

Abstract
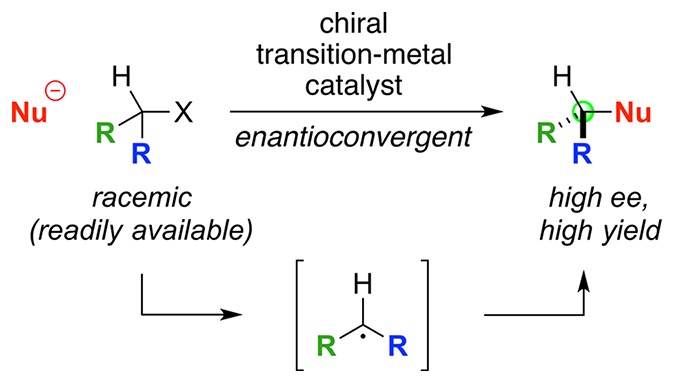
Classical methods for achieving nucleophilic substitutions of alkyl electrophiles (SN1 and SN2) have limited scope and are not generally amenable to enantioselective variants that employ readily available racemic electrophiles. Radical-based pathways catalyzed by chiral transition-metal complexes provide an attractive approach to addressing these limitations.
Short abstract
Radical-based pathways catalyzed by chiral transition-metal complexes address limitations of classical SN1 and SN2 reactions.
Unsolved Challenges in Nucleophilic Substitution Reactions of Alkyl Electrophiles
Nucleophilic substitution of an alkyl electrophile is an extremely useful strategy in organic synthesis (Figure 1). SN1 and SN2 reactions are the two classical pathways for achieving this process; both have significant limitations.1,2
Figure 1.
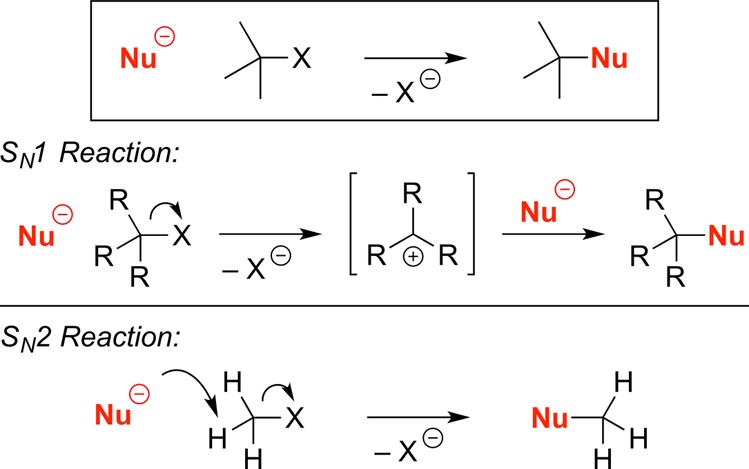
Nucleophilic substitution reactions of alkyl electrophiles: SN1 and SN2 reactions.
In the case of the SN1 reaction, this pathway is typically only useful for tertiary or for activated (e.g., benzylic or allylic) electrophiles, due to the need to access a carbocationic intermediate (C in Figure 2); other (e.g., unactivated primary and secondary) electrophiles are generally not suitable substrates. Furthermore, in the case of electrophiles that are able to form a carbocation, elimination of a proton to generate an olefin, as well as hydride and alkyl rearrangements, may occur in preference to capture by the nucleophile. Finally, because SN1 reactions often require the presence of a Brønsted acid or a Lewis acid to facilitate dissociation of the leaving group (X), the range of nucleophiles that can be utilized is limited, due to the susceptibility of many potential nucleophiles to undergo protonation or to form a Lewis acid–base complex.
Figure 2.
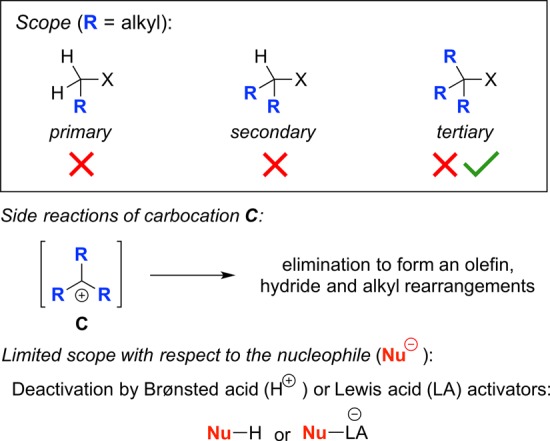
Limitations of the SN1 reaction.
Similarly, the SN2 reaction has substantial limitations. In particular, it is sensitive to the steric demands of the nucleophile and the electrophile, and it is therefore often unsuccessful in the case of hindered primary (e.g., neopentyl) and secondary electrophiles (Figure 3). Furthermore, SN2 reactions are generally conducted under Brønsted-basic conditions, which can lead to elimination of H–X to form an olefin, rather than substitution.
Figure 3.
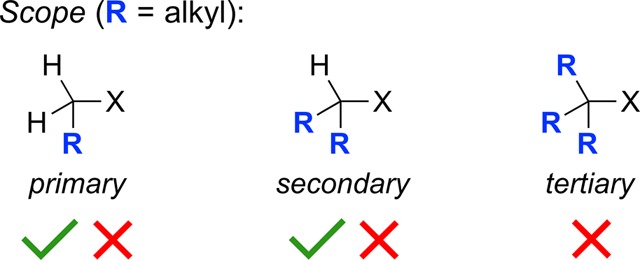
Limitations of the SN2 reaction.
In addition to considerable limitations in scope, another critical shortcoming of classical substitution reactions is the difficulty in controlling the stereochemistry at the carbon undergoing substitution (for chiral secondary and tertiary electrophiles). In the case of the SN1 reaction, the intermediacy of an achiral carbocation (C in Figure 2) generally leads to racemic product, regardless of whether an enantiopure or a racemic electrophile is employed.3,4 In the case of the SN2 reaction, the formation of enantioenriched product typically requires the use of an enantioenriched electrophile, and the scope of such stereospecific processes is somewhat limited due to the modest electrophilicity of unactivated secondary alkyl electrophiles.
Thus, expanding the breadth of nucleophilic substitution reactions of alkyl electrophiles, while simultaneously providing products with high enantiomeric excess (ee) from readily accessible racemic electrophiles (e.g., eq 1), would enormously empower organic synthesis.
 |
1 |
Approach: Radical Intermediates and Transition-Metal Catalysis
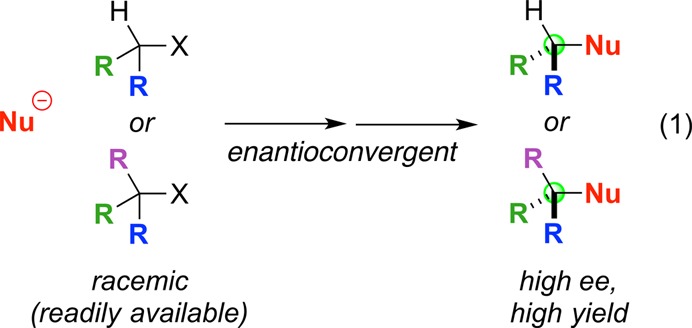
Whereas it can be challenging to generate carbocations from alkyl halides under synthetically useful conditions, a spectrum of radicals (primary, secondary, and tertiary) can be formed under mild conditions through pathways such as outer-sphere electron transfer and halogen-atom abstraction (Figure 4).5 Furthermore, whereas carbocations can readily undergo 1,2-hydride and 1,2-alkyl shifts, radicals are not prone to corresponding rearrangements (Figure 4). Consequently, the development of nucleophilic substitution reactions that proceed via radical intermediates seems attractive.
Figure 4.
Attributes of radicals as intermediates in substitution reactions.
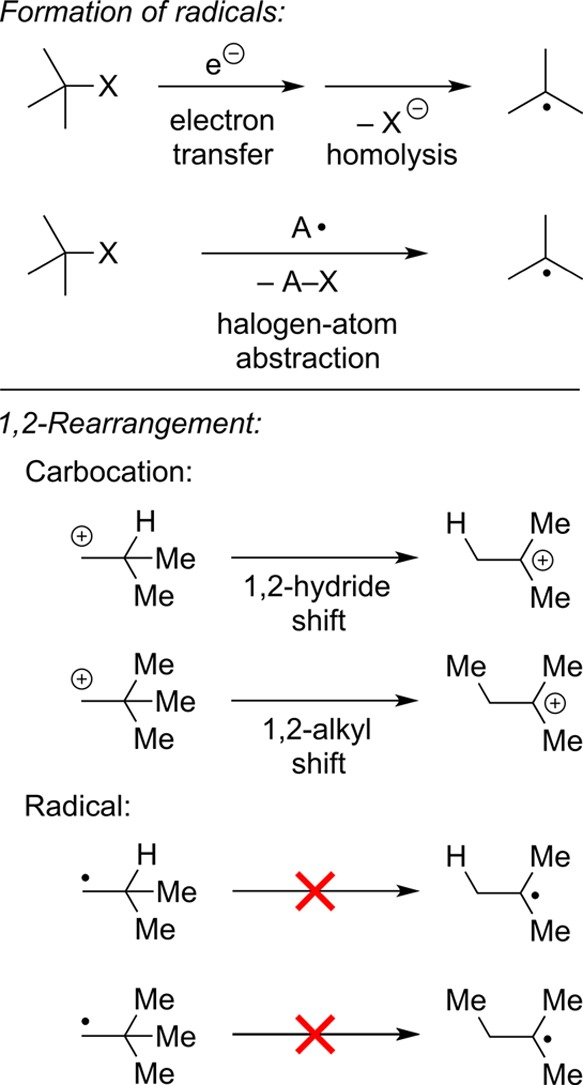
In fact, substitution reactions of alkyl halides via a radical pathway are well-established, including reductions and allylations (Figure 5); in these processes, the key bond formation occurs through an elementary step such as atom abstraction or addition to a π system by an alkyl radical.5 However, because direct SH2 reactions at first-row atoms such as carbon are not generally accessible, the scope of such processes is limited. Furthermore, direct reaction of an alkyl radical with a first-row nucleophile that bears a localized lone pair is typically unfavorable (Figure 5).
Figure 5.
Substitution reactions via radical intermediates (without a transition-metal catalyst).
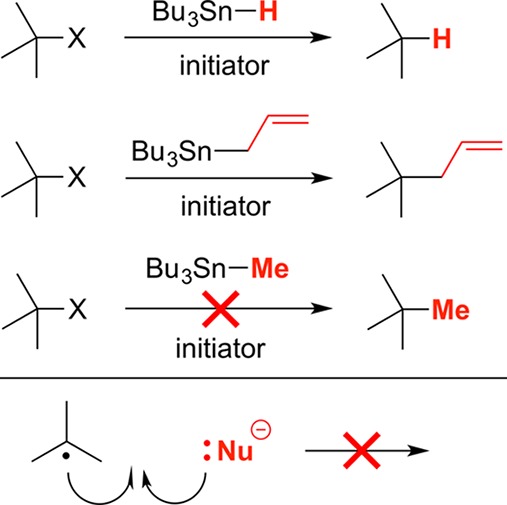
Catalysis by transition metals has provided an attractive solution to a variety of important problems in organic synthesis, including nucleophilic substitution reactions of aryl and alkenyl electrophiles; a generalized pathway for the palladium-catalyzed cross-coupling of an aryl electrophile is outlined in Figure 6.6,7 In this process, oxidative addition of the C–X bond of the aryl electrophile is believed to typically occur via direct insertion of palladium(0). Due to steric considerations, the corresponding insertion into the C–X bond of a secondary or tertiary alkyl halide is challenging at best.
Figure 6.
Outline of a mechanism for palladium-catalyzed cross-couplings (nucleophilic substitutions) of aryl electrophiles.
Fortunately, the oxidative addition of alkyl halides to transition metals can be achieved by mechanisms other than direct insertion of the metal into the C–X bond, for example, a two-step radical pathway of halogen atom abstraction by the transition metal, followed by capture of the alkyl radical by the transition metal.8 This then opens the door to metal-catalyzed substitutions of alkyl halides through catalytic cycles analogous to those previously reported for aryl halides (e.g., Figure 6).9
In addition to expanding the scope of nucleophilic substitution reactions of alkyl halides, transition-metal catalysis via radical intermediates could enable, through the use of a chiral catalyst, enantioconvergent reactions of racemic halides (eq 2). Thus, the generation of a radical from the alkyl halide provides a ready mechanism for stereoconvergence, since both enantiomers of the racemic halide are transformed into the same radical intermediate. This radical could, for example, combine with an enantiopure transition-metal catalyst (cat*) to afford a single stereoisomer of an alkylmetal complex (I), which could proceed to form a single enantiomer of the substitution product.
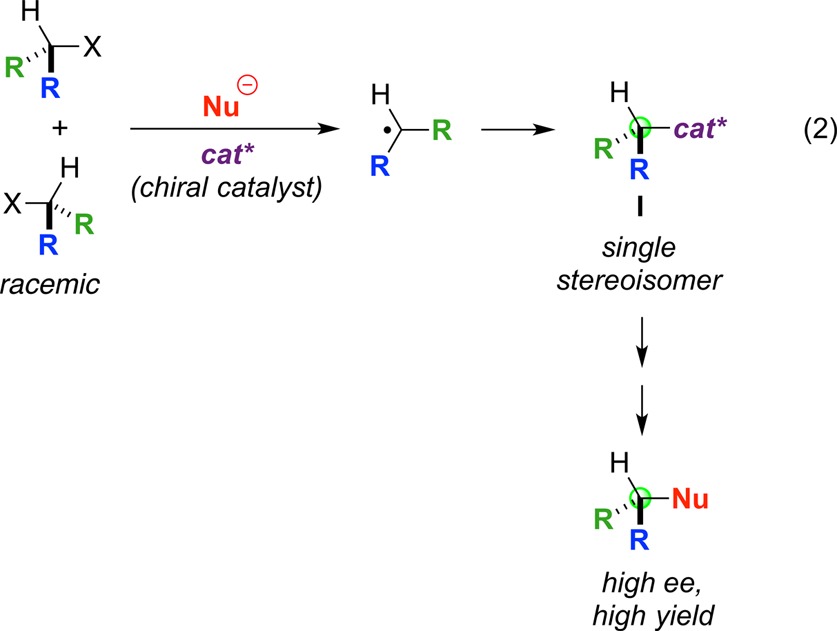 |
2 |
In this Outlook, we describe how the combination of radical chemistry and transition-metal catalysis has indeed opened the door to addressing the challenges of reactivity and of enantioselectivity in nucleophilic substitution reactions of secondary and tertiary alkyl electrophiles. Because this area of research has expanded extremely rapidly during the past decade or so,10−12 a comprehensive review is not possible in the context of an Outlook. Consequently, we focus on studies from our lab to provide illustrative examples of just a few of the recent developments in this field.
Expanding the Scope of Substitution Reactions by Carbon-Based Nucleophiles
Due to the ubiquity of carbon–carbon bonds in organic molecules, our initial studies of radical-based, transition-metal-catalyzed substitution reactions of alkyl electrophiles focused on the use of carbon nucleophiles. Prior to 2000, there had been limited success in achieving metal-catalyzed substitutions with unactivated alkyl electrophiles as substrates.10−12 It was widely believed that a primary impediment to accomplishing such processes is facile β-hydride elimination of the metal–alkyl intermediate that is generated upon oxidative addition of the alkyl electrophile to the transition metal. For example, in the case of palladium, the most commonly used catalyst for cross-coupling reactions of aryl halides, β-hydride elimination of palladium(II)–alkyls is often very rapid; indeed, this is an essential elementary step for powerful transformations such as the Wacker oxidation13 and the Heck reaction (eq 3).14
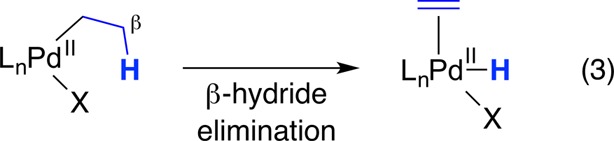 |
3 |
Nevertheless, by 2000, important pioneering studies by a variety of researchers had established that metal-catalyzed couplings of organometallic nucleophiles with unactivated alkyl electrophiles that contain β-hydrogens can in fact be achieved, although the methods generally employed highly reactive nucleophiles with poor functional-group compatibility (e.g., Grignard reagents) or were limited to primary alkyl electrophiles.10−12 We sought to expand the scope of such processes to include substitutions by nucleophiles that have good functional-group tolerance (e.g., organoboron and organozinc reagents) with secondary and tertiary alkyl electrophiles.
In our initial work, we explored the use of palladium-based catalysts, but the methods that we developed were only effective for primary alkyl electrophiles.15 Our subsequent mechanistic studies suggested that these palladium-catalyzed couplings proceed through an SN2, rather than a radical, pathway for oxidative addition of the C–X bond, which accounts for the inability to apply these methods to unactivated secondary and tertiary electrophiles.
In order to expand the scope of our metal-catalyzed nucleophilic substitution processes to secondary alkyl electrophiles, we turned to the use of nickel-based catalysts. The greater propensity of nickel, an earth-abundant first-row transition metal, as compared to its congeners (palladium or platinum) to access an array of oxidation states (e.g., M0, MI, MII, and MIII),16 along with the important studies of Knochel on coupling reactions of primary alkyl electrophiles with organozinc reagents,17 made nickel an attractive choice for the development of radical-based, metal-catalyzed substitutions of alkyl electrophiles by organometallic nucleophiles.18
Because organometallic nucleophiles based on different metals may be generated from different precursors and may have different functional-group compatibility, it is useful in organic synthesis to have as many options for nucleophilic partners as possible. We have determined that nickel catalysts can achieve substitutions of an array of unactivated primary and, more significantly, secondary alkyl halides with organozinc, -boron, -silicon, and -tin reagents (Figure 7).19−26 Although only a fraction of the possible permutations have been examined to date, it is clear that nickel catalysts enable the substitution of a useful range of combinations of reaction partners. In each case, essentially no carbon–carbon bond formation is observed in the absence of nickel.
Figure 7.
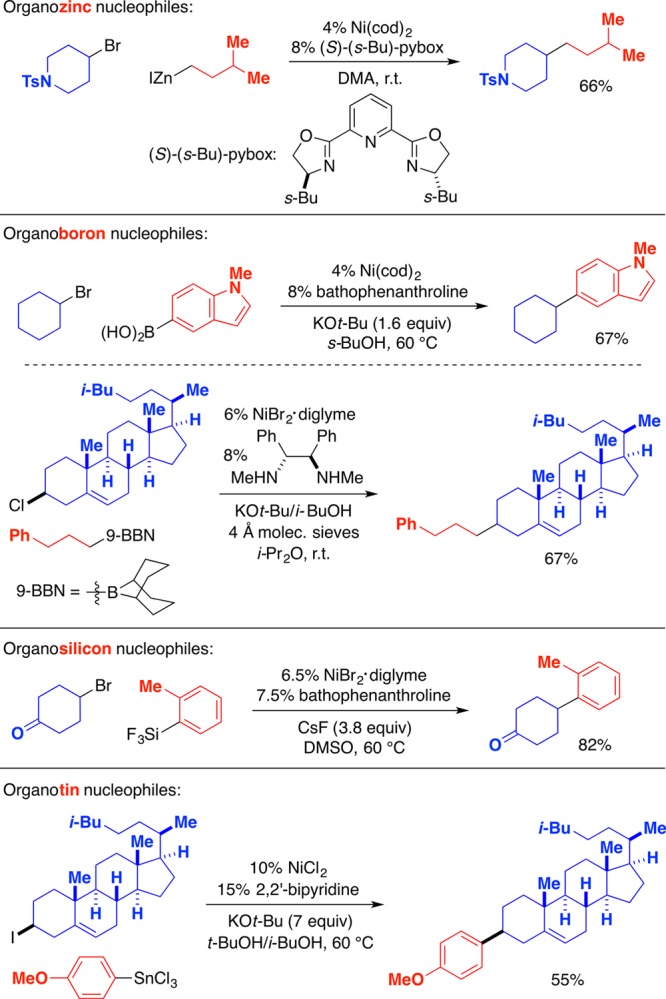
Representative examples of nickel-catalyzed substitution reactions of unactivated secondary alkyl halides by organozinc, -boron, -silicon, and -tin nucleophiles.
A variety of observations are consistent with the formation of a radical intermediate from the alkyl halide in these nickel-catalyzed substitution processes (Figure 8).22,27 For example, endo- and exo-2-bromonorbornane couple with phenylboronic acid to form the exo product with the same diastereoselectivity (>20:1), as expected for a common intermediate. Furthermore, when the five-membered cyclic bromides illustrated at the bottom of Figure 8 are subjected to the nickel-catalyzed coupling conditions, in both cases the bicyclic product is generated with the same stereoselectivity as for the corresponding Bu3SnH-mediated reductive cyclizations of these alkyl bromides; this is consistent with the same intermediate undergoing cyclization in the nickel-catalyzed and the Bu3SnH-mediated processes, specifically, an alkyl radical. Finally, as expected for a radical pathway, the use of an alkyl tosylate, rather than an alkyl halide, does not lead to the desired substitution.
Figure 8.
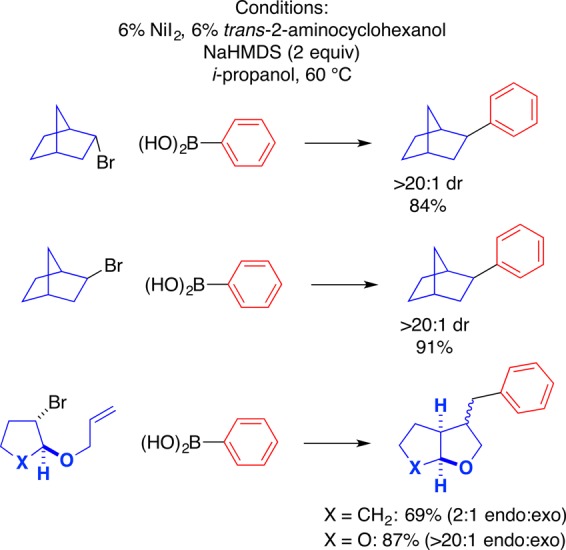
Observations consistent with the intermediacy of an alkyl radical.
We have also begun to explore the possibility of achieving metal-catalyzed substitution reactions of unactivated tertiary alkyl electrophiles. Although we have not yet developed highly general methods, we have determined that nickel-catalyzed couplings of arylboron reagents with unactivated tertiary alkyl bromides are viable (Figure 9).28
Figure 9.
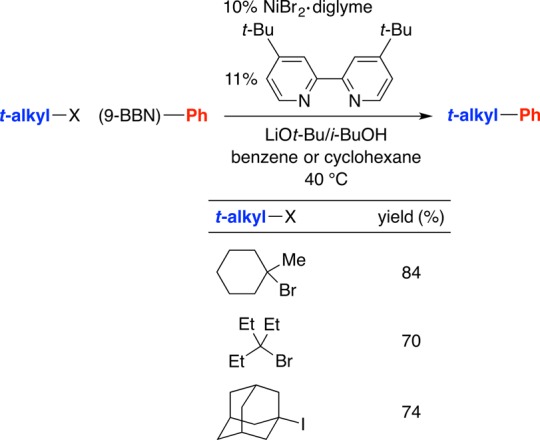
Nickel-catalyzed substitution reactions of unactivated tertiary alkyl halides by arylboron nucleophiles.
Enantioselective Carbon–Carbon Bond Formation
The studies described in the previous section set the stage for the pursuit of metal-catalyzed enantioconvergent substitution reactions of carbon nucleophiles with racemic alkyl electrophiles (eq 2). It is essential to note that, prior to this work, there were a number of reports of such processes that largely involve activated electrophiles and proceed through nonradical pathways; palladium-catalyzed substitutions of allylic electrophiles (mostly with stabilized carbon nucleophiles such as malonate anions) are an especially prominent example.29,30
Indeed, our first advance in metal-catalyzed enantioconvergent substitution reactions also involved an activated alkyl electrophile, specifically, the coupling of racemic secondary α-haloamides with organozinc reagents (top of Figure 10).31−33 Thus, a chiral nickel/pybox catalyst achieves an array of alkyl–alkyl couplings with good enantioselectivity and good yield (e.g., 96% ee and 90% yield). The fact that both values are high establishes that this is not a kinetic resolution in which the chiral catalyst selectively reacts with one enantiomer of the electrophile and leaves the other enantiomer unreacted; instead, it is an enantioconvergent reaction in which both enantiomers of the racemic starting material are being converted into the desired product.
Figure 10.
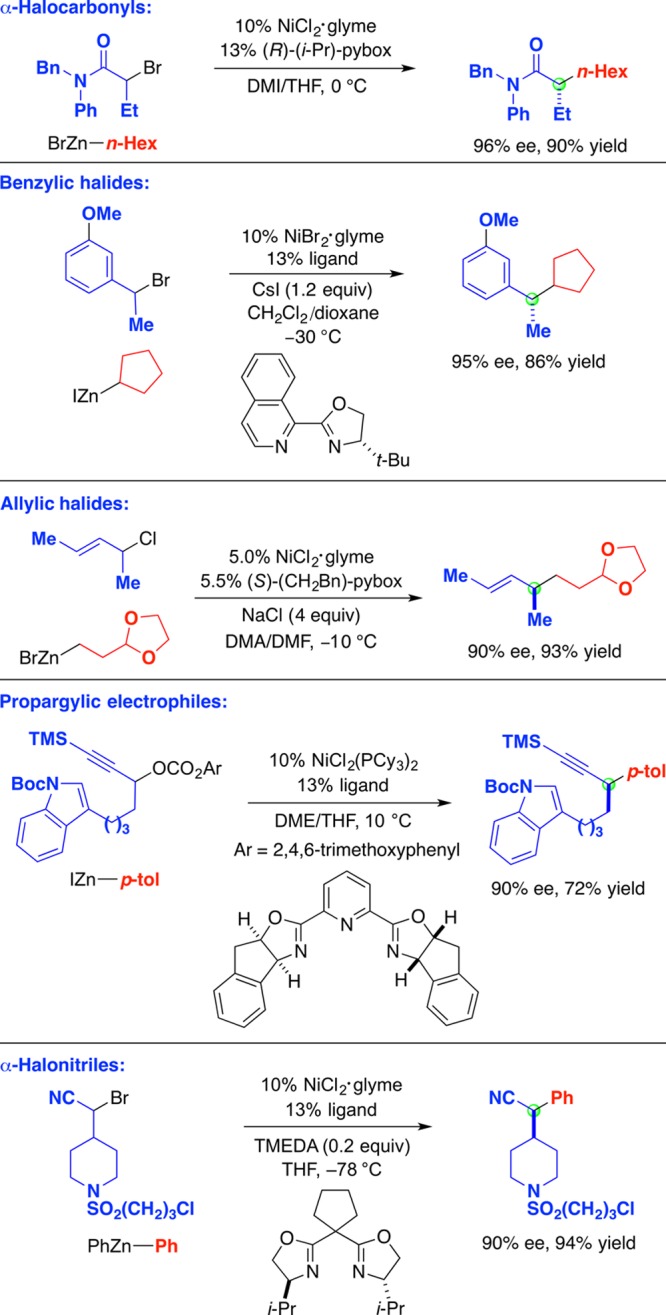
Enantioconvergent substitution reactions of activated electrophiles: Organozinc reagents as nucleophiles.
Chiral nickel catalysts can achieve enantioconvergent substitution reactions of an array of other activated electrophiles, including benzylic, allylic, and propargylic electrophiles, as well as α-halonitriles, with alkyl-, aryl-, and alkenylzinc nucleophiles (Figure 10).34−40 Again, only a fraction of the possible permutations of electrophiles and nucleophiles have been examined to date. Nevertheless, these methods have found application in the catalytic asymmetric synthesis of diverse families of natural products (Figure 11).41−43
Figure 11.
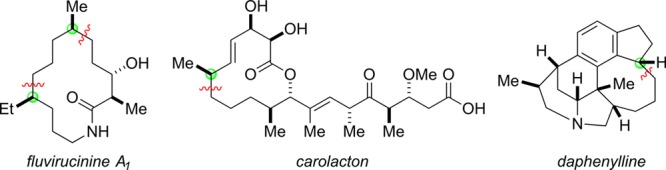
Applications of nickel-catalyzed enantioconvergent substitution reactions to the synthesis of natural products. The chiral catalyst controls the stereocenters indicated by the green balls and forms the bonds indicated by the wavy red lines.
The enantioconvergent arylation of propargylic carbonates that is illustrated in Figure 10 is worthy of comment. In the case of our nickel-catalyzed nucleophilic substitution reactions of alkyl halides, we hypothesize that formation of an alkyl radical from the alkyl halide occurs through an inner-sphere electron-transfer reaction with a nickel(I) complex (SH2 reaction). In the case of propargylic carbonates, for which a direct SH2 reaction is not viable, we have suggested that the nickel(I) complex adds to the carbonyl group to generate a (nickel) ketyl, which then fragments to form the alkyl radical.38 Related strategies that generate alkyl radicals through electron-transfer reactions of carbonyl compounds have recently been applied in other interesting nickel-catalyzed coupling processes.44
Significantly, enantioconvergent substitution reactions of activated electrophiles are not limited to the use of organozinc reagents as nucleophiles. Thus, chiral nickel complexes catalyze couplings of an array of organomagnesium, -silicon, -zirconium, and -boron (aryl and alkenyl) nucleophiles with a variety of racemic α-halocarbonyl compounds (ketones, esters, and amides; Figure 12).45−49
Figure 12.
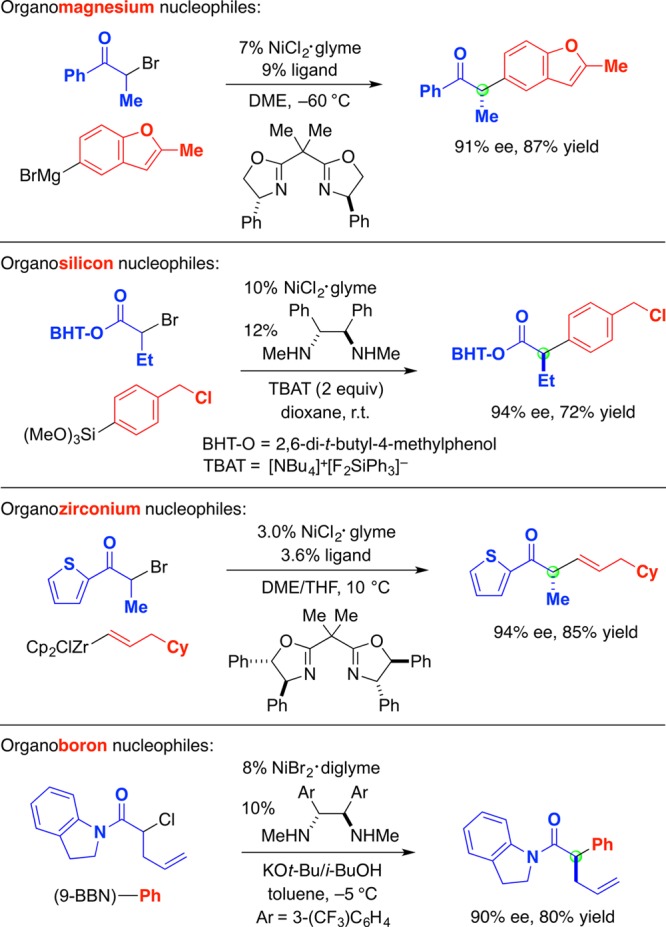
Enantioconvergent substitution reactions of activated electrophiles: Other families of organometallic nucleophiles.
Clearly, it would be useful to achieve enantioconvergent substitution reactions not only of activated but also of unactivated secondary alkyl electrophiles. In terms of the substituents attached to the electrophilic carbon, this requires that the chiral catalyst be able to differentiate between two alkyl (sp3-hybridized) groups and a hydrogen. We have established that this is possible, although to date a suitably positioned directing group, which likely binds to the catalyst in the stereochemistry-determining step of the coupling process, has generally been necessary in order to obtain good enantioselectivity (Figure 13).50−54 A variety of directing groups have proved to be effective, including an aromatic ring, a carbonyl group, the nitrogen of an aniline, and a sulfonamide.
Figure 13.
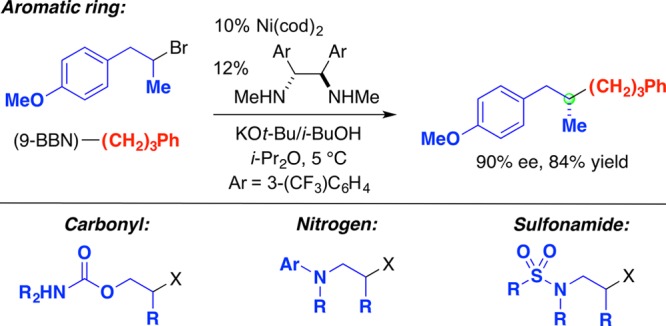
Enantioconvergent substitution reactions of unactivated electrophiles: Alkylboron reagents as nucleophiles.
If one of the two alkyl substituents of the racemic secondary alkyl electrophile is perfluorinated, nickel-catalyzed substitution by an arylzinc nucleophile can proceed with very good ee and yield (eq 4).55 It has not yet been determined if the enantioselectivity arises from fluorine serving as a directing group or from an electronic effect of the perfluorinated alkyl substituent.
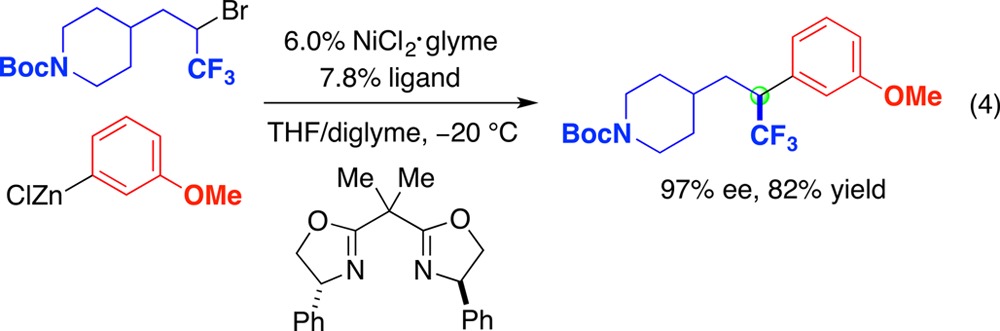 |
4 |
Furthermore, racemic α-haloboronate esters serve as suitable electrophiles in nickel-catalyzed enantioconvergent substitution reactions with alkylzinc reagents (eq 5).56 Due to the wide range of stereospecific transformations of the C–B bond (e.g., to C–C, C–N, C–O, C–Cl, C–Br, and C–I bonds), this asymmetric coupling reaction may be particularly useful in asymmetric synthesis.
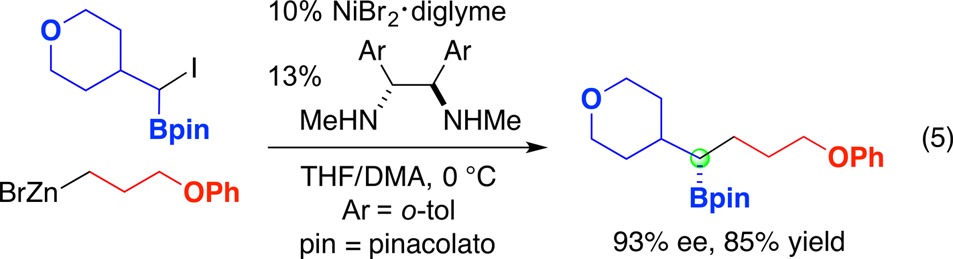 |
5 |
Substitution by Heteroatom-Based Nucleophiles: Boron, Silicon, and Nitrogen
Boron and Silicon Nucleophiles
Organoboron and organosilicon compounds serve as useful intermediates in organic synthesis and as important target molecules in a wide variety of fields.57,58 As for carbon-based nucleophiles, nickel can serve as an effective catalyst for substitution reactions of unactivated alkyl electrophiles with appropriate boron and silicon nucleophiles (Figure 14).59,60 Not only secondary but also tertiary alkyl halides serve as suitable electrophiles. As in the case of the corresponding C–C bond-forming processes, a variety of mechanistic studies (stereochemical, reactivity, and trapping) are consistent with the hypothesis that these substitutions proceed via the generation of an alkyl radical intermediate from the electrophile. It is important to note that, in the case of C–B bond formation, Liu and Ito independently described copper-catalyzed borylations of primary and secondary (but not tertiary) alkyl electrophiles.61,62
Figure 14.
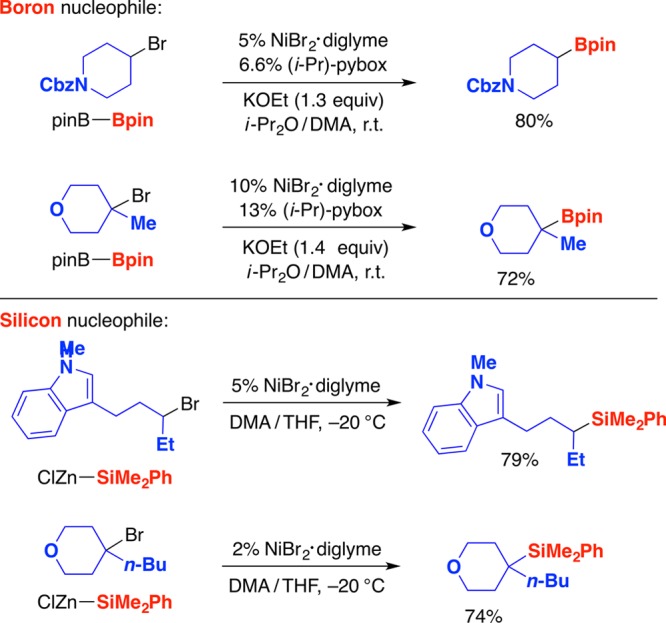
Nickel-catalyzed substitution reactions of unactivated secondary and tertiary alkyl halides by boron and silicon nucleophiles.
Nitrogen Nucleophiles
A substantial fraction of bioactive molecules include an N–alkyl bond.63 In principle, substitution reactions of alkyl electrophiles by nitrogen nucleophiles are a straightforward approach to constructing these bonds, but in practice such processes suffer from many of the limitations described in the introduction. For example, nitrogen nucleophiles are typically deactivated under the acidic conditions that are often employed in SN1 reactions. And, in the case of electrophiles that are suitably reactive for SN2 processes, overalkylation of the nucleophile can be a significant side reaction. Unfortunately, our attempts to apply nickel-based catalysts, which have proved useful for substitution reactions of alkyl electrophiles by several other families of nucleophiles (see preceding sections), to couplings by nitrogen nucleophiles have been unsuccessful. More broadly, until a few years ago, there had been virtually no systematic studies of transition-metal catalysis of substitution reactions of alkyl electrophiles by nitrogen nucleophiles.64−66
In 2011, we began a collaboration with the laboratory of Prof. Jonas Peters that was focused initially on copper-catalyzed substitution reactions of aryl electrophiles by nitrogen nucleophiles, specifically, a photoinduced variant of the Ullmann coupling reaction.67,68 As part of a mechanistic study of this process, we observed the formation of an N–alkyl, rather than an N–aryl, bond in the coupling of a nitrogen nucleophile with an aryl halide that bears a pendant olefin (eq 6). A stereochemical investigation was consistent with the suggestion that an alkyl radical is an intermediate in this N-alkylation process. This stimulated us to pursue the possibility that photoinduced copper catalysis might be an effective strategy for substitution reactions by nitrogen nucleophiles not only of aryl but also of alkyl halides.
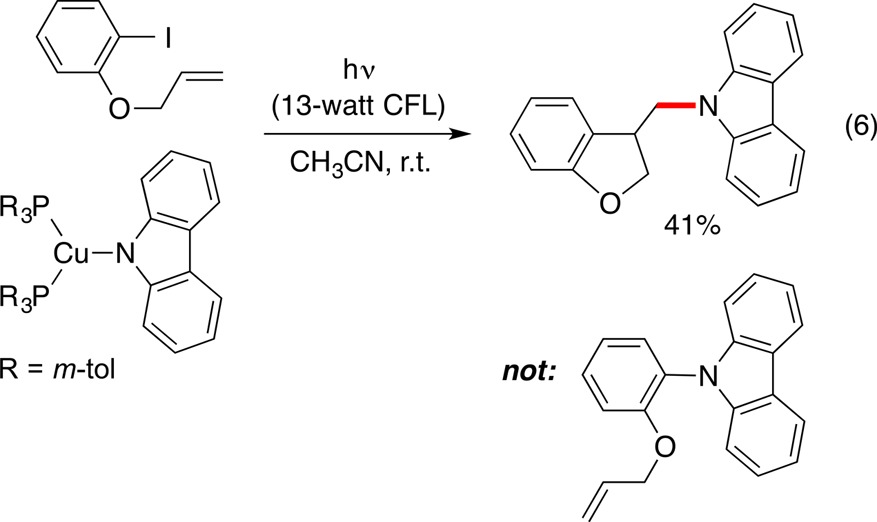 |
6 |
In an initial study, we established that the combination of a copper catalyst and light enables the alkylation of carbazoles by unactivated secondary alkyl iodides at 0 °C (eq 7).69 In the case of a trans-substituted cyclohexyl iodide, retention of stereochemistry is observed in the photoinduced, copper-catalyzed substitution reaction (eq 8); in contrast, under conditions commonly used for SN2 reactions (e.g., NaH/DMF), the alkyl iodide is consumed without formation of the substitution product (<1%).
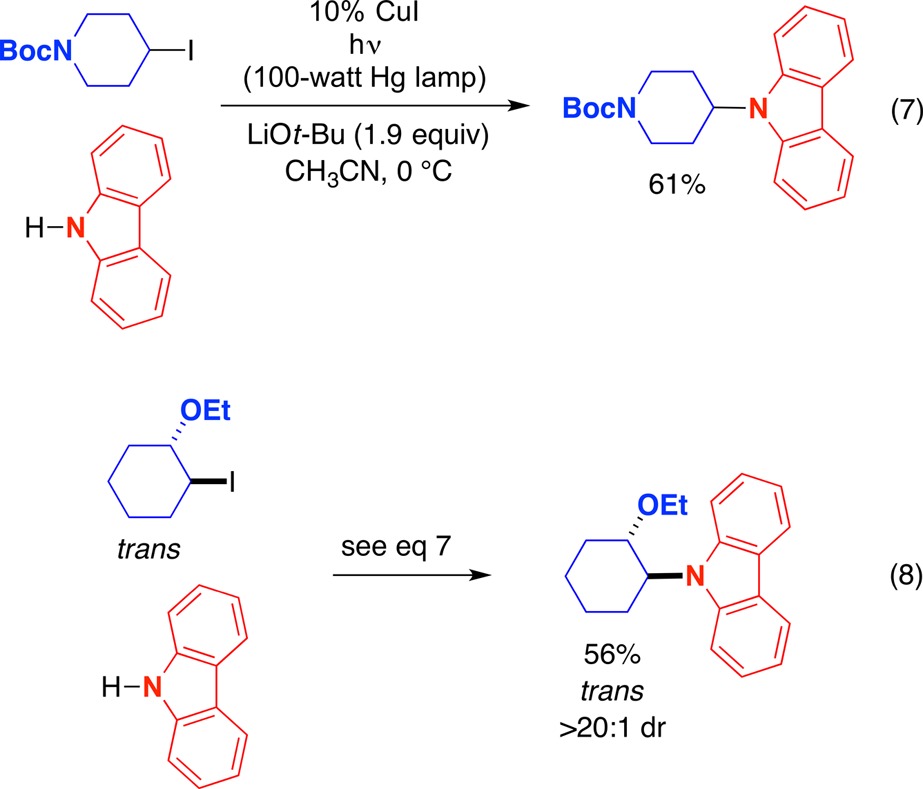 |
7 |
Recognizing that carbazole subunits are not frequently encountered in bioactive compounds, we turned our attention to the use of a different family of nitrogen nucleophiles, specifically, amides, since amides are widespread.70 We have determined that photoinduced, copper-catalyzed substitution can be achieved at room temperature with a variety of primary amides and alkyl electrophiles, including alkyl iodides, bromides, and chlorides (eq 9).71 In the absence of light or copper, essentially no alkylation is observed (<2%). Furthermore, overalkylation to the tertiary amide is not a significant side reaction (<2%).
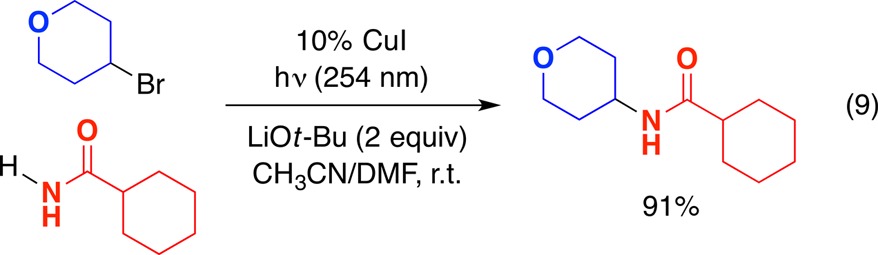 |
9 |
Next, we examined catalytic enantioselective substitution reactions of racemic alkyl electrophiles by nitrogen nucleophiles. Through a combination of CuI, a chiral monodentate phosphine, and blue LED lamps, enantioconvergent couplings of racemic tertiary alkyl chlorides can be achieved with good ee and yield (eq 10 and eq 11).72 Both carbazoles and indoles serve as suitable nucleophiles.73 To deliver high enantiomeric excess in these substitution reactions, the chiral catalyst must differentiate between three carbon-based substituents, which is often a difficult challenge in asymmetric synthesis.74,75
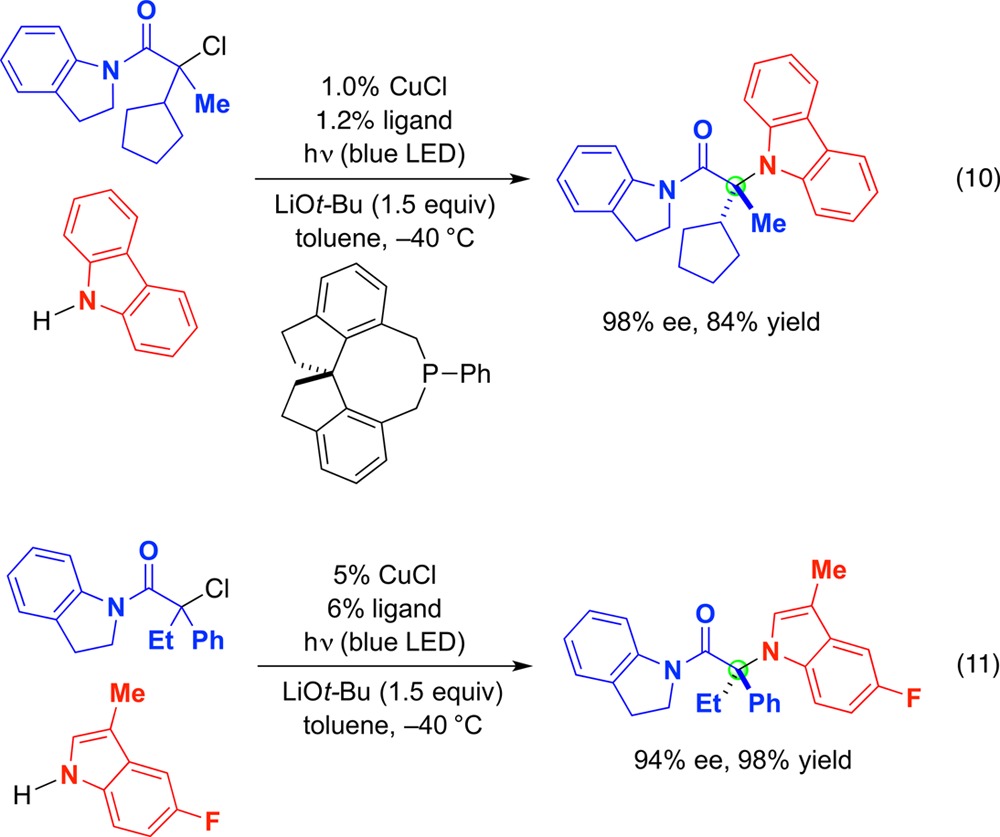 |
10 |
Conclusions
The ability to achieve, at will, any nucleophilic substitution reaction of an alkyl electrophile (and to control stereochemistry when relevant) would have a dramatic impact on organic synthesis. Although classical SN1 and SN2 pathways are unlikely to meet this challenge, radical-based processes that exploit transition-metal catalysis have considerable potential.
Thus, in recent years, radical-based transition-metal catalysis has enabled substitution reactions of unactivated secondary and tertiary alkyl electrophiles by carbon- as well as by heteroatom-based (e.g., boron, silicon, and nitrogen) nucleophiles. Furthermore, with respect to asymmetric catalysis, a broad array of enantioconvergent substitution processes have been described, using readily available racemic electrophiles. Clearly, however, given the enormous range of conceivable nucleophilic substitution processes, many interesting challenges have yet to be addressed. Consequently, the development of radical-based, metal-catalyzed reactions will no doubt be a fertile area of investigation for years to come.
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences: R01-GM062871 and R01-GM109194). All group members past and present, as well as Prof. Jonas C. Peters and his group, are gratefully acknowledged for their contributions to this research program.
The author declares no competing financial interest.
References
- Hartshorn S. R.Aliphatic Nucleophilic Substitution; Cambridge University Press: London, 1973. [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Substitutions at Aliphatic Centers and Thermal Isomerizations/Rearrangements. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; Chapter 11. [Google Scholar]
- For examples of interesting exceptions (albeit with limited scope with respect to the nucleophile and the electrophile), see:Reisman S. E.; Doyle A. G.; Jacobsen E. N. Enantioselective Thiourea-Catalyzed Additions to Oxocarbenium Ions. J. Am. Chem. Soc. 2008, 130, 7198–7199. 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Kaib P. S. J.; List B. Asymmetric Catalysis via Cyclic, Aliphatic Oxocarbenium Ions. J. Am. Chem. Soc. 2017, 139, 2156–2159. 10.1021/jacs.6b11993. [DOI] [PubMed] [Google Scholar]
- Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu C., Studer A., Eds.; John Wiley & Sons, Chichester, U.K., 2012; Vols. 1 and 2. [Google Scholar]
- Metal-Catalyzed Cross-Coupling Reactions and More; de Meijere A., Bräse S., Oestreich M., Eds.; Wiley–VCH: Weinheim, Germany, 2014. [Google Scholar]
- New Trends in Cross-Coupling; Colacot T. J., Ed.; Royal Society of Chemistry: Cambridge, U.K., 2015. [Google Scholar]
- For leading references, see:Labinger J. A. Tutorial on Oxidative Addition. Organometallics 2015, 34, 4784–4795. 10.1021/acs.organomet.5b00565. [DOI] [Google Scholar]
- For leading references, see:Jahn U. Radicals in Transition Metal Catalyzed Reactions? Transition Metal Catalyzed Radical Reactions?: A Fruitful Interplay Anyway. Top. Curr. Chem. 2011, 320, 323–452. 10.1007/128_2011_288. [DOI] [PubMed] [Google Scholar]
- For leading references, see:Iwasaki T.; Kambe N.. Coupling Reactions between sp3-carbon centers. In Comprehensive Organic Synthesis; Knochel P., Molander G. A., Eds.; Elsevier: Amsterdam, 2014; Vol. 3, pp 337–391. [Google Scholar]
- Manolikakes G.Coupling Reactions between sp3 and sp2 carbon centers. In Comprehensive Organic Synthesis; Knochel P., Molander G. A., Eds.; Elsevier: Amsterdam, 2014; Vol. 3, pp 392–464. [Google Scholar]
- Choi J.; Fu G. C. Transition metal–catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 2017, 356, eaaf7230. 10.1126/science.aaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B. W.; Steffens L. D.; Sigman M. S. The Wacker Oxidation. Org. React. 2014, 84, 75–413. 10.1002/0471264180.or084.02. [DOI] [Google Scholar]
- The Mizoroki–Heck Reaction; Oestreich M., Ed.; John Wiley & Sons: Chichester, U.K., 2009. [Google Scholar]
- For leading references to synthetic and mechanistic studies, see:Netherton M. R.; Fu G. C.. Palladium-Catalyzed Cross-Coupling Reactions of Unactivated Alkyl Electrophiles with Organometallic Compounds. Topics in Organometallic Chemistry: Palladium in Organic Synthesis; Tsuji J., Ed.; Springer: New York, 2005; pp 85–108. [Google Scholar]
- For a discussion and leading references, see:Tasker S. Z.; Standley E. A.; Jamison T. F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For example, see:Devasagayaraj A.; Stüdemann T.; Knochel P. A New Nickel-Catalyzed Cross-Coupling Reaction between sp3 Carbon Centers. Angew. Chem., Int. Ed. Engl. 1996, 34, 2723–2725. 10.1002/anie.199527231. [DOI] [Google Scholar]
- For leading references on the intermediacy of radicals in certain nickel-catalyzed couplings of aryl electrophiles, see ref (9).
- Organozinc nucleophiles:Zhou J.; Fu G. C. Cross-Couplings of Unactivated Secondary Alkyl Halides: Room-Temperature Nickel-Catalyzed Negishi Reactions of Alkyl Bromides and Iodides. J. Am. Chem. Soc. 2003, 125, 14726–14727. 10.1021/ja0389366. [DOI] [PubMed] [Google Scholar]
- Organoboron nucleophiles:Zhou J.; Fu G. C. Suzuki Cross-Couplings of Unactivated Secondary Alkyl Bromides and Iodides. J. Am. Chem. Soc. 2004, 126, 1340–1341. 10.1021/ja039889k. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Fu G. C. Alkyl–Alkyl Suzuki Cross-Coupling of Unactivated Secondary Alkyl Chlorides. Angew. Chem., Int. Ed. 2010, 49, 6676–6678. 10.1002/anie.201003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See also:González-Bobes F.; Fu G. C. Amino Alcohols as Ligands for Nickel-Catalyzed Suzuki Reactions of Unactivated Alkyl Halides, Including Secondary Alkyl Chlorides, with Arylboronic Acids. J. Am. Chem. Soc. 2006, 128, 5360–5361. 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]
- Saito B.; Fu G. C. Alkyl–Alkyl Suzuki Cross-Couplings of Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc. 2007, 129, 9602–9603. 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organosilicon nucleophiles:Powell D. A.; Fu G. C. Nickel-Catalyzed Cross-Couplings of Organosilicon Reagents with Unactivated Secondary Alkyl Bromides. J. Am. Chem. Soc. 2004, 126, 7788–7789. 10.1021/ja047433c. [DOI] [PubMed] [Google Scholar]
- See also:Strotman N. A.; Sommer S.; Fu G. C. Hiyama Reactions of Activated and Unactivated Secondary Alkyl Halides Catalyzed by a Nickel/Norephedrine Complex. Angew. Chem., Int. Ed. 2007, 46, 3556–3558. 10.1002/anie.200700440. [DOI] [PubMed] [Google Scholar]
- Organotin nucleophiles:Powell D. A.; Maki T.; Fu G. C. Stille Cross-Couplings of Unactivated Secondary Alkyl Halides Using Monoorganotin Reagents. J. Am. Chem. Soc. 2005, 127, 510–511. 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]
- For a mechanistic study that implicates radical intermediates in a nickel-catalyzed enantioconvergent coupling of an activated electrophile, see:Schley N. D.; Fu G. C. Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zultanski S. L.; Fu G. C. Nickel-Catalyzed Carbon–Carbon Bond-Forming Reactions of Unactivated Tertiary Alkyl Halides: Suzuki Arylations. J. Am. Chem. Soc. 2013, 135, 624–627. 10.1021/ja311669p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen G.; Kazmaier U.; Förster S.. Enantioselective allylic substitutions with carbon nucleophiles. In Catalytic Asymmetric Synthesis; Ojima I., Ed.; John Wiley & Sons: Hoboken, NJ, 2010; pp 497–641. [Google Scholar]
- For leading references, see:Cherney A. H.; Kadunce N. T.; Reisman S. E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds. Chem. Rev. 2015, 115, 9587–9652. 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer C.; Fu G. C. Asymmetric Nickel-Catalyzed Negishi Cross-Couplings of Secondary α-Bromo Amides with Organozinc Reagents. J. Am. Chem. Soc. 2005, 127, 4594–4595. 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- See also:Lundin P. M.; Esquivias J.; Fu G. C. Catalytic Asymmetric Cross-Couplings of Racemic α-Bromoketones with Arylzinc Reagents. Angew. Chem., Int. Ed. 2009, 48, 154–156. 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Fu G. C. Catalytic Asymmetric Synthesis of Tertiary Alkyl Fluorides: Negishi Cross-Couplings of Racemic α,α-Dihaloketones. J. Am. Chem. Soc. 2014, 136, 5520–5524. 10.1021/ja501815p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzylic halides:Binder J. T.; Cordier C. J.; Fu G. C. Catalytic Enantioselective Cross-Couplings of Secondary Alkyl Electrophiles with Secondary Alkylmetal Nucleophiles: Negishi Reactions of Racemic Benzylic Bromides with Achiral Alkylzinc Reagents. J. Am. Chem. Soc. 2012, 134, 17003–17006. 10.1021/ja308460z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See also:Arp F. O.; Fu G. C. Catalytic Enantioselective Negishi Reactions of Racemic Secondary Benzylic Halides. J. Am. Chem. Soc. 2005, 127, 10482–10483. 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- Do H.-Q.; Chandrashekar E. R. R.; Fu G. C. Nickel/Bis(oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc. 2013, 135, 16288–16291. 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allylic halides:Son S.; Fu G. C. Nickel-Catalyzed Asymmetric Negishi Cross-Couplings of Secondary Allylic Chlorides with Alkylzincs. J. Am. Chem. Soc. 2008, 130, 2756–2757. 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- Propargylic electrophiles:Oelke A. J.; Sun J.; Fu G. C. Nickel-Catalyzed Enantioselective Cross-Couplings of Racemic Secondary Electrophiles That Bear an Oxygen Leaving Group. J. Am. Chem. Soc. 2012, 134, 2966–2969. 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See also:Smith S. W.; Fu G. C. Nickel-Catalyzed Asymmetric Cross-Couplings of Racemic Propargylic Halides with Arylzinc Reagents. J. Am. Chem. Soc. 2008, 130, 12645–12647. 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- α-Halonitriles:Choi J.; Fu G. C. Catalytic Asymmetric Synthesis of Secondary Nitriles via Stereoconvergent Negishi Arylations and Alkenylations of Racemic α-Bromonitriles. J. Am. Chem. Soc. 2012, 134, 9102–9105. 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluvirucinine A1: ref (37).
- Carolacton:Schmidt T.; Kirschning A. Total Synthesis of Carolacton, a Highly Potent Biofilm Inhibitor. Angew. Chem., Int. Ed. 2012, 51, 1063–1066. 10.1002/anie.201106762. [DOI] [PubMed] [Google Scholar]
- Daphenylline:Yamada R.; Adachi Y.; Yokoshima S.; Fukuyama T. Total Synthesis of (−)-Daphenylline. Angew. Chem., Int. Ed. 2016, 55, 6067–6070. 10.1002/anie.201601958. [DOI] [PubMed] [Google Scholar]
- For example, see:Qin T.; Cornella J.; Li C.; Malins L. R.; Edwards J. T.; Kawamura S.; Maxwell B. D.; Eastgate M. D.; Baran P. S. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organomagnesium nucleophiles:Lou S.; Fu G. C. Nickel/Bis(oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. J. Am. Chem. Soc. 2010, 132, 1264–1266. 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organosilicon nucleophiles:Dai X.; Strotman N. A.; Fu G. C. Catalytic Asymmetric Hiyama Cross-Couplings of Racemic α-Bromo Esters. J. Am. Chem. Soc. 2008, 130, 3302–3303. 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- Organozirconium nucleophiles:Lou S.; Fu G. C. Enantioselective Alkenylation via Nickel-Catalyzed Cross-Coupling with Organozirconium Reagents. J. Am. Chem. Soc. 2010, 132, 5010–5011. 10.1021/ja1017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See also:Choi J.; Martín-Gago P.; Fu G. C. Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones. J. Am. Chem. Soc. 2014, 136, 12161–12165. 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organoboron nucleophiles:Lundin P. M.; Fu G. C. Asymmetric Suzuki Cross-Couplings of Activated Secondary Alkyl Electrophiles: Arylations of Racemic α-Chloroamides. J. Am. Chem. Soc. 2010, 132, 11027–11029. 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromatic ring:Saito B.; Fu G. C. Enantioselective Alkyl–Alkyl Suzuki Cross-Couplings of Unactivated Homobenzylic Halides. J. Am. Chem. Soc. 2008, 130, 6694–6695. 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- Carbonyl:Owston N. A.; Fu G. C. Asymmetric Alkyl–Alkyl Cross-Couplings of Unactivated Secondary Alkyl Electrophiles: Stereoconvergent Suzuki Reactions of Racemic Acylated Halohydrins. J. Am. Chem. Soc. 2010, 132, 11908–11909. 10.1021/ja105924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See also:Zultanski S. L.; Fu G. C. Catalytic Asymmetric γ-Alkylation of Carbonyl Compounds via Stereoconvergent Suzuki Cross-Couplings. J. Am. Chem. Soc. 2011, 133, 15362–15364. 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitrogen:Lu Z.; Wilsily A.; Fu G. C. Stereoconvergent Amine-Directed Alkyl–Alkyl Suzuki Reactions of Unactivated Secondary Alkyl Chlorides. J. Am. Chem. Soc. 2011, 133, 8154–8157. 10.1021/ja203560q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulfonamide:Wilsily A.; Tramutola F.; Owston N. A.; Fu G. C. New Directing Groups for Metal-Catalyzed Asymmetric Carbon–Carbon Bond-Forming Processes: Stereoconvergent Alkyl–Alkyl Suzuki Cross-Couplings of Unactivated Electrophiles. J. Am. Chem. Soc. 2012, 134, 5794–5797. 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Fu G. C. Stereoconvergent Negishi Arylations of Racemic Secondary Alkyl Electrophiles: Differentiating between a CF3 and an Alkyl Group. J. Am. Chem. Soc. 2015, 137, 9523–9526. 10.1021/jacs.5b04725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Choi J.; Liu A. T.; Slusarczyk M.; Fu G. C. A general, modular method for the catalytic asymmetric synthesis of alkylboronate esters. Science 2016, 354, 1265–1269. 10.1126/science.aai8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron Science: New Technologies and Applications; Hosmane N. S., Ed.; CRC Press: Boca Raton, FL, 2012. [Google Scholar]
- Organosilicon Chemistry; Auner N., Weis J., Eds.; Wiley–VCH: Weinheim, Germany, 2005. [Google Scholar]
- Boron nucleophile:Dudnik A. S.; Fu G. C. Nickel-Catalyzed Coupling Reactions of Alkyl Electrophiles, Including Unactivated Tertiary Halides, To Generate Carbon–Boron Bonds. J. Am. Chem. Soc. 2012, 134, 10693–10697. 10.1021/ja304068t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silicon nucleophile:Chu C. K.; Liang Y.; Fu G. C. Silicon–Carbon Bond Formation via Nickel-Catalyzed Cross-Coupling of Silicon Nucleophiles with Unactivated Secondary and Tertiary Alkyl Electrophiles. J. Am. Chem. Soc. 2016, 138, 6404–6407. 10.1021/jacs.6b03465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-T.; Zhang Z.-Q.; Tajuddin H.; Wu C.-C.; Liang J.; Liu J.-H.; Fu Y.; Czyzewska M.; Steel P. G.; Marder T. B.; Liu L. Alkylboronic esters from copper-catalyzed borylation of primary and secondary alkyl halides and pseudohalides. Angew. Chem., Int. Ed. 2012, 51, 528–532. 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]
- Ito H.; Kubota K. Copper(I)-Catalyzed Boryl Substitution of Unactivated Alkyl Halides. Org. Lett. 2012, 14, 890–893. 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- For example, see:Alkaloids: A Treasure of Poisons and Medicines; Funayama S., Cordell G. A., Eds.; Academic Press: Waltham, MA, 2015. [Google Scholar]
- There were isolated reports of N-alkylations of amines with unactivated alkyl halides that proceed in the presence of a substoichiometric quantity of a transition metal. For example, see:Aydin A.; Kaya I. Synthesis and characterization of yellow and green light emitting novel polymers containing carbazole and electroactive moieties. Electrochim. Acta 2012, 65, 104–114. (165 °C; primary alkyl bromide) 10.1016/j.electacta.2012.01.028. [DOI] [Google Scholar]
- Tu X.; Fu X.; Jiang Q.; Liu Z.; Chen G. The synthesis and electrochemical properties of cathodic–anodic composite electrochromic materials. Dyes Pigm. 2011, 88, 39–43. (80 °C; primary alkyl bromide) 10.1016/j.dyepig.2010.04.012. [DOI] [Google Scholar]
- Kozuka M.; Tsuchida T.; Mitani M. The direct synthesis of tertiary amines with three different substituents via the reaction of primary amines, alkyl halides, and α-chlorine substituted allylsilanes catalyzed by Lewis acids. Tetrahedron Lett. 2005, 46, 4527–4530. (83 °C; primary alkyl bromide) 10.1016/j.tetlet.2005.05.022. [DOI] [Google Scholar]
- Creutz S. E.; Lotito K. J.; Fu G. C.; Peters J. C. Photoinduced Ullmann C-N coupling: demonstrating the viability of a radical pathway. Science 2012, 338, 647–651. 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- For an overview of Ullmann reactions, see: Copper-Mediated Cross-Coupling Reactions; Evano G., Blanchard N., Eds.; John Wiley & Sons: Hoboken, NJ, 2014. [Google Scholar]
- Bissember A. C.; Lundgren R. J.; Creutz S. E.; Peters J. C.; Fu G. C. Transition-metal-catalyzed alkylations of amines with alkyl halides: photoinduced, copper-catalyzed couplings of carbazoles. Angew. Chem., Int. Ed. 2013, 52, 5129–5133. 10.1002/anie.201301202. [DOI] [PubMed] [Google Scholar]
- For example, amides are the most frequently occurring functional group in the IBEX database of patents in the area of medicinal chemistry:Brown D. G.; Boström J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?. J. Med. Chem. 2016, 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- Do H.-Q.; Bachman S.; Bissember A. C.; Peters J. C.; Fu G. C. Photoinduced, Copper-Catalyzed Alkylation of Amides with Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc. 2014, 136, 2162–2167. 10.1021/ja4126609. [DOI] [PubMed] [Google Scholar]
- Kainz Q. M.; Matier C. M.; Bartoszewicz A.; Zultanski S. L.; Peters J. C.; Fu G. C. Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 2016, 351, 681–684. (mechanistic studies of this process are underway) 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble G. W., Ed.; Springer–Verlag: Berlin, 2010. [Google Scholar]
- Quasdorf K. W.; Overman L. E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516, 181–191. 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis; Christoffers J., Baro A., Eds.; Wiley–VCH: New York, 2005. [Google Scholar]