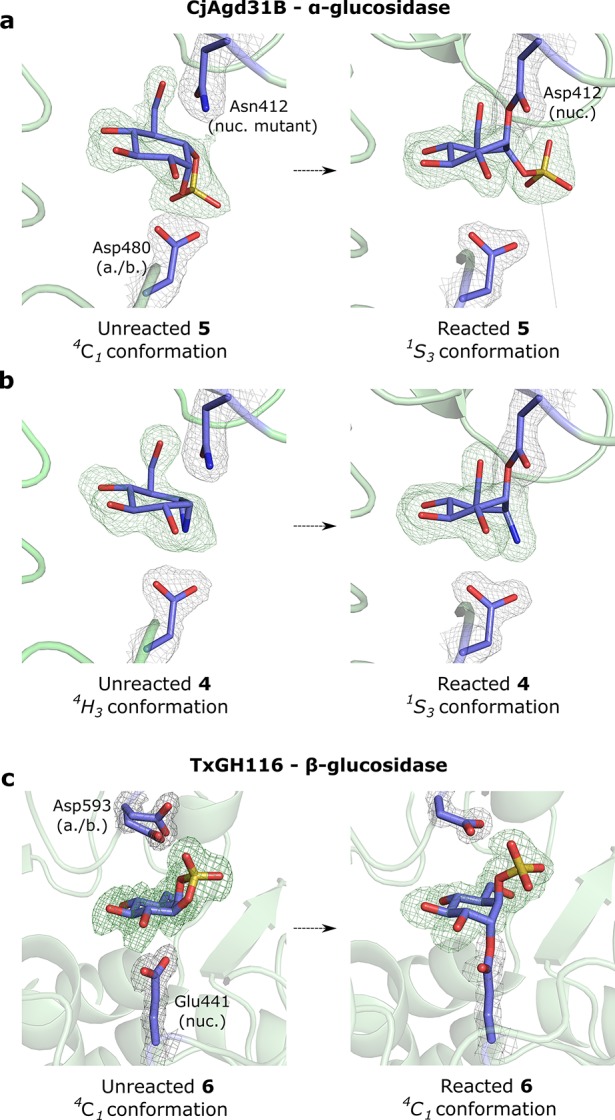
Structures of reacted 4 and 5 bound to wild type CjAgd31B and reacted 6 bound to wild type TxGH116. (a) Unreacted (left) and reacted (right) 5 in complex with CjAgd31B D412N nucleophile mutant and wt CjAgd31B respectively. Unreacted 5 adopts a 4C1 ring conformation in the active site of CjAgd31B, mimicking the Michaelis complex conformation of GH31 α-glucosidase substrates. Reacted 5 adopts a 1S3 covalent intermediate conformation. (b) Unreacted (left) and reacted (right) 4 in complex with CjAgd31B D412N nucleophile mutant and wt CjAgd31B respectively. Unreacted 4 adopts a 4H3 TS conformation. Reacted 4 adopts the same 1S3 intermediate conformation as observed for 5. (c) Unreacted (left) and reacted (right) β-cyclosulfate 6 with TxGH116. Unreacted 6 in complex with GH116 β-glucosidase TxGH116 adopts a 4C1 conformation in the enzyme active site, which is poorly poised for attack by the enzyme nucleophile Glu441, and thus reacts extremely slowly. (Two conformations for the enzyme catalytic acid/base Asp593 can be seen in this complex.) Reacted 6 can be observed after extended soaking, and also adopts a 4C1 conformation, covalently bound to the enzyme nucleophile. Electron density for protein side chains is REFMAC maximum-likelihood/σA-weighted 2Fo – Fc contoured to 0.44–0.51 and 0.40–0.42 electron/Å3 for CjAgd31B and TxGH116 complexes, respectively. Electron density for ligand is Fo – Fc maps calculated just prior to building in ligand, contoured to 0.17–0.26 and 0.27–0.35 electron/Å3 for CjAgd31B and TxGH116 complexes, respectively. nuc. = nucleophile; a./b. = acid/base.
We then utilized the β-glucosidase from Thermoanaerobactrium xylanolyticum TxGH116 for structural studies with 6. Due to its slow reactivity as an irreversible β-glucosidase inhibitor, we were able to capture an unreacted complex of 6 with crystals of wild type TxGH116 by utilizing shorter ligand soaking times. After 10–20 min soaks, a single molecule of unreacted 6 was observed within the active site of TxGH116, adopting a 4C1 conformation in line with FEL calculations and NMR spectra. This 4C1 conformation renders the equatorial C1–O bond of 6 poorly poised for in-line attack by the enzyme nucleophile Glu441, thus providing a structural rationale for the slow reactivity of this inhibitor against β-glucosidases. Extended ligand soaking times (∼24 h) were required to produce a reacted covalent complex of 6 with TxGH116. Reacted 6 was observed bound to the TxGH116 catalytic nucleophile (Glu540) in a 4C1 covalent intermediate conformation, consistent with the typical retaining β-glucosidase conformational itinerary utilized by this enzyme and matching the 4C1 conformation previously observed for reacted 2-deoxy-2-fluoroglucoside (PDB code 5BX2).
We also investigated the structural basis for “off-target” inhibition by cyclophellitol aziridines, which we had observed to competitively inhibit glucosidases of “opposing” anomeric specificity. A crystal structure of 2 in complex with CjAgd31B showed a single molecule of inhibitor occupying the enzyme active site, with the aziridine nitrogen H-bonding to the catalytic nucleophile carboxylate (∼2.6 Å between aziridine N and Asp O Oδ; Figure S5). The presence of an unreacted aziridine between the nucleophile and the plane of the cyclitol ring induces an E3 conformation for the inhibitor, rather than the 4H3 observed for an unreacted “correct” aziridine (Figure 4b). It is likely that the competitive inhibition of α-glucosidases by 4 is based around a similar interaction between the unreacted aziridine nitrogen and catalytic nucleophile.
Competitive ABPP of α-Glucosidases by Cyclosulfate 5
To assess the utility of cyclosulfates as enzyme inhibitors in complex biological samples, we examined the activity and selectivity of α-cyclosulfate 5 by competitive activity-based protein profiling (ABBP), against 13(10) (Figure S1), a fluorescent retaining α-glucosidase aziridine probe which labels GAA at pH 4 (isoforms at 70 and 76 kDa) and both isoforms of GANAB (∼100 kDa) at pH 7.
We first preincubated fibroblast lysates with varying concentrations (1, 0.3, 0.1, and 0.03 μM) of 5 for 15, 30, or 60 min at pH 4 or pH 7, and subsequently labeled with 13 (1 μM) for 30 min. SDS–PAGE followed by fluorescence scanning showed time dependent inhibition of both GAA and GANAB by 5 at pH 4 and 7 respectively (Figure 5a). Competitive ABPP in GBA2 and GBA3 overexpressing HEK293T lysates (GBA2+ and GBA3+) against β-glucosidase probe 14 (Figure S1) showed no inhibition of retaining β-glucosidases by 5, demonstrating the high selectivity of this inhibitor class (Figure S5).
Figure 5.

In vitro and in situ inhibition of GAA and GANAB. (a) 5 inhibits labeling of GAA and GANAB by fluorescent ABP 13 in fibroblast lysates in a concentration and time dependent manner. (b) 5 inhibits labeling of several α-glucosidases in mouse intestine lysate by 13. Sucrase-isomaltase (Sis), maltase-glucoamylase (MGAM), GAA, and GANAB are labeled by 13, as well as some off-target β-glucosidase labeling (LPH and GBA). α-Glucosidase labeling can be abrogated by preincubation with 5, while β-glucosidase labeling persists, demonstrating the superior selectivity of 5 compared to 13. (c) In situ inhibition of GAA and GANAB in fibroblasts at pH 4.0 and 7.0 respectively by 5 at incubation times of 2 and 24 h, followed by labeling of GAA and GANAB by cyclophellitol aziridine Cy5 probe 13. (d) Apparent IC50s for in situ inhibition of GAA and GANAB enzyme activity by 5. Reported IC50s are mean ± standard deviation from two biological replicates, each with three technical replicates.
Acarbose, miglitol, and voglibose are α-glucosidase inhibitors (AGIs) widely used in diabetes mellitus type II patients. These inhibitors delay the absorption of carbohydrates, decrease postprandial hyperglycemia and hyperinsulinemia, and thus improve insulin sensitivity and release stress on beta cells.33−35 Prompted by the potential of AGIs as leads for diabetes mellitus type II drug development, we investigated the inhibition of a group of key metabolic α-glucosidases in murine gastrointestinal tract tissues using competitive ABPP. Our studies showed that, in addition to GAA and GANAB, 5 inhibited two α-glucosidases expressed specifically in intestinal tissue: sucrase-isomaltase (SIS) and maltase-glycoamylase (MGAM) (Figure 5b). Significantly, β-glucosidases lactase-phlorizin hydrolase (LPH) and GBA1, which are labeled in an off-target fashion by 13, are not outcompeted by preincubation with 5, clearly demonstrating superior selectivity of cyclosulfate inhibitors compared to aziridines.
Lastly, we investigated whether α-cyclosulfate 5 is able to cross the cell membrane. Human fibroblasts were exposed to varying concentrations (10, 1, 0.1, 0.01, 0.001, and 0 μM) of 5 for 2 and 24 h, followed by extensive washing and lysate preparation. Lysates were incubated with 13 (0.5 μM) for 30 min and then separated by SDS–PAGE, followed by fluorescence scanning. Time dependent abrogation of ABP 13 fluorescence was observed, illustrating that 5 could cross the cell membrane to act in situ as α-glucosidase inhibitor (Figure 5c). Loss of ABP labeling was also accompanied by a corresponding loss of α-glucosidase enzymatic activity in harvested fibroblast lysates. Activity assays utilizing 4-methylumbelliferyl-α-glucose suggested robust in situ inhibition of both GAA and GANAB by 5, with IC50s of 3.0 and 1.6 μM respectively (Figure 5d).
Conclusion
In this study, we have presented a new class of irreversible glycosidase inhibitors: cyclophellitol cyclosulfates. We have shown that their irreversible action is specific for α- and β-glucosidases in a manner reflecting both the “anomeric” stereochemistry of the cyclophellitol cyclosulfates and the conformational itinerary of the target enzyme.
We have demonstrated, through ab initio metadynamics approaches and 3-D structures of enzyme ligand complexes, that unreacted α-cyclosulfate 5 favors a 4C1 “Michaelis complex like” conformation which is perfectly poised for nucleophilic attack by α-glucosidases utilizing a 4C1 → 4H3 → 1S3 glycosylation itinerary. Thus, 5 is a nanomolar mechanism-based retaining α-glucosidase inactivator, which follows pseudo first order kinetics, and displays superior selectivity compared to existing mechanism-based inhibitors due to its unique conformational behavior. We have also shown that 5 inhibits the intestinal retaining α-glucosidases SIS, MGAM, GAA, and GANAB, without affecting β-glucosidases intestinal LPH or GBA1. Thus, 5, which is also able to cross the cell membrane, may be a good starting point for the development of agents for the treatment of type II diabetes.
β-Cyclosulfate 6 also adopts a 4C1 conformation in its unreacted state, which does not match the 1S3 conformation of typical β-glucosidase Michaelis complexes. When applied to retaining β-glucosidases following a 1S3 → 4H3 → 4C1 conformational itinerary, 6 reacts extremely slowly compared to its congener 5.
FEL calculations show that the ground state conformation of 6 is centered around 4C1, a conformation with an equatorial C1–O bond that precludes in-line attack from the nucleophile. Crystal structures of unreacted 6 in complex with β-glucosidase TxGH116 show that the β-cyclosulfate continues to adopt this 4C1 conformation even within the enzyme active site. However, FEL calculations also suggest that it is energetically possible for 6 to access a TS-like 4H3 conformation. Transient sampling of this TS conformation may allow for occasional reaction of 6 with β-glucosidases, as the cyclosulfate adopts a conformation better suited to nucleophilic attack by the enzyme. However, the slow rate of irreversible inhibition observed for 6 suggests that its predominant conformation within the enzyme active site is one where nucleophilic attack from the enzyme is disfavored.
The last 10–20 years have provided us a deep appreciation of reaction coordinates of diverse glycosidases.6,7,10,36 This conformational canvas inspires the design and application of chemical species to mimic specific species along the reaction coordinate. Much existing work has focused on the design of noncovalent transition state mimics, or conformationally restricted species that mimic a species along only certain reaction trajectories, such as the specific inhibition of GH47 α-mannosidases by kifunensine.37,38 Recent years have, however, seen a resurgence in the development of irreversible enzyme inhibitors as clinical probes and diagnostics.10,11,16,39 With such applications in mind, the development of conformationally restrained irreversible inhibitors primed for nucleophilic attack through mimicry of the Michaelis complex, coupled to more electrophilic reaction centers, inspires creation of conceptually new, potent and selective glycosidase inhibitors. We envision that our design strategy will also be applied for other glycosidases, both those following a similar 4C1 → 4H3 → 1S3 conformational itinerary and those following other conformational trajectories, guided by the conformational preference of the reaction pathway.
Materials and Methods
All chemicals were obtained from Sigma-Aldrich, unless otherwise stated. Pompe disease fibroblasts, HEK293T cells (ATCC-CRL-3216), and normal fibroblast cell lines were obtained from the American Type Culture Collection (ATCC) and transfected for GBA2 and GBA3 overexpression (Supporting Information). Cell lines were cultured in DMEM/F-12 (Ham) medium (Invitrogen) supplemented with 10% (v/v) fetal calf serum (FCS; Sigma) and 1% penicillin/streptomycin (Sigma). Mouse tissues were isolated according to guidelines approved by the ethical committee of Leiden University (DEC#13191). Human recombinant enzymes rGBA1 (Cerezyme) and rGAA (Myozyme) were donated by Genzyme, GBA2 was overexpressed in HEK293T lysates, and GANAB was obtained from fibroblasts of Pompe patients diagnosed on the basis of absence of GAA (Supporting Information). Bacterial enzymes TmGH1,29TxGH116,30 and CjAgd31B31 were expressed as previously described. All cell or tissue lysates were prepared in KPI buffer (25 mM potassium phosphate pH 6.5, supplemented with protease inhibitor 1× cocktail (Roche)) via homogenization on ice with SilentCrusher S equipped with Typ 7 F/S head (30 × 1000 rpm, 3 × 7 s). Lysate protein concentrations were determined with BCA Protein Assay Kit (Pierce). Lysates and proteins were stored in small aliquots at −80 °C until use.
IC50 and Inhibition Kinetics Determination
Detailed protocols for IC50 and inhibition kinetics measurements can be found in the Supporting Information.
In brief, apparent IC50 values were determined by preincubating enzymes with a range of inhibitor concentrations in a 25 μL volume for 30 min at 37 °C (for human enzymes) or 25 °C (for bacterial enzymes). Following preincubation, 25 μL of the enzyme–inhibitor mixture was transferred into 100 μL of the appropriate 4-MU–Glc substrate mixture to determine residual activity. Reactions were quenched with 1 M NaOH–glycine (pH 10.3) upon completion, and 4-MU fluorescence was measured with a LS55 fluorescence spectrophotometer (PerkinElmer) (λEX 366 nm; λEM 445 nm). IC50 values reported are the mean values from three technical replicates.
In situ IC50 values were determined by incubating normal human dermal fibroblasts (grown to confluency) with 5 for 2 and 24 h. Cells were washed three times with PBS and harvested by scraping into KPI buffer supplemented with 0.1% (v/v) Triton X-100 and 1× cOmplete protease inhibitor cocktail (Roche). Residual GAA and GANAB activity was measured on the basis of hydrolysis of 4-MU−α-Glc at pH 4 or 7. In situ IC50 values are mean values from two biological replicates, each with 3 technical repeats (Figure 5d).
For kinetics, enzyme and relevant concentrations of inhibitor were preincubated for 0.5, 2, 3.5, 5, and 8 min for fast inhibitors and 0.5, 20, 60, 120, 180, 240, and 360 min for slow inhibitors. At relevant time points, 5 μL of this enzyme–inhibitor mixture was added to 2,4-DNP–Glc substrate, and release of 2,4-dinitrophenolate was monitored via absorbance at 400 nm to determine the rate of hydrolysis after inhibition (V) compared to a no inhibitor control (V0).
Pseudo first order rate constants (kobs) were obtained from a linear fit of −ln V/V0 against time for each value of [I]. A plot of kobs against [I] fitted to the hyperbolic equation kobs = (kinact[I]/KI + [I]) was used to determine kinact and KI. Where fast inhibition was observed (>50% after 30 s), the kinact/KI ratio was determined at low values of [I] (≪KI), using the approximation kobs ≈ kinact[I]/KI, where kinact/KI is the slope of a linear fit of kobs vs [I]. Where reversible inhibition was observed (no variation of −ln V/V0 with time), KI was determined by use of Lineweaver–Burk plots at different values of [I].
Time and Concentration Dependent in Vitro Inhibition of GAA and GANAB
Homogenates of human normal dermal fibroblast (17.6 μg total protein) were preincubated on ice for 5 min in 150 mM McIlvaine buffer pH 4 or 7 in a total volume of 10 μL. Samples were then incubated for 15, 30, and 60 min at 37 °C with 2.5 μL inhibitor dilutions in McIlvaine buffer pH 4 or 7 to obtain a final concentration of 5, 1.5, 0.5, 0.15, and 0 μM. Afterward, the samples were further incubated with 2.5 μL of ABP 13 (1 μM), diluted in McIlvaine buffer pH 4 or 7 for 30 min at 37 °C. Finally, samples were denatured with 5 μL of 4× Laemmli sample buffer for 5 min at 98 °C, and resolved by 10% (w/v) SDS–PAGE. Wet slab gels were subjected to Cy5 fluorescence scaning (Typhoon FLA9500, GE, λEX ≥ 635 nm, λEM ≥ 665 nm).
In Vitro Inhibition of α-Glucosidases in Mouse Gastrointestinal Lysate
Mixtures of mouse duodenum, jejunum, and ileum homogenates containing 40 μg total protein were incubated on ice for 5 min with McIlvaine buffer at pH 4 or 7, in a total volume of 10 μL. Samples were subsequently incubated with 2.5 μL of 5 for 1 h at 37 °C at various inhibitor dilutions in McIlvaine buffer at pH 4.0 or 7.0 to obtain a final concentration of 150, 50, 15, 5, 1.5, 0.5, 0.15, 0.05, 0.015, 0.005, and 0 μM. Then, the samples were further incubated with 2.5 μL of 13 (6 μM, diluted in McIlvaine buffer at pH 4.0 or 7.0) for 30 min at 37 °C. Samples were denatured by boiling with 3.75 μL of 4× Laemmli sample buffer for 5 min, and resolved by 10% (w/v) SDS–PAGE. Wet slab gels were subjected to Cy5 fluorescence scanning (Typhoon FLA9500, GE, λEX ≥ 635 nm, λEM ≥ 665 nm).
In Situ inhibition of GAA and GANAB
Human normal dermal fibroblasts were grown to confluence and in situ treated with inhibitor 5 at various concentrations (10, 1, 0.1, 0.01, 0.001, and 0 μM) in duplicates for 2 or 24 h. Cells were harvested by first washing three times with PBS and subsequently lysed with KPI buffer (25 mM K2HPO4/KH2PO4, pH 6.5, 0.1% (v/v) Triton X-100, protease inhibitor cocktail (Roche)) on ice for 30 min. Homogenates were collected by scraping, vortexed, and stored at −80 °C. Lysates containing 4 μg total protein were equilibrated to pH 4 or 7 in 150 mM McIlvaine buffer for 5 min on ice, and incubated with probe 13 (0.5 μM) for 30 min at 37 °C at either pH 4 or 7 in a total volume of 15 μL. Samples were denatured, resolved by SDS–PAGE, and subjected to fluorescent scan (Typhoon FLA9500, GE, λEX ≥ 635 nm, λEM ≥ 665 nm)
Acknowledgments
We thank The Netherlands Organization for Scientific Research (NWO-CW, ChemThem grant to J.M.F.G.A. and H.S.O.), the European Research Council (ERC-2011-AdG-290836 “Chembiosphing”, to H.S.O., and ERC-2012-AdG-322942 “Glycopoise”, to G.J.D.), Sanofi Genzyme (research grant to J.M.F.G.A. and H.S.O. and postdoctoral contract to M.A.), the Spanish Ministry of Economy and Competitiveness (CTQ2014-55174-P), and the Generalitat de Catalunya (2014SGR-987) for financial support. We thank the Diamond Light Source for access to beamline i02 and i04 (proposal number mx-13587) and the Barcelona Supercomputing Center (BSC-CNS, RES-QCM-2017-1-00014) for technical support and computational resources provided, which contributed to the results presented here. L.R. acknowledges a Ph.D. fellowship from the University of Barcelona (APIF-UB). G.J.D. is supported by the Royal Society though the Ken Murray Research Professorship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00214.
Synthesis and characterization of 5 and 6, synthesis of all intermediates, molecular modeling, labeling of glucosidases, SDS–PAGE analysis and fluorescence scanning, and crystallographic data collection and refinement statistics (PDF)
Author Contributions
M.A. synthesized the inhibitors under the guidance of J.D.C.C. and G.A.v.d.M. M.A., M.J.F., and C.-L.K. conducted the enzyme inhibition and biochemistry assays under the guidance of J.M.F.G.A.. L.W., W.A.O., I.Z.B., and G.J.D. designed and performed the X-ray diffraction experiments. L.R. performed metadynamics simulations under guidance of C.R. H.S.O. and G.J.D. conceived the idea, supervised the project, and with M.A. and L.W. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lombard V.; Ramulu H. G.; Drula E.; Coutinho P. M.; Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongkees S. A. K.; Withers S. G. Unusual Enzymatic Glycoside Cleavage Mechanisms. Acc. Chem. Res. 2014, 47, 226–235. 10.1021/ar4001313. [DOI] [PubMed] [Google Scholar]
- Gloster T. M.; Davies G. J. Glycosidase inhibition: assessing mimicry of the transition state. Org. Biomol. Chem. 2010, 8, 305–20. 10.1039/B915870G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasella A.; Davies G. J.; Bohm M. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2002, 6, 619–629. 10.1016/S1367-5931(02)00380-0. [DOI] [PubMed] [Google Scholar]
- Vocadlo D. J.; Davies G. J. Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 2008, 12, 539–555. 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Ardevol A.; Rovira C. Reaction Mechanisms in Carbohydrate-Active Enzymes: Glycoside Hydrolases and Glycosyltransferases. Insights from ab Initio Quantum Mechanics/Molecular Mechanics Dynamic Simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. 10.1021/jacs.5b01156. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Williams S. J. Carbohydrate-active enzymes: sequences, shapes, contortions and cells. Biochem. Soc. Trans. 2016, 44, 79–87. 10.1042/BST20150186. [DOI] [PubMed] [Google Scholar]
- Kallemeijn W. W.; Witte M. D.; Wennekes T.; Aerts J. M. Mechanism-based inhibitors of glycosidases: design and applications. Adv. Carbohydr. Chem. Biochem. 2014, 71, 297–338. 10.1016/B978-0-12-800128-8.00004-2. [DOI] [PubMed] [Google Scholar]
- Beenakker T. J. M.; Wander D. P. A.; Offen W. A.; Artola M.; Raich L.; Ferraz M. J.; Li K. Y.; Houben J.; van Rijssel E. R.; Hansen T.; van der Marel G. A.; Codee J. D. C.; Aerts J.; Rovira C.; Davies G. J.; Overkleeft H. S. Carba-cyclophellitols Are Neutral Retaining-Glucosidase Inhibitors. J. Am. Chem. Soc. 2017, 139, 6534–6537. 10.1021/jacs.7b01773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte M. D.; Kallemeijn W. W.; Aten J.; Li K. Y.; Strijland A.; Donker-Koopman W. E.; van den Nieuwendijk A. M.; Bleijlevens B.; Kramer G.; Florea B. I.; Hooibrink B.; Hollak C. E.; Ottenhoff R.; Boot R. G.; van der Marel G. A.; Overkleeft H. S.; Aerts J. M. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. 10.1038/nchembio.466. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Beenakker T. J.; Kallemeijn W. W.; van der Marel G. A.; van den Elst H.; Codee J. D.; Aerts J. M.; Overkleeft H. S. Comparing Cyclophellitol N-Alkyl and N-Acyl Cyclophellitol Aziridines as Activity-Based Glycosidase Probes. Chem. - Eur. J. 2015, 21, 10861–10869. 10.1002/chem.201501313. [DOI] [PubMed] [Google Scholar]
- Withers S. G.; Umezawa K. Cyclophellitol: a naturally occurring mechanism-based inactivator of beta-glucosidases. Biochem. Biophys. Res. Commun. 1991, 177, 532–537. 10.1016/0006-291X(91)92016-D. [DOI] [PubMed] [Google Scholar]
- Gloster T. M.; Madsen R.; Davies G. J. Structural basis for cyclophellitol inhibition of a beta-glucosidase. Org. Biomol. Chem. 2007, 5, 444–446. 10.1039/B616590G. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Beenakker T. J.; Murray B.; Scheij S.; Kallemeijn W. W.; Boot R. G.; Verhoek M.; Donker-Koopman W. E.; Ferraz M. J.; van Rijssel E. R.; Florea B. I.; Codee J. D.; van der Marel G. A.; Aerts J. M.; Overkleeft H. S. Potent and selective activity-based probes for GH27 human retaining alpha-galactosidases. J. Am. Chem. Soc. 2014, 136, 11622–11625. 10.1021/ja507040n. [DOI] [PubMed] [Google Scholar]
- Jiang J. B.; Kallemeijn W. W.; Wright D. W.; van den Nieuwendijk A. M. C. H.; Rohde V. C.; Folch E. C.; van den Elst H.; Florea B. I.; Scheij S.; Donker-Koopman W. E.; Verhoek M.; Li N.; Schurmann M.; Mink D.; Boot R. G.; Codee J. D. C.; van der Marel G. A.; Davies G. J.; Aerts J. M. F. G.; Overkleeft H. S. In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 alpha-L-fucosidases. Chem. Sci. 2015, 6, 2782–2789. 10.1039/C4SC03739A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. Y.; Jiang J.; Witte M. D.; Kallemeijn W. W.; Donker-Koopman W. E.; Boot R. G.; Aerts J. M.; Codee J. D.; van der Marel G. A.; Overkleeft H. S. Exploring functional cyclophellitol analogues as human retaining beta-glucosidase inhibitors. Org. Biomol. Chem. 2014, 12, 7786–7791. 10.1039/C4OB01611D. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Sharpless K. B. Vicinal Diol Cyclic Sulfates - Like Epoxides Only More Reactive. J. Am. Chem. Soc. 1988, 110, 7538–7539. 10.1021/ja00230a045. [DOI] [Google Scholar]
- Ernst B.; Magnani J. L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discovery 2009, 8, 661–677. 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi S.; Umezawa K.; Iinuma H.; Naganawa H.; Nakamura H.; Iitaka Y.; Takeuchi T. Production, isolation and structure determination of a novel beta-glucosidase inhibitor, cyclophellitol, from Phellinus sp. J. Antibiot. 1990, 43, 49–53. 10.7164/antibiotics.43.49. [DOI] [PubMed] [Google Scholar]
- Tatsuta K.; Niwata Y.; Umezawa K.; Toshima K.; Nakata M. Syntheses and enzyme inhibiting activities of cyclophellitol analogs. J. Antibiot. 1991, 44, 912–914. 10.7164/antibiotics.44.912. [DOI] [PubMed] [Google Scholar]
- McCarter J. D.; Withers S. G. 5-fluoro glycosides: A new class of mechanism-based inhibitors of both alpha- and beta-glucosidases. J. Am. Chem. Soc. 1996, 118, 241–242. 10.1021/ja952732a. [DOI] [Google Scholar]
- Withers S. G.; Street I. P.; Bird P.; Dolphin D. H. 2-Deoxy-2-Fluoroglucosides - a Novel Class of Mechanism-Based Glucosidase Inhibitors. J. Am. Chem. Soc. 1987, 109, 7530–7531. 10.1021/ja00258a047. [DOI] [Google Scholar]
- Rempel B. P.; Withers S. G. Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology 2008, 18, 570–586. 10.1093/glycob/cwn041. [DOI] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen F. G.; Bundgaard E.; Madsen R. A short synthesis of (+)-cyclophellitol. J. Org. Chem. 2005, 70, 10139–10142. 10.1021/jo051645q. [DOI] [PubMed] [Google Scholar]
- Li K.-Y.; Jiang J.; Witte M. D.; Kallemeijn W. W.; van den Elst H.; Wong C.-S.; Chander S. D.; Hoogendoorn S.; Beenakker T. J. M.; Codée J. D. C.; Aerts J. M. F. G.; van der Marel G. A.; Overkleeft H. S. Synthesis of Cyclophellitol, Cyclophellitol Aziridine, and Their Tagged Derivatives. Eur. J. Org. Chem. 2014, 2014, 6030–6043. 10.1002/ejoc.201402588. [DOI] [Google Scholar]
- Jiang J.; Kuo C. L.; Wu L.; Franke C.; Kallemeijn W. W.; Florea B. I.; van Meel E.; van der Marel G. A.; Codee J. D.; Boot R. G.; Davies G. J.; Overkleeft H. S.; Aerts J. M. Correction to ″Detection of Active Mammalian GH31 alpha-Glucosidases in Health and Disease Using In-Class, Broad-Spectrum Activity-Based Probes″. ACS Cent. Sci. 2016, 2, 351–358. 10.1021/acscentsci.6b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster T. M.; Meloncelli P.; Stick R. V.; Zechel D.; Vasella A.; Davies G. J. Glycosidase inhibition: An assessment of the binding of 18 putative transition-state mimics. J. Am. Chem. Soc. 2007, 129, 2345–2354. 10.1021/ja066961g. [DOI] [PubMed] [Google Scholar]
- Charoenwattanasatien R.; Pengthaisong S.; Breen I.; Mutoh R.; Sansenya S.; Hua Y.; Tankrathok A.; Wu L.; Songsiriritthigul C.; Tanaka H.; Williams S. J.; Davies G. J.; Kurisu G.; Cairns J. R. Bacterial Beta-Glucosidase Reveals the Structural and Functional Basis of Genetic Defects in Human Glucocerebrosidase 2 (GBA2). ACS Chem. Biol. 2016, 11, 1891–1900. 10.1021/acschembio.6b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J.; Izumi A.; Hemsworth G. R.; Davies G. J.; Brumer H. Structural Enzymology of Cellvibrio japonicus Agd31B Protein Reveals alpha-Transglucosylase Activity in Glycoside Hydrolase Family 31. J. Biol. Chem. 2012, 287, 43288–43299. 10.1074/jbc.M112.416511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. R.; Grubb J. H.; Sly W. S. C-terminal processing of human beta-glucuronidase. The propeptide is required for full expression of catalytic activity, intracellular retention, and proper phosphorylation. J. Biol. Chem. 1993, 268, 22627–22633. [PubMed] [Google Scholar]
- Scheen A. J. Is there a role for alpha-glucosidase inhibitors in the prevention of type 2 diabetes mellitus?. Drugs 2003, 63, 933–951. 10.2165/00003495-200363100-00002. [DOI] [PubMed] [Google Scholar]
- van de Laar F. A.; Lucassen P. L.; Akkermans R. P.; van de Lisdonk E. H.; Rutten G. E.; van Weel C. Alpha-glucosidase inhibitors for patients with type 2 diabetes: results from a Cochrane systematic review and meta-analysis. Diabetes Care 2005, 28, 154–163. 10.2337/diacare.28.1.154. [DOI] [PubMed] [Google Scholar]
- Wardrop D. J.; Waidyarachchi S. L. Synthesis and biological activity of naturally occurring alpha-glucosidase inhibitors. Nat. Prod. Rep. 2010, 27, 1431–1468. 10.1039/b914958a. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Planas A.; Rovira C. Conformational analyses of the reaction coordinate of glycosidases. Acc. Chem. Res. 2012, 45, 308–316. 10.1021/ar2001765. [DOI] [PubMed] [Google Scholar]
- Vallee F.; Karaveg K.; Herscovics A.; Moremen K. W.; Howell P. L. Structural basis for catalysis and inhibition of N-glycan processing class I alpha 1,2-mannosidases. J. Biol. Chem. 2000, 275, 41287–41298. 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Dabin J.; Iglesias-Fernandez J.; Ardevol A.; Dinev Z.; Williams S. J.; Bande O.; Siriwardena A.; Moreland C.; Hu T. C.; Smith D. K.; Gilbert H. J.; Rovira C.; Davies G. J. The reaction coordinate of a bacterial GH47 alpha-mannosidase: a combined quantum mechanical and structural approach. Angew. Chem., Int. Ed. 2012, 51, 10997–11001. 10.1002/anie.201205338. [DOI] [PubMed] [Google Scholar]
- Schultheis P. J.; Fleming S. M.; Clippinger A. K.; Lewis J.; Tsunemi T.; Giasson B.; Dickson D. W.; Mazzulli J. R.; Bardgett M. E.; Haik K. L.; Ekhator O.; Chava A. K.; Howard J.; Gannon M.; Hoffman E.; Chen Y.; Prasad V.; Linn S. C.; Tamargo R. J.; Westbroek W.; Sidransky E.; Krainc D.; Shull G. E. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.