

Abstract
The antimalarial artemisinin (ART) possesses anticancer activity, but its underlying mechanism remains largely unclear. Using a chemical proteomics approach with artemisinin-based activity probes, we identified over 300 specific ART targets. This reveals an anticancer mechanism whereby ART promiscuously targets multiple critical biological pathways and leads to cancer cell death. The specific cytotoxicity of ART against colorectal cancer (CRC) cells rather than normal colon epithelial cells is due to the elevated capacity of heme synthesis in the cancer cells. Guided by this mechanism, the specific cytotoxicity of ART toward CRC cells can be dramatically enhanced with the addition of aminolevulinic acid (ALA), a clinically used heme synthesis precursor, to increase heme levels. Importantly, this novel ART/ALA combination therapy proves to be more effective than an ART monotherapy in a mouse xenograft CRC model. Thus, ART can be repurposed and potentiated by exploitation of its mechanism of action and the metabolic features of the CRC cells.
Short abstract
Chemical proteomics reveals a role of heme in the activation and anticancer activity of artemisinin. Based on this mechanism, we propose a novel combination treatment to enhance its efficacy.
Introduction
Artemisinin is an active compound of a Chinese herbal medicine that has been used to treat chills and fever for at least 2000 years.1 Currently, artemisinin and its derivatives (herein collectively referred to as ART) are important frontline drugs against uncomplicated malarial infections.2,3 Because of their potency and rapid onset of action with few documented adverse effects, they are effective in eliminating the otherwise multidrug resistant parasite, Plasmodium falciparum.4 Beyond these well-established antimalarial properties, there is accumulating evidence demonstrating that artemisinin and its derivatives possess cytotoxic effects against many human cancer cell types both in vitro and in animal experiments in vivo.5−10 As an extension of these studies, our group in the United Kingdom has recently conducted a randomized double-blind pilot clinical phase II trial using oral artesunate (the salt form of artesunic acid, a derivative of artemisinin) neoadjuvant therapy in colorectal cancer (CRC) patients. It was shown that artesunate is generally well tolerated and has antiproliferative properties in CRC patients.11 These preliminary data suggest artesunate as a promising drug in CRC treatment regimens, and a more thorough study is being conducted with a larger patient sample. Yet, the detailed anticancer mechanism of action (MOA) of ART has not been fully understood.12 Artemisinin and its derivatives such as artesunate are prodrugs which require cellular activation to cleave the endoperoxide moiety for antimalarial and anticancer efficacy.13−17 However, the identity of the activator remains inconclusive. On one hand, previous studies have proposed the role of iron18−24 in the in vitro decay of ART endoperoxide. On the other hand, other studies have pointed out a similar function for heme depending on the context of the cellular environment.13−17,25−27 Recently, using chemical proteomics approaches, we28−31 and other groups32−34 have independently shown that ART can be activated by heme before indiscriminately alkylating multiple proteins. However, given the biological differences between cancer cells and malaria parasites, the postactivation or downstream mechanisms by which ART exerts its cytotoxicity are likely to differ from its antimalaria mechanism.12,28−35 Here, we sought to understand the following in the specific context of cancer cells: (i) What are the detailed pathways affected by ART treatment and the mechanisms explaining its anticancer effects? (ii) How can ART specifically kill cancer cells but not normal cells? (iii) Is it possible to further enhance the anticancer effect of ART, given that ART derivatives have been selected for antimalarial potency but not anticancer efficacy?
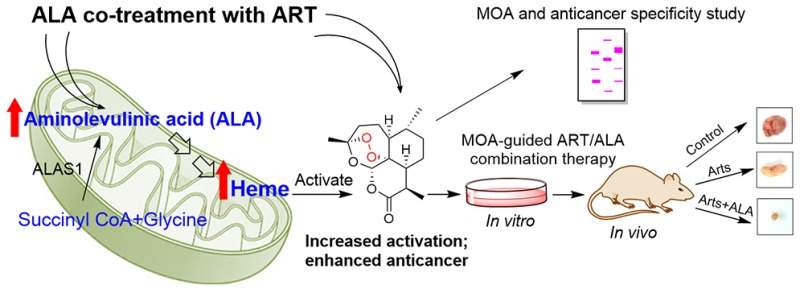
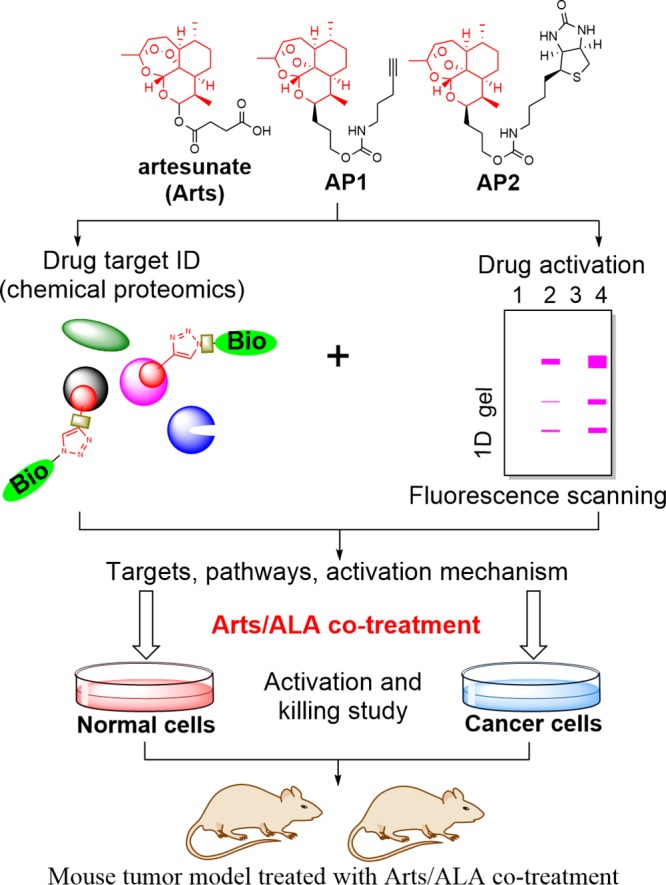
To address these questions, we first used alkyne (AP1)- and biotin (AP2)-tagged ART probes (Figure 1) to specifically identify protein targets and their implicated biological pathways in CRC cells. Several pathways that were disrupted by ART treatment were examined in detail. Importantly, we found that the specificity of ART against CRC cells could stem from the elevated capacity of heme synthesis in the cancer cells versus the normal cells. Based on this distinctive metabolic feature of CRC cells, we introduced a clinically used heme synthesis precursor, aminolevulinic acid (ALA), as a sensitizer of artesunate to specifically increase the heme level and thus dramatically enhance its anticancer effects. The efficacy of this newly developed ART/ALA combination therapy was further validated in a mouse xenograft CRC model (Figure 1).
Figure 1.
An outline of the quantitative chemical proteomics study of the molecular mechanism for ART’s specific anticancer property and its combination with aminolevulinic acid (ALA).
Results and Discussion
To characterize the direct binding targets of ART and its MOA in CRC cells, an alkyne-tagged artemisinin-based probe AP1 was used to enrich and visualize its interacting protein profile on an SDS–PAGE gel.28,29 To further increase the stringency of protein target identification, we also synthesized a biotin-tagged ART probe AP2 (Scheme 1). Only the targets that were consistently identified with both probes were considered as specific ART targets in CRC cells. The in vitro growth inhibitory assay confirmed that both probes largely retained the anti-CRC effects as shown in Figure S1, indicating that our tagging strategies do not interfere with drug activation or targeting (Figure 1).
Scheme 1. Structure and Synthetic Scheme of Alkyne-Tagged Artemisinin Probe AP1 and Biotin-Tagged Artemisinin Probe AP2.

Of note, AP1 has been published in our previous report.28
The protein binding targets of ART in CRC cells were labeled with AP1 and visualized by conjugating AP1 with a fluorescence dye through click chemistry. Live CRC cells treated with increasing concentrations of AP1 showed increasing levels of drug target labeling upon in-gel fluorescent scanning (Figure 2b). The direct targets of ART were enriched by streptavidin affinity purification and identified with mass spectrometry. Nonspecifically bound proteins were filtered out by comparing the enrichment ratio of the pull-down group versus the control group, with a stringent enrichment ratio of 2 as the cutoff (with a minimum of two quantified peptides). In total, 304 proteins were consistently identified by both probes as specific targets of ART (the full list is shown in Table S1). Seven representative proteins from the target list were selected, and their interactions with AP1 were further verified by immunoblotting (Figures 2a and 2c).
Figure 2.

(a) Selective targets of ART in HCT116 cells. (b) In situ fluorescence labeling of HCT 116 cells to show the cellular targets of ART. (c) Western blot validation of pulled-down fractions of HCT116 by AP1 (or DMSO as negative control) with respective antibodies.
Gene ontology (GO) analysis revealed that ART’s cellular targets are widely distributed in different subcellular compartments (Figure 3a). To visualize the cellular localization of ART binding targets, we employed a fluorescence (TAMRA)-tagged ART probe. Probe-treated CRC cells showed high level of fluorescent signal evenly distributed throughout the cells, consistent with the GO analysis. No fluorescence could be observed in the DMSO-treated control cells (Figure 3b). Ingenuity pathway analysis (IPA) further suggested that ART may exert its cytotoxicity on cancer cells by targeting multiple critical functions and pathways including the EIF2, mitochondrial functions, phagosome maturation, and eIF4/p70S6K pathways (Figure 3c), with possible influences on the fatty acid metabolism, protein synthesis, autophagy, and free radical scavenging process (Figure 3d). Based on these predictions, we next carried out validation studies on possible functional pathways affected by ART treatment.
Figure 3.

(a) Cellular components of the ART targets determined by GO analysis. (b) Images of HCT116 cells treated with red fluorescent ART. Cells were treated with TAMRA-tagged ART probe (20 μM) for 30 min and were subjected to analysis by confocal microscopy. (c) Top canonical pathways with the ART protein targets significantly over-represented. (d) Top cellular and molecular functions that are associated with ART-targeted proteins.
First, we examined the changes in the levels of protein synthesis (Figures 4a, 3d, and S2) in HCT116 cells upon artesunate treatment using the artificial amino acid azidohomoalanine (AHA), a methionine analogue that is incorporated into proteins that are newly synthesized and allows the dynamic monitoring of de novo protein synthesis.36,37 Newly synthesized proteins with AHAs incorporated were subsequently conjugated with a fluorescence tag through a click chemistry reaction, and flow cytometry was used to measure the fluorescence intensity of the labeled cells to quantify the relative level of newly synthesized proteins. It was shown that artesunate inhibited the de novo protein synthesis in a dose dependent manner, and 2 μM artesunate treatment for 12 h led to a 50% reduction of the protein synthesis in HCT116 cells (Figure 4a).
Figure 4.

(a) Dose-dependent reduction of protein synthesis by artesunate (Arts) (0.5 μM, 1 μM, 2 μM, 12 h) in HCT116 cells. Data shown are mean ± SD from triplicate experiments (*p < 0.05, **p < 0.01, Student’s t-test). (b) Cell viability of HCT116 cells treated with Arts (2 μM) for 24 h with or without the presence of palmitate. (c) Arts increases autophagic flux in HCT116 cells. HCT116 cells were treated with Arts (5 μM) for 12 h with or without chloroquine (CQ; 25 μM), and cell lysates were prepared for Western blot. (d) HeLa cells stably expressing GFP-LC3 were treated with Arts (5 μM) for 12 h. GFP-LC3 puncta were observed under confocal microscopy. (e) ART activates lysosomal function in HCT116 cells. HCT116 cells were treated with Arts (5 μM) for 12 h and then stained with LysoTracker Red DND-99 (50 nM) for 15 min. Confocal microscopy was performed to analyze. (f) ROS level increases in HCT116 cells upon Arts treatment. Cells were treated with 5 μM Arts for 12 h, followed by DCFH-DA staining and flow cytometry analysis.
In cancer pathogenesis, endogenous fatty acid biosynthesis is substantially upregulated to provide essential building blocks for phospholipid membranes, conferring growth advantages to cancer cells.38 Our chemical proteomics data and pathway analysis identified fatty acid synthase (FASN) as well as various other mediators of lipid metabolism as ART binding targets (Figures 2a and S3). FASN inhibition has been investigated as a potential therapeutic avenue for specific cancers.38−40 To investigate ART’s effect on FASN, palmitate—the end product of FASN’s catalytic reaction—was used to treat HCT116 cells with or without the presence of artesunate. As shown in Figure 4b, the antiproliferative effect of artesunate was partially rescued by palmitate addition. This suggests that artesunate may inhibit fatty acid synthesis through binding with FASN and other targets (Figures S3), partially contributing to its anticancer effect.
Autophagy is a catabolic process that results in degradation of bulk cytoplasmic contents, abnormal protein aggregates, and excess or damaged organelles through autophagosome–lysosome fusion,41 which is negatively regulated by the kinase mTOR. mTOR activation by Akt or MAPK signaling inhibits autophagy whereas mTOR inhibition by AMPK or p53 signaling enhances autophagy.42 As reflected in our pathway analysis, EIF2, phagosome maturation, and eIF4/p70S6K pathways were involved in ART-treated cells. These pathways are highly related to the autophagy process. Here, we examined the effect of ART on autophagy. As shown in Figure 4c, in the presence of chloroquine, a lysosomal inhibitor,43 artesunate significantly enhanced the levels of autophagic marker LC3-II and reduced levels of autophagy substrate p62, indicating that artesunate causes cellular damage and increases autophagic flux in HCT116 cells. Consistent with this, confocal microscopy showed that artesunate also increased the abundance of GFP-LC3 puncta in Hela cells (Figure 4d). Moreover, lysoTracker staining in HCT116 cells showed that artesunate significantly enhanced lysosomal activation (Figure 4e), which further promotes the autophagosomal degradation during later stages of autophagy.
Our pathway analysis also suggested mitochondrial dysfunction upon ART treatment, which might lead to elevated reactive oxygen species (ROS) production. We found that artesunate significantly increased ROS levels (Figure 4f) in HCT116 cells while cotreatment with ROS scavengers could partially rescue the cell death induced by artesunate (Figure S4). The increase of ROS not only directly contributes to cancer cell death but also leads to autophagosome formation and autolysosomal degradation, which further overwhelms the cellular system.6,35,44
After detailed dissection of the functional pathways for ART’s anticancer effects, we next attempted to uncover the molecular mechanism of the specificity of ART against cancer cells versus normal cells. Previous research by us and other groups have shown that artesunate is activated by heme to generate reactive free radicals that can promiscuously alkylate various cellular targets.14,28−34 Heme, as an essential cofactor of many important cellular enzymes, is involved in several critical steps of cell metabolism.45,46 We hypothesize that cancer cells, with higher metabolic rate for rapid growth, may also require higher levels of heme synthesis. Thus, ART activation could be specifically enhanced in this context. The heme levels of HCT116 (colon cancer cell line) and CCD841 (normal colon epithelial cell line) were first measured and compared. Consistent with our hypothesis, the cancerous HCT116 cells have higher heme level than the noncancerous CCD841 cells (Figure 5a). In mammalian cells, heme is synthesized in both the mitochondria and cytosol by a series of metabolic reactions (Figure 5b).45 The rate-limiting step of the heme synthesis pathway is the production of aminolevulinic acid (ALA), catalyzed by ALA synthase (ALAS1).47 We therefore examined the expression level of this key enzyme by immunoblotting. Our results demonstrated that the ALAS1 level in HCT116 cells was substantially higher than that of CCD841 cells (Figure 5c and 5d), suggesting an enhanced capacity of heme synthesis in cancer cells to sustain faster growth. Notably, we found that the amount of ALAS1 was significantly higher in HCT116 cells when treated with artesunate, while minimal change was found in the normal cells (Figures 5c and 5d). Consistent with enhanced heme synthesis in HCT116 cells, a substantially higher level of ART activation and targeting was observed by in-gel fluorescence scanning (Figure 5e). Importantly, this correlated well with the viability of HCT116 cells and CCD 841 cells upon ART treatment (Figures 5f and 5g). The elevated heme level in cancer cells thus corresponds to the enhanced activation of ART and its subsequent multipathway targeting, and eventually cell death. In contrast, heme levels in normal cells and tissues are strictly controlled and maintained at lower levels, minimizing ART’s activation, which could possibly explain the specificity and low toxicity of ART.46 However, we cannot rule out the potential contribution of the intracellular iron inside cancer cells to ART activation, the broad targeting spectrum, and its downstream events.20−22,24,48,49
Figure 5.

(a) Assessment of heme levels in normal (CCD841) and colorectal cancer cells (HCT116) in the absence (basal) and presence of 1 mM ALA treatment for 24 h. (b) Schematic diagram of the effects of exogenous modulators on the heme biosynthetic pathway. (c) The protein levels of heme biosynthetic enzyme ALAS1 in CCD841 and HCT116 in the absence (basal) and presence of artesunate (Arts) treatment. (d) Normalized protein expression levels of ALAS1 for the results from panel c. Tubulin level was used for normalization. (e) The fluorescence labeling (4 h) of HCT116 and CCD841 cells with and without (basal) 1 mM ALA treatment. (f) Effects of Arts and ALA combination treatment on HCT116 cell viability. (g) Effects of Arts and ALA combination treatment on CCD 841 cell viability. Error bars represent SD of triplicate experiments in panels a, d, f, and g.
Although ART has shown some promising anticancer effects, its potency is somewhat limited compared to its remarkable antimalarial effects and the IC50 of ART against cancer is in the micromolar range.5 Artesunate is transformed to dihydroartemisinin (DHA), and DHA has a relatively short elimination half-life (∼45m) with Cmax values in the low micromolar range when used in antimalarial therapies.50 The high potency of artesunate in antimalarial parasite therapy (with IC50 values in the low nanomolar range) is due to rapid drug activation triggered by high concentration of heme released by hemoglobin digestion.14 Thus, the antimalarial effect is not compromised given the limited drug exposure time.51 Although ART can be similarly activated by heme in cancer cells, the heme level is much lower compared to the case of malaria parasites which undergo massive hemoglobin digestion.3 Previously, we have developed a mitochondrial targeting ART analogue that can target ART to the sites of heme synthesis in mammalian cells.29 In this manner, ART activation can be enhanced by directly delivering the drug to heme-concentrated regions of the cell. Thus, we hypothesized that another possible strategy to enhance ART’s anticancer effect is to increase the level of intracellular heme, thereby enhancing ART activation and its cytotoxicity. As mentioned above, we have found that the ALA synthase enzyme was highly expressed in cancer cells in comparison with normal cells, implying a higher capacity of cancer cells to utilize ALA for heme synthesis. ALA itself is a clinically used drug in photodynamic therapy for imaging-guided surgery or tumor treatment.52 It has been reported to have minimal side effects and is clinically safe for cancer treatment.52−54 Therefore, we introduced ALA as a potential sensitizer to enhance the efficacy of artesunate by increasing heme levels to enhance artesunate activation. We examined cancer cell viability after treatments with artesunate, artesunate + ALA, and artesunate + ALA + SA (succinylacetone, an inhibitor of heme synthesis) respectively. As shown in Figure 5f, artesunate treatment alone for 24 h had an IC50 of approximately 2 μM, while the addition of ALA lowered the IC50 approximately 10-fold to around 200 nM. The addition of the heme synthesis inhibitor SA fully blocked the cytotoxicity of artesunate treatments, regardless of the presence of ALA, strongly supporting a heme-centric mechanism for both single and combination treatments. In normal cells, no significant killing is observed in either ART or ART + ALA treatment even when the concentration of ART was as high as 20 μM (Figure 5g). Consistently, addition of ALA indeed increased intracellular heme level and enhanced ART activation in cancer cells but had negligible effect in normal cells (Figures 5a and 5e). This may explain the specific enhancement of the anticancer effects of the ART + ALA cotreatment.
Finally, to determine whether the efficacy of artesunate and ALA combined treatment against CRC cells (HCT116) observed in vitro can be recapitulated in vivo in mouse xenograft models, the following treatments were administered to male nude mice inoculated with visible tumors: (1) control (PBS), (2) ALA, (3) artesunate, and (4) artesunate + ALA for 8 days. Consistent with previous reports, artesunate administration exhibited inhibitory effects against tumor growth (Figures 6a and 6b; Figures S5a and S5b).55,56 Strikingly, the combination of artesunate and ALA showed significant tumor growth delay in comparison to both the control and the artesunate or ALA single treatment groups, supporting the notion that ALA can potentiate the anticancer effects of artesunate. Interestingly, ALA treatment also inhibited tumor growth to some extent, which might be related to its potential photodynamic efficacy on tumors.52−54 As a control, the tolerance of the combined treatment of artesunate and ALA was confirmed by the comparable body weight reduction among different groups (Figure S5c). Consistently, the ART activation level in the mouse tumor tissue was shown to be elevated by the combination treatment, as revealed by in-gel fluorescence scan (Figure 6c).
Figure 6.

(a, b) In vivo efficacy comparison of artesunate (Arts), ALA, and combined treatment in human colon cancer xenograft models. Five days after subcutaneous implantation of 3 × 106 HCT116 cells in PBS, male nude mice received one dose of the following treatments: (1) Ctrl (PBS), (2) ALA (100 mg/kg), (3) Arts (50 mg/kg), or (4) Arts (50 mg/kg) plus ALA (100 mg/kg) via intraperitoneal (ip) injection every day. After 8 days of administration, tumors were isolated, and tumor volumes were examined and estimated every day. Each data point represents mean tumor volume of at least 7 tumors in each group ± SE. p value, *<0.05; **<0.01. (c) The mice were treated for 4 h with AP1 probe or other treatments, and the tumors were harvested and lysed for in-gel fluorescence scanning.
Conclusions
In conclusion, we have comprehensively characterized the targets and biological pathways disrupted by ART in the context of colorectal cancer. We propose a heme-centric mechanism to explain ART activation and specificity in cancers. Based on this heme-centric activation model, we further designed a novel ART combination therapy by incorporation of a heme precursor ALA as a potent enhancer of ART’s anticancer effects, and demonstrated the efficacy of this combination in both cancer cell lines and animal models. Since both artesunate and ALA are clinically used and well-tolerated,52 this combination has the potential to be safely applied to subsequent clinical testing, pending further study into its pharmacokinetics and pharmacodynamics profiles. As the level of free ferrous iron is relatively high in cancer cells and it can catalyze the breakage of the endoperoxide bridge of ART,18−24 its contribution both to ART activation and to downstream events in cell lines and animal models should be further investigated. Iron-based strategies, such as the well-reported coadministration of ART with holotransferrin,57,58 might also turn out to be an interesting avenue of exploration in conjunction with the presently described heme-based combination. Finally, alternative formulations such as several artemisinin-derived dimers7−10,22,59,60 with higher anticancer potency have been developed recently, and these promising artemisinin derivatives can also be tested as combination therapy in the future.
Acknowledgments
We thank the National Medical Research Council Singapore (NMRC/CIRG/1373/2013), the Chinese National Natural Sciences Foundation (81630092, 81421091), and the Doctoral Station Science Foundation from the Chinese Ministry of Education of China (20130091130003) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00156.
Author Contributions
J.W. conceived the idea and supervised this project, designed and conducted experiments, and wrote the manuscript. J.Z. and Y.S. performed part of the experiments. C.X. contributed the idea and helped with the manuscript writing. C.Z. assisted with the design of the probes. Y.K.W., Y.M.L., Y.H., T.K.L., and W.S. assisted with the mass spectrometry experiments. S.K. and Z.-C.H. guided the data analysis and manuscript writing. J.W., H.-M.S. and Q.L. supervised this project.
Author Contributions
⊥ J.W., J.Z., Y.S., and C.X. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Tu Y. Artemisinin-A Gift from Traditional Chinese Medicine to the World (Nobel Lecture). Angew. Chem., Int. Ed. 2016, 55, 10210–10226. 10.1002/anie.201601967. [DOI] [PubMed] [Google Scholar]
- Eastman R. T.; Fidock D. A. Artemisinin-Based Combination Therapies: A Vital Tool in Efforts to Eliminate Malaria. Nat. Rev. Microbiol. 2009, 7, 864–874. 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow C. J.; White N. J. The Clinical Impact of Artemisinin Resistance in Southeast Asia and the Potential for Future Spread. FEMS Microbiol. Rev. 2017, 41, 34–48. 10.1093/femsre/fuw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S.; Bustamante L.; Haynes R. K.; Staines H. M. Artemisinins: Their Growing Importance in Medicine. Trends Pharmacol. Sci. 2008, 29, 520–527. 10.1016/j.tips.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T.; Dunstan H.; Sauerbrey A.; Miyachi H.; Chitambar C. R. The Anti-Malarial Artesunate Is Also Active against Cancer. Int. J. Oncol. 2001, 18, 767–773. 10.3892/ijo.18.4.767. [DOI] [PubMed] [Google Scholar]
- Ho W. E.; Peh H. Y.; Chan T. K.; Wong W. S. F. Artemisinins: Pharmacological Actions beyond Anti-Malarial. Pharmacol. Ther. 2014, 142, 126–139. 10.1016/j.pharmthera.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Ndreshkjana B.; Muenzner J. K.; Reiter C.; Hofmeister E.; Mederer S.; Fatfat M.; El-Baba C.; Gali-Muhtasib H.; Schneider-Stock R.; Tsogoeva S. B. Synthesis of Novel Hybrids of Thymoquinone and Artemisinin with High Activity and Selectivity Against Colon Cancer. ChemMedChem 2017, 12, 226–234. 10.1002/cmdc.201600594. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Fröhlich T.; Gruber L.; Hutterer C.; Marschall M.; Voigtländer C.; Friedrich O.; Kappes B.; Efferth T.; Tsogoeva S. B. Highly Potent Artemisinin-Derived Dimers and Trimers: Synthesis and Evaluation of Their Antimalarial, Antileukemia and Antiviral Activities. Bioorg. Med. Chem. 2015, 23, 5452–5458. 10.1016/j.bmc.2015.07.048. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Capcı Karagöz A.; Fröhlich T.; Klein V.; Zeino M.; Viertel K.; Held J.; Mordmüller B.; Emirdağ Öztürk S.; Anıl H.; Efferth T.; Tsogoeva S. B. Synthesis and Study of Cytotoxic Activity of 1,2,4-Trioxane- and Egonol-Derived Hybrid Molecules against Plasmodium Falciparum and Multidrug-Resistant Human Leukemia Cells. Eur. J. Med. Chem. 2014, 75, 403–412. 10.1016/j.ejmech.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Fröhlich T.; Zeino M.; Marschall M.; Bahsi H.; Leidenberger M.; Friedrich O.; Kappes B.; Hampel F.; Efferth T.; Tsogoeva S. B. New Efficient Artemisinin Derived Agents against Human Leukemia Cells, Human Cytomegalovirus and Plasmodium Falciparum: 2nd Generation 1,2,4-Trioxane-Ferrocene Hybrids. Eur. J. Med. Chem. 2015, 97, 164–172. 10.1016/j.ejmech.2015.04.053. [DOI] [PubMed] [Google Scholar]
- Krishna S.; Ganapathi S.; Ster I. C.; Saeed M. E. M.; Cowan M.; Finlayson C.; Kovacsevics H.; Jansen H.; Kremsner P. G.; Efferth T.; Kumar D. A Randomised, Double Blind, Placebo-Controlled Pilot Study of Oral Artesunate Therapy for Colorectal Cancer. EBioMedicine 2015, 2, 82–90. 10.1016/j.ebiom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P. M.; Barton V. E.; Ward S. A. The Molecular Mechanism of Action of Artemisinin - The Debate Continues. Molecules 2010, 15, 1705–1721. 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A.; Meunier B. Is Alkylation the Main Mechanism of Action of the Antimalarial Drug Artemisinin?. Chem. Soc. Rev. 1998, 27, 273–279. 10.1039/a827273z. [DOI] [Google Scholar]
- Meunier B.; Robert A. Heme as Trigger and Target for Trioxane-Containing Antimalarial Drugs. Acc. Chem. Res. 2010, 43, 1444–1451. 10.1021/ar100070k. [DOI] [PubMed] [Google Scholar]
- Robert A.; Dechy-Cabaret O.; Cazelles J.; Meunier B. From Mechanistic Studies on Artemisinin Derivatives to New Modular Antimalarial Drugs. Acc. Chem. Res. 2002, 35, 167–174. 10.1021/ar990164o. [DOI] [PubMed] [Google Scholar]
- Robert A.; Meunier B. Characterization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968–5969. 10.1021/ja970412g. [DOI] [Google Scholar]
- Robert A.; Benoit-Vical F.; Claparols C.; Meunier B. The Antimalarial Drug Artemisinin Alkylates Heme in Infected Mice. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13676–13680. 10.1073/pnas.0500972102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein-Ludwig U.; Webb R. J.; Van Goethem I. D. A.; East J. M.; Lee A. G.; Kimura M.; O’Neill P. M.; Bray P. G.; Ward S. a; Krishna S. Artemisinins Target the SERCA of Plasmodium Falciparum. Nature 2003, 424, 957–961. 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- Stocks P. a; Bray P. G.; Barton V. E.; Al-Helal M.; Jones M.; Araujo N. C.; Gibbons P.; Ward S. a; Hughes R. H.; Biagini G. a; Davies J.; Amewu R.; Mercer A. E.; Ellis G.; O'Neill P. M. Evidence for a Common Non-Heme Chelatable-Iron-Dependent Activation Mechanism for Semisynthetic and Synthetic Endoperoxide Antimalarial Drugs. Angew. Chem., Int. Ed. 2007, 46, 6278–6283. 10.1002/anie.200604697. [DOI] [PubMed] [Google Scholar]
- Wu W.-M.; Wu Y.; Wu Y.-L.; Yao Z.-J.; Zhou C.-M.; Li Y.; Shan F. Unified Mechanistic Framework for the Fe(II)-Induced Cleavage of Qinghaosu and Derivatives/Analogues. The First Spin-Trapping Evidence for the Previously Postulated Secondary C-4 Radical. J. Am. Chem. Soc. 1998, 120, 3316–3325. 10.1021/ja973080o. [DOI] [Google Scholar]
- Haynes R. K.; Chan W. C.; Lung C.-M.; Uhlemann A.-C.; Eckstein U.; Taramelli D.; Parapini S.; Monti D.; Krishna S. The Fe2+-Mediated Decomposition, PfATP6 Binding, and Antimalarial Activities of Artemisone and Other Artemisinins: The Unlikelihood of C-Centered Radicals as Bioactive Intermediates. ChemMedChem 2007, 2, 1480–1497. 10.1002/cmdc.200700108. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Çapcı Karagöz A.; Reiter C.; Tsogoeva S. B. Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. 10.1021/acs.jmedchem.5b01380. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A.; Stein H. A.; Turschner S.; Toegel I.; Mora R.; Jennewein N.; Efferth T.; Eils R.; Brady N. R. Artesunate Activates Mitochondrial Apoptosis in Breast Cancer Cells via Iron-Catalyzed Lysosomal Reactive Oxygen Species Production. J. Biol. Chem. 2011, 286, 6587–6601. 10.1074/jbc.M110.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N.-D.; Tan S.-H.; Ng S.; Shi Y.; Zhou J.; Tan K. S. W.; Wong W.-S. F.; Shen H.-M. Artesunate Induces Cell Death in Human Cancer Cells via Enhancing Lysosomal Function and Lysosomal Degradation of Ferritin. J. Biol. Chem. 2014, 289, 33425–33441. 10.1074/jbc.M114.564567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Gosser D. K.; Meshnick S. R. Hemin-Catalyzed Decomposition of Artemisinin (Qinghaosu). Biochem. Pharmacol. 1992, 43, 1805–1809. 10.1016/0006-2952(92)90713-S. [DOI] [PubMed] [Google Scholar]
- Meshnick S. R.; Thomas A.; Ranz A.; Xu C. M.; Pan H. Z. Artemisinin (Qinghaosu): The Role of Intracellular Hemin in Its Mechanism of Antimalarial Action. Mol. Biochem. Parasitol. 1991, 49, 181–189. 10.1016/0166-6851(91)90062-B. [DOI] [PubMed] [Google Scholar]
- Benoit-Vical F.; Robert A.; Meunier B. Potentiation of Artemisinin Activity against Chloroquine-Resistant Plasmodium Falciparum Strains by Using Heme Models. Antimicrob. Agents Chemother. 1999, 43, 2555–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhang C.-J.; Chia W. N.; Loh C. C. Y.; Li Z.; Lee Y. M.; He Y.; Yuan L.-X.; Lim T. K.; Liu M.; Liew C. X.; Lee Y. Q.; Zhang J.; Lu N.; Lim C. T.; Hua Z.-C.; Liu B.; Shen H.-M.; Tan K. S. W.; Lin Q. Haem-Activated Promiscuous Targeting of Artemisinin in Plasmodium Falciparum. Nat. Commun. 2015, 6, 10111. 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.-J.; Wang J.; Zhang J.; Lee Y. M.; Feng G.; Lim T. K.; Shen H.-M.; Lin Q.; Liu B. Mechanism-Guided Design and Synthesis of a Mitochondria-Targeting Artemisinin Analogue with Enhanced Anticancer Activity. Angew. Chem., Int. Ed. 2016, 55, 13770–13774. 10.1002/anie.201607303. [DOI] [PubMed] [Google Scholar]
- Ravindra K. C.; Ho W. E.; Cheng C.; Godoy L. C.; Wishnok J. S.; Ong C. N.; Wong W. S. F.; Wogan G. N.; Tannenbaum S. R. Untargeted Proteomics and Systems-Based Mechanistic Investigation of Artesunate in Human Bronchial Epithelial Cells. Chem. Res. Toxicol. 2015, 28, 1903–1913. 10.1021/acs.chemrestox.5b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Lin Q. Chemical Proteomics Approach Reveals the Direct Targets and the Heme-Dependent Activation Mechanism of Artemisinin in Plasmodium Falciparum Using an Artemisinin-Based Activity Probe. Microb. Cell 2016, 3, 230–231. 10.15698/mic2016.05.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail H. M.; Barton V.; Phanchana M.; Charoensutthivarakul S.; Wong M. H. L.; Hemingway J.; Biagini G. A.; O’Neill P. M.; Ward S. A. Artemisinin Activity-Based Probes Identify Multiple Molecular Targets within the Asexual Stage of the Malaria Parasites Plasmodium Falciparum 3D7. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 2080–2085. 10.1073/pnas.1600459113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Li W.; Xiao Y. Profiling of Multiple Targets of Artemisinin Activated by Hemin in Cancer Cell Proteome. ACS Chem. Biol. 2016, 11, 882–888. 10.1021/acschembio.5b01043. [DOI] [PubMed] [Google Scholar]
- Ismail H. M.; Barton V. E.; Panchana M.; Charoensutthivarakul S.; Biagini G. A.; Ward S. A.; O’Neill P. M. A Click Chemistry-Based Proteomic Approach Reveals That 1,2,4-Trioxolane and Artemisinin Antimalarials Share a Common Protein Alkylation Profile. Angew. Chem., Int. Ed. 2016, 55, 6401–6405. 10.1002/anie.201512062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.; Li J.; Cao Y.; Long G.; Zhou B. Two Distinct and Competitive Pathways Confer the Cellcidal Actions of Artemisinins. Microb. Cell 2015, 2, 14–25. 10.15698/mic2015.01.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhang J.; Lee Y.-M.; Koh P.-L.; Ng S.; Bao F.; Lin Q.; Shen H.-M. Quantitative Chemical Proteomics Profiling of de Novo Protein Synthesis during Starvation-Mediated Autophagy. Autophagy 2016, 12, 1931–1944. 10.1080/15548627.2016.1196317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhang J.; Lee Y. M.; Ng S.; Shi Y.; Hua Z.; Lin Q.; Shen H. Nonradioactive Quantification of Autophagic Protein Degradation with L-Azidohomoalanine Labeling. Nat. Protoc. 2017, 12, 279–288. 10.1038/nprot.2016.160. [DOI] [PubMed] [Google Scholar]
- Flavin R.; Peluso S.; Nguyen P.; Loda M. Fatty Acid Synthase as a Potential Therapeutic Target in Cancer. Future Oncol. 2010, 6, 551–562. 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E.; Schulze A.; Zechner R.; Walther T. C.; Farese R. V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima T.; Seimiya H.; Tsuruo T. De Novo Fatty-Acid Synthesis and Related Pathways as Molecular Targets for Cancer Therapy. Br. J. Cancer 2009, 100, 1369–1372. 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codogno P.; Mehrpour M.; Proikas-Cezanne T. Canonical and Non-Canonical Autophagy: Variations on a Common Theme of Self-Eating?. Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- Alers S.; Löffler A. S.; Wesselborg S.; Stork B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T.; Klionsky D. J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad M. B.; Chen Y.; Gibson S. B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signaling 2009, 11, 777–790. 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- Hooda J.; Cadinu D.; Alam M. M.; Shah A.; Cao T. M.; Sullivan L. A.; Brekken R.; Zhang L. Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells. PLoS One 2013, 8, e63402. 10.1371/journal.pone.0063402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa S. Why Heme Needs to Be Degraded to Iron, Biliverdin IXalpha, and Carbon Monoxide?. Antioxid. Redox Signaling 2004, 6, 819–824. 10.1089/ars.2004.6.819. [DOI] [PubMed] [Google Scholar]
- Ponka P. Cell Biology of Heme. Am. J. Med. Sci. 1999, 318, 241–256. 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- Ooko E.; Saeed M. E. M.; Kadioglu O.; Sarvi S.; Colak M.; Elmasaoudi K.; Janah R.; Greten H. J.; Efferth T. Artemisinin Derivatives Induce Iron-Dependent Cell Death (Ferroptosis) in Tumor Cells. Phytomedicine 2015, 22, 1045–1054. 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A.; Stein H. A.; Turschner S.; Toegel I.; Mora R.; Jennewein N.; Efferth T.; Eils R.; Brady N. R. Artesunate Activates Mitochondrial Apoptosis in Breast Cancer Cells via Iron-Catalyzed Lysosomal Reactive Oxygen Species Production. J. Biol. Chem. 2011, 286, 6587–6601. 10.1074/jbc.M110.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshnick S. R. Artemisinin: Mechanisms of Action, Resistance and Toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. 10.1016/S0020-7519(02)00194-7. [DOI] [PubMed] [Google Scholar]
- ter Kuile F.; White N. J.; Holloway P.; Pasvol G.; Krishna S. Plasmodium Falciparum: In Vitro Studies of the Pharmacodynamic Properties of Drugs Used for the Treatment of Severe Malaria. Exp. Parasitol. 1993, 76, 85–95. 10.1006/expr.1993.1010. [DOI] [PubMed] [Google Scholar]
- Wachowska M.; Muchowicz A.; Firczuk M.; Gabrysiak M.; Winiarska M.; Wańczyk M.; Bojarczuk K.; Golab J. Aminolevulinic Acid (ALA) as a Prodrug in Photodynamic Therapy of Cancer. Molecules 2011, 16, 4140–4164. 10.3390/molecules16054140. [DOI] [Google Scholar]
- Peng Q.; Warloe T.; Berg K.; Moan J.; Kongshaug M.; Giercksky K. E.; Nesland J. M. 5-Aminolevulinic Acid-Based Photodynamic Therapy. Clinical Research and Future Challenges. Cancer 1997, 79, 2282–2308. . [DOI] [PubMed] [Google Scholar]
- Kawczyk-Krupka A.; Sieroń-Stołtny K.; Latos W.; Czuba Z. P.; Kwiatek B.; Potempa M.; Wasilewska K.; Król W.; Stanek A. ALA-Induced Photodynamic Effect on Vitality, Apoptosis, and Secretion of Vascular Endothelial Growth Factor (VEGF) by Colon Cancer Cells in Normoxic Environment in Vitro. Photodiagn. Photodyn. Ther. 2016, 13, 308–315. 10.1016/j.pdpdt.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Dell’Eva R.; Pfeffer U.; Vené R.; Anfosso L.; Forlani A.; Albini A.; Efferth T. Inhibition of Angiogenesis in Vivo and Growth of Kaposi’s Sarcoma Xenograft Tumors by the Anti-Malarial Artesunate. Biochem. Pharmacol. 2004, 68, 2359–2366. 10.1016/j.bcp.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Du J.-H.; Zhang H.-D.; Ma Z.-J.; Ji K.-M. Artesunate Induces Oncosis-like Cell Death in Vitro and Has Antitumor Activity against Pancreatic Cancer Xenografts in Vivo. Cancer Chemother. Pharmacol. 2010, 65, 895–902. 10.1007/s00280-009-1095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. P.; Lai H. Selective Toxicity of Dihydroartemisinin and Holotransferrin toward Human Breast Cancer Cells. Life Sci. 2001, 70, 49–56. 10.1016/S0024-3205(01)01372-8. [DOI] [PubMed] [Google Scholar]
- Lai H.; Sasaki T.; Singh N. P.; Messay A. Effects of Artemisinin-Tagged Holotransferrin on Cancer Cells. Life Sci. 2005, 76, 1267–1279. 10.1016/j.lfs.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Herrmann A.; Çapci A.; Efferth T.; Tsogoeva S. B. New Artesunic Acid Homodimers: Potent Reversal Agents of Multidrug Resistance in Leukemia Cells. Bioorg. Med. Chem. 2012, 20, 5637–5641. 10.1016/j.bmc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Horwedel C.; Tsogoeva S. B.; Wei S.; Efferth T. Cytotoxicity of Artesunic Acid Homo- and Heterodimer Molecules toward Sensitive and Multidrug-Resistant CCRF-CEM Leukemia Cells. J. Med. Chem. 2010, 53, 4842–4848. 10.1021/jm100404t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.