

Abstract
Objective:
To describe the autopsy case of a patient with a homozygous 2-base deletion, c171_172delGA (p.N58fs), in the C12orf65 gene.
Methods:
We described the clinical history, neuroimaging data, neuropathology, and genetic analysis of the patients with C12orf65 mutations.
Results:
The patient was a Japanese woman with a history of delayed psychomotor development, primary amenorrhea, and gait disturbance in her 20s. She was hospitalized because of respiratory failure at the age of 60. Pectus excavatum, long fingers and toes, and pes cavus were revealed by physical examination. Her IQ score was 44. Neurologic examination revealed ophthalmoplegia, optic atrophy, dysphagia, distal dominant muscle weakness and atrophy, hyperreflexia at patellar tendon reflex, hyporeflexia at Achilles tendon reflex, and extensor plantar reflexes. At age 60, she died of pneumonia. Lactate levels were elevated in the patient's serum and CSF. T2-weighted brain MRI showed symmetrical hyperintense brainstem lesions. At autopsy, axial sections exposed symmetrical cyst formation with brownish lesions in the upper spinal cord, ventral medulla, pons, dorsal midbrain, and medial hypothalamus. Microscopic analysis of these areas demonstrated mild gliosis with rarefaction. Cell bodies in the choroid plexuses were eosinophilic and swollen. Electron microscopic examination revealed that these cells contained numerous abnormal mitochondria. Whole-exome sequencing revealed the 2-base deletion in C12orf65.
Conclusions:
We report an autopsy case of the C12orf65 mutation, and findings suggest that mitochondrial dysfunction may underlie the unique clinical presentations.
The C12orf65 gene encodes a mitochondrial matrix protein that has a role in releasing peptides from mitochondrial ribosomes.1 To date, 2 phenotypes have been associated with C12orf65 gene mutations: combined oxidative phosphorylation deficiency type 7 (COXPD7)1 and autosomal recessive spastic paraplegia 55 (SPG55).2 While C12orf65 defects exhibit a wide spectrum of phenotypes, the 3 primary clinical features are optic atrophy, peripheral neuropathy, and spastic paraparesis.3 Although biochemical studies suggest that these C12orf65 mutations cause mitochondrial dysfunction,1,2,4 very few pathologic analyses and no autopsy cases supporting these findings have been reported. In this article, we report an autopsy case associated with 2-nucleotide deletion in the C12orf65 gene. Our patient presented with optic atrophy, peripheral neuropathy, pyramidal signs, mental retardation, and several additional features, including long fingers and toes, pectus excavatum, lack of secondary sexual characteristics, primary amenorrhea, osteoporosis, and late onset respiratory insufficiency.
METHODS
Standard protocol approvals, registrations, and patient consents.
The protocol of the studies was reviewed and approved by the Institutional Review Board of Kagoshima University. The proband (IV:1) and her younger sister (IV:2) provided written informed consent to participate in this study.
Postmortem study, histology, light microscopy, and immunohistochemistry.
An autopsy was performed 12 hours after the patient's death. After a thorough macroscopic inspection, conventional histologic techniques were performed to make specimens as mentioned previously.5–7 Tissue sections were then stained with hematoxylin and eosin (H&E). At the time of autopsy, several small pieces of fresh brain tissue (right frontal lobe) were dissected, immediately frozen in dry ice, and stored at −80°C for future studies.
The remainder of the brain was fixed, dissected, and stained as previously reported.5–8 The 3-μm-thick sections were stained with H&E for nuclei and eosinophilic structures, modified Gallyas-Braak silver staining for fibrils, and Klüver-Barrera for myelin. We performed immunohistochemical studies using the following monoclonal antibodies: amyloid Aβ (11–28) (12B8, 1:100; IBL, Gunma, Japan), phospho-tau (AT8, 1:3,000, Innogenetics, Ghent, Belgium), phosphorylated α-synuclein (1:7,000, pSyn#64, monoclonal, Wako), glial fibrillary acidic protein (1:200, monoclonal; DAKO, Tokyo, Japan), aquaporin 4 (1:1,000, polyclonal; Merck Millipore, Tokyo, Japan), neurofilament (SMI31) and Schwann/2E (1:5000, monoclonal; COSMO BIO, Tokyo, Japan), and phospho-TDP-43 (s409/410, 1:7,000; COSMO BIO).8
Peripheral nerve specimens were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer, postfixed in 1% osmium tetroxide, embedded in Epon, and stained with toluidine blue. Muscle samples were snap frozen in liquid nitrogen–cooled isopentane. Serial 8-μm-thick cryosections were stained with H&E, nicotinamide adenine dinucleotide tetrazolium reductase, adenosine triphosphatase (ATPase, preincubation at pH 4.6 and 10.8), modified Gomori trichrome, succinate dehydrogenase (SDH), and cytochrome oxidase (COX).
Electron microscopy.
Choroid plexus specimens from formalin-fixed tissues were dissected and refixed in 2.5% glutaraldehyde. Muscle samples were directly fixed in 2.5% glutaraldehyde. Electron microscopy was performed as previously reported.7 Ultrathin sections were examined under a Hitachi H-7500 transmission electron microscope.
Genetic analysis.
We used the same methodology as was used in a previous study.9 The captured exome library was sequenced using a HiSeq 2000 (Illumina, San Diego, CA). Sequences were aligned to the human reference genome (NCBI37/hg19) using the Burrows-Wheeler Aligner, and variant calling was performed using SAMtools.10,11 Variants were annotated using in-house scripts, which provided the list of variants. The mutation in the C12orf65 gene was validated using Sanger sequencing on samples from the patient and her younger sister.
RESULTS
Description of the patient.
The proband (IV:1), a 61-year-old Japanese woman at the time of death, was born of consanguineous parents (figure 1). Based on the information obtained from her parents, no other family members had neurologic disorders. The patient was delivered without any physical abnormalities at birth. However, she showed delayed psychomotor development and could not walk until she was 1 year and 10 months old. She was admitted to elementary school at age 7 and needed special support education because of mental retardation. At age 26, she was diagnosed with primary amenorrhea. According to medical records, neurologic examinations revealed bilateral optic atrophy, exotropia, weakness in both tibialis anterior muscles, steppage gait, and hyperreflexia at patellar reflex. The patient was developmentally disabled and placed in a nursing home when she was in her 30s. In her early 40s, she was diagnosed with osteoporosis. Leg weakness was exacerbated, and she needed aid for walking at age 55. At 60 years of age, she was unable to eat and was admitted to a local hospital where she was diagnosed as having CO2 narcosis with infection. Although she recovered from the infections, she still had hypercapnia and was transferred to Yamaguchi University Hospital. Physical examination revealed an abnormally long arm span (162 cm) compared with her height (159 cm), long fingers and toes, pes cavus, pectus excavatum, and hypertelorism. Neurologic examination showed bilateral optic atrophy, ophthalmoplegia, dysphagia, significant distal dominant muscle weakness and wasting, hyperreflexia at patellar tendon reflex, hyporeflexia at Achilles tendon reflex, and extensor plantar reflexes. Pain and temperature sensations were almost normal. Neuropsychological evaluation was indicative of an intellectual disability (Wechsler Adult Intelligence Scale–Revised: Verbal IQ 50, Performance IQ 47, and Full-Scale IQ 44). Arterial blood gas analysis showed hypercapnia (60.7 mm Hg), hypoxia (65.4 mm Hg), and hyperbicarbonemia (35.6 mmol/L) under ambient conditions. Lactate levels in the plasma and CSF were slightly elevated (1.51 and 2.02 mmol/L, respectively). Exercise and pulmonary function tests could not be performed because of her respiratory and cognitive status. Brain MRI revealed symmetric T2 prolongation involving the bilateral thalamus, which extended inferiorly along the midbrain pontine tegmentum and terminating in the ventral aspect of the medulla (figure 2, A and B). Nerve conduction studies showed distal dominant motor sensory axonal dysfunction. Although her respiratory status temporarily improved under noninvasive positive pressure ventilation assistance, respiratory dysfunction gradually worsened, and the patient died of pneumonia at age 61 years.
Figure 1. Family pedigree and genetic studies.
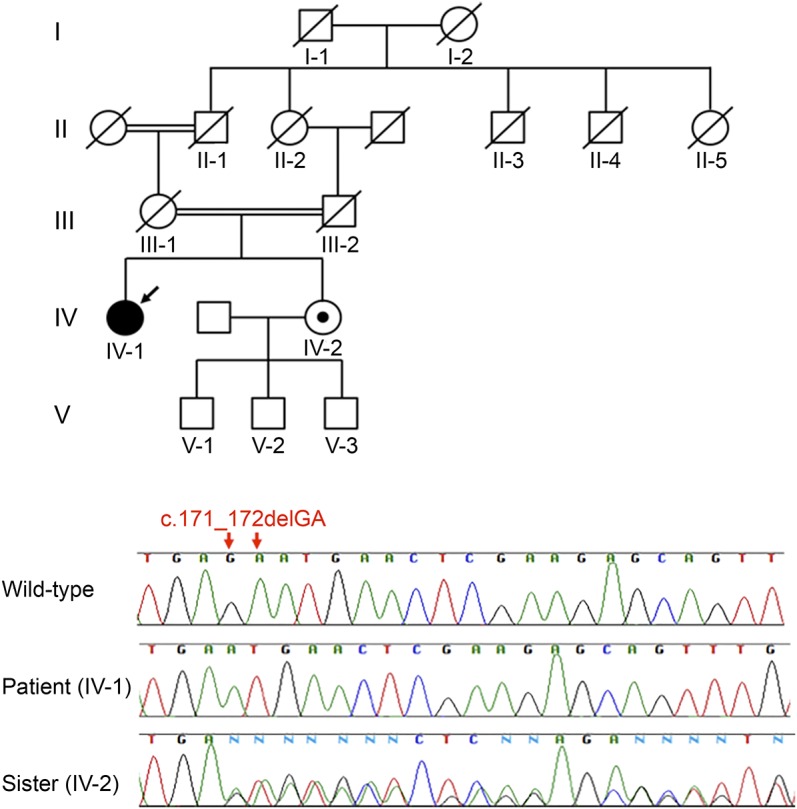
Arrow indicates the proband. Her parents were first cousins. There was no family history of similar illness. Sequencing chromatogram of the 2-base deletion in the C12orf65 gene of the proband (IV:1; arrow) and her sister, a wild-type, heterozygous carrier (IV:2).
Figure 2. Brain MRI.

(A) Sagittal image of T2-weighted brain MRI. Arrows indicate hyperintense lesions in the brainstem. (B) Axial images demonstrating symmetrical T2 high signals (arrows) in the bilateral thalamus, extending inferiorly along the midbrain pontine tegmentum, terminating in the ventral aspect of the medulla.
Genetic analysis.
Exome sequencing identified a novel homozygous 2-base deletion, c171_172delGA (p.N58fs), in the patient's C12orf65 gene (figure 1). This loss-of-function mutation in exon 2, which encodes the C12orf65 RF-1 domain, could introduce a premature stop codon that leads to messenger RNA degradation through the nonsense-mediated decay mechanism. The patient's younger sister (figure 1, IV:2) was heterozygous for the 2-base deletion, c171_172delGA (p.N58fs) in C12orf65. Gene analysis of their parents could not be performed because they were deceased. We confirmed that this mutation was not found in public databases, including the 1000 Genomes project databases (browser.1000genomes.org), the Exome Aggregation Consortium (exac.broadinstitute.org/), and the Human Genetic Variation Database comprising the exome sequencing results of 1,208 Japanese individuals (genome.med.kyoto-u.ac.jp/SnpDB/) using same methodology as previously reported.9 No other causative mutations were identified among the known disease-causing genes, including those related to Charcot-Marie-Tooth disease, hereditary sensory and autonomic neuropathies, hereditary motor neuropathy, and amyotrophic lateral sclerosis.
Postmortem study, histology, light microscopy, immunohistochemistry, and electron microscopy.
General autopsy.
The patient was 159.2 cm in height and 31 kg in body weight. Her skeletal muscles were extremely atrophic in a distal dominant manner. The patient also had long fingers and toes, pectus excavatum, and pes cavus. Her organs revealed mild atrophy. The uterus showed extreme hypoplasia with a length of 35 mm and a width of 20 mm.
Brain pathology.
Postfixed brain weight was 1,150 g, and the pituitary gland was enlarged. A thin membrane with brownish discoloration was observed on the inner surface of the dura mater, which is consistent with a previous subdural hematoma. The arachnoid membrane was cloudy at the level of the frontal lobes. The frontal lobes also exhibited mild atrophy. The brainstem and cerebellum were preserved. Axial sections exposed symmetrical brownish lesions with cyst formation in the upper spinal cord, ventral medulla, pons, dorsal midbrain, and medial hypothalamus (figure 3A). Using microscopy, we found that these areas showed moderate-to-severe rarefaction and mild-to-moderate gliosis (figure 3, B–E). Numerous macrophages, as well as proliferation of small vessel channels, were observed around the cystic lesions (figure 3F). Ischemic neurons were not found around the lesions, nor was there evidence of occluded vessels. Microscopic examination revealed markedly swollen and eosinophilic cytoplasm in the choroid plexus cells of the lateral ventricle (figure 3G). In the H&E sections, those cell bodies were markedly swollen and eosinophilic compared with the control cases. These morphological features suggested the presence of underlying mitochondrial abnormalities. Therefore, the choroid plexus cells were subjected to electron microscopic analysis. Ultrastructurally, these cells contained numerous abnormally enlarged mitochondria (figure 3I). We also identified numerous large mitochondria in the pituitary gland and skeletal muscles. Additional findings included hyperplasia of the pituitary gland and small petechial hemorrhages in the posterior cingulate cortex.
Figure 3. Pathologic findings.

(A) Gross pathology of the brainstem. Axial sections revealed symmetrical brownish lesions with cyst formation (arrows) at the level of the medial hypothalamus and dorsal midbrain and pons. These cystic lesions were also present in the ventral medulla and upper spinal cord. (B–D) Hematoxylin and eosin (H&E) staining of the midbrain (B), pons (C), and medulla (D). (E) Enlarged image defined by a square in B (bar = 500 μm). These T2WI high-signal areas showed rarefaction with gliosis. (F) A photomicrograph around a cystic lesion of the inferior olivary nucleus. Numerous macrophages and proliferation of small vessel channels were observed (H&E staining). (G) H&E staining of the patient's choroid plexus cells. Microscopic examination demonstrated the markedly swollen and eosinophilic cytoplasm of the lateral ventricle choroid plexus cells (bar = 10 μm). (I) Electron micrograph of the patient's choroid plexus cells. The cells contained numerous enlarged abnormal mitochondria (bar = 2 μm). (H, J) H&E staining (H, bar = 10 μm) and electron micrograph (J, bar = 2 μm) of the choroid plexus cells in normal subjects. (K, L) Immunostaining of cytochrome c oxidase of the patient's skeletal muscle (K) and age-matched control's (L). Cytochrome oxidase activity in whole fibers was low in the patient's skeletal muscle.
Peripheral nerves and muscles.
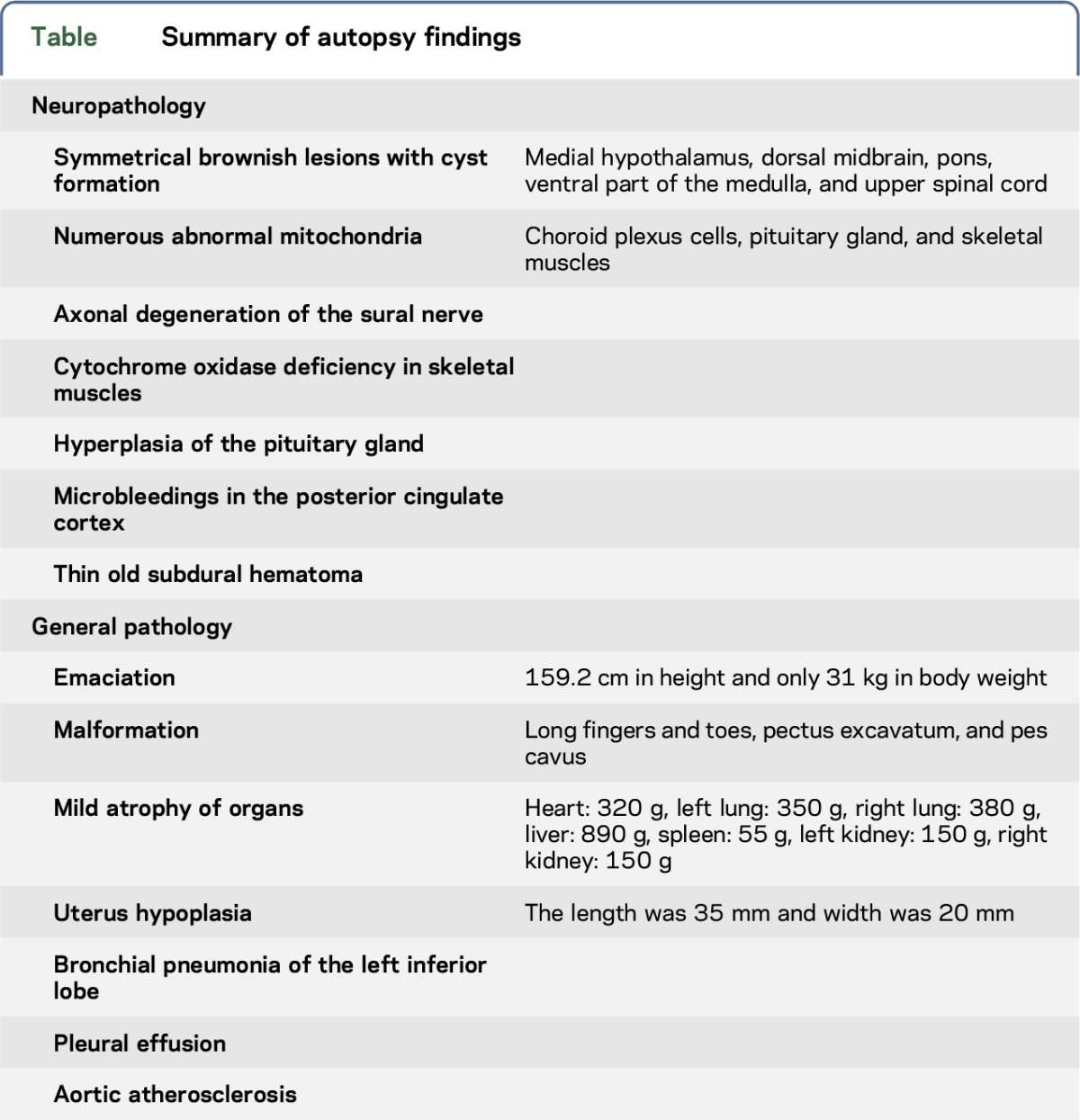
A sural nerve biopsy specimen showed a mild loss of myelinated fibers without myelin ovoids, which was indicative of longstanding axonal degeneration. The biceps brachii muscle displayed mildly atrophic fibers and slight fat replacement. ATPase staining revealed mild small group atrophy and fiber type grouping. There were no ragged-red fibers or strongly SDH-reactive blood vessels. COX activity in whole fibers was low compared with the age-matched control, and there was no focal COX deficiency (figure 3, K and L). Most of the diaphragm muscle fibers were small in size (20–30 μm); however, fat replacement was minimal and large group atrophy was absent. The pathologic findings are summarized in the table.
Table.
Summary of autopsy findings
DISCUSSION
We have described clinical, neuroradiologic, and pathologic studies of an autopsy case with a homozygous mutation (c171_172delGA [p.N58fs]) in the C12orf65 gene. In 2010, C12orf65 gene mutations were reported in 3 individuals who developed Leigh syndrome, optic atrophy, and ophthalmoplegia. This clinical phenotype was designated COXPD7.1 In 2012, the second phenotype of the C12orf65 gene mutation was reported. The individual showed autosomal recessive hereditary spastic paraplegias with optic atrophy and neuropathy. This clinical condition, designated SPG55, has milder clinical presentations than COXPD7.2
Because the number of mutation sites has increased recently, genotype-phenotype heterogeneities have been reported.3,4,12–15 The typical phenotypes of a C12orf65 mutation includes optic atrophy, peripheral neuropathy, pyramidal signs, and cognitive disorders. Although our patient showed these typical features of a C12orf65 mutation, she also developed endocrine abnormalities such as a lack of secondary sexual characteristics, primary amenorrhea, and osteoporosis, all of which are features of mitochondrial diseases.16–18 The endocrine abnormalities of our patient may have been caused by mitochondrial dysfunction because morphological abnormalities were also found in mitochondria of the pituitary gland cells. Although an endocrine function test was not performed, these findings suggest dyspituitarism.
Disease severity and mortality are thought to be related to C12orf65 protein length and whether the mutant site contains the glycine-glycine-glutamine motif, which interacts with the large ribosomal subunit to release the polypeptide chain from the P-site bound peptidyl transfer RNA.3,13 Previous findings suggest that pathogenesis in C12orf65-mutant patients is mediated by mitochondrial dysfunctions.1 Fibroblasts cultured from 2 patients with C12orf65 mutations showed global and uniform defects in the translation of mitochondrial DNA-encoded proteins, resulting in a severe decrease in oxidative phosphorylation complexes I, IV, and V and a small decrease in complex III. Patients with SPG55 showed a relatively larger C12orf65 protein, possibly due to a comparatively preserved complex V and unaffected complex III activity.2 These previous data suggest that C12orf65 protein length is related to mitochondrial respiratory chain function and thereby disease severity. The expected protein length in our patient was 57 amino acids, the shortest of those previously reported, and the same length as the protein studied by Heidary et al.12 Moreover, immunohistochemical analysis revealed that our patient's cytochrome c oxidase activities were extremely low, possibly pointing to mitochondrial respiratory chain complex abnormalities. Several patients with the COXPD7 phenotype and a short C12orf65 protein experienced respiratory failure in the first or second decade of life.1 In comparison, our patient's respiratory ability was well preserved, and the prognosis was relatively good, suggesting that protein length is not always correlated with disease severity.
Until now, only 3 reports included histologic information from C12orf65-mutant patients, 2 using a sural nerve specimen and the other a skin biopsy specimen. The 2 sural nerve biopsies showed markedly reduced numbers of myelinated fibers accompanied by numerous small myelinated fiber clusters, suggesting regeneration. A decrease in the number of unmyelinated fibers was also confirmed.4,15 Our patient also showed large myelinated fiber loss and no acute changes such as myelin ovoids. Therefore, C12orf65 mutations may cause chronic axonal degeneration in peripheral nerves. Electron photomicrographs of the 1 skin biopsy revealed enlarged mitochondria engorged by an abnormally large number of densely packed cristae.12 Previous reports suggested that mitochondrial dysfunctions are related to certain features of a C12orf65 mutation such as peripheral neuropathy, optic atrophy, and spastic paraplegia.1,2,4,19 Our investigation found evidence of mitochondrial abnormalities not only on the skin but also in the choroid plexus, pituitary gland, and skeletal muscles, demonstrating that mitochondrial dysfunction was widespread and could be related to the pathogenesis and observed clinical manifestations.
Our patient also showed symmetrical MRI abnormalities in the hypothalamus over the upper cervical spinal cord. The same abnormalities were reported in previous cases, but histologic analysis had not been performed.12,14 Pathologic analysis of the present patient showed bilateral rarefaction in part of the medial hypothalamus, mesencephalic tegmentum, pontine tegmentum, ventral medulla, and upper cervical spinal cord tissues, surrounded by mild gliosis. These tissues contained white matter and gray matter, which suggests that there is no association with specific anatomical tracts and structures. In addition, the pathologic changes of the choroid plexuses again allude to a diagnosis of mitochondrial disease. In fact, the characteristic MRI findings, pathologic features of the cystic lesions, and choroid plexus pathology strongly suggest the presence of mitochondrial abnormalities and led to molecular analysis. Although the pathogenesis of bilateral rarefaction in the brainstem is unclear, the same MRI abnormalities have been reported in other mitochondrial disorders such as Leber optic neuropathy and MFN2 mutation.20,21 Symmetrical brainstem abnormalities should be considered a key indicator of possible mitochondrial dysfunction.
We reported an autopsy case with a confirmed C12orf65 gene mutation. The patient, with a novel homozygous loss-of-function deletion, presented with peripheral neuropathy, optic atrophy, mental retardation, pyramidal signs, and primary amenorrhea. Pathologic findings revealed numerous and enlarged abnormal mitochondria, which might be related to the pathogenesis of the various phenotypes.
ACKNOWLEDGMENT
The authors are grateful to all study individuals and their relatives. They also thank Mitsutoshi Tano for technical assistance.
GLOSSARY
- ATPase
adenosine triphosphatase
- COX
cytochrome oxidase
- COXPD7
combined oxidative phosphorylation deficiency type 7
- H&E
hematoxylin and eosin
- SDH
succinate dehydrogenase
- SPG55
spastic paraplegia 55
AUTHOR CONTRIBUTIONS
Dr. Nishihara performed the experiments, analyzed and interpreted the data, and wrote the manuscript. Dr. Omoto, Prof. Takao, Prof. Kawano, and Prof. Ikeda performed the pathologic experiments, analyzed the data, and edited the manuscript. Dr. Higuchi and Prof. Takashima performed the genetic experiments, analyzed the data, and edited the manuscript. Dr. Koga and Dr. Kawai evaluated the data, provided advice about the experiments, and edited the manuscript. Prof. Kanda designed and supervised the study, evaluated the data, and wrote the manuscript. The study was planned by Prof. Kanda.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
H. Nishihara has received research grants (16H07008) from the Japan Society for the Promotion of Science and has received research support from the Uehara Memorial Foundation and the Japan Multiple Sclerosis Society. M. Omoto reports no disclosures. M. Takao has received Grants-in-Aid for Scientific Research on Innovative Areas (Comprehensive Brain Science Network, 221S0003) and the Platform of Supporting Cohort Study and Biospecimen Analysis (JSPS KAKENHI JP 16H06277) and KIBAN C (JSPS KAKENHI 26430060). Y. Higuchi, M. Koga, M. Kawai, H. Kawano, and E. Ikeda report no disclosures. H. Takashima has received speaker honoraria from Kyushu University, GlaxoSmithKline, Bayer, Eisai Co., Novartis, Tanabe Mitsubishi, Biogen Idec, Benesis Co., Takeda, Teijin, Pfizer, Otsuka, Ono, Hisamitsu, Shionogi, Asteras, FP, Boehringer, and Dainippon Sumitomo Pharma; has been a consultant for Teijin; has received research support from Nervous and Mental Disorders, Research Committee for Charcot-Marie-Tooth Disease, Neuropathy, Ataxic Disease, Applying Health and Technology, and the Ministry of Health, Labour, and Welfare (Japan); and has received royalty payments from Athena Diagnostics. T. Kanda has served as the Editor-in-Chief for Clinical and Experimental Neuroimmunology and was on the editorial advisory broad for Neuropathology and has received research grants (Nos. 23659457, 25293203, and 26670443) from the Japan Society for the Promotion of Science (Tokyo, Japan), a Research Grant for Neuroimmunological Diseases from the Ministry of Health, Labour, and Welfare of Japan (K2002528), and an Intramural Research Grant (25-4) for Neurological and Psychiatric Disorders of National Center of Neurology and Psychiatry. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Antonicka H, Ostergaard E, Sasarman F, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 2010;87:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimazaki H, Takiyama Y, Ishiura H, et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55). J Med Genet 2012;49:777–784. [DOI] [PubMed] [Google Scholar]
- 3.Spiegel R, Mandel H, Saada A, et al. Delineation of C12orf65-related phenotypes: a genotype-phenotype relationship. Eur J Hum Genet 2014;22:1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tucci A, Liu YT, Preza E, et al. Novel C12orf65 mutations in patients with axonal neuropathy and optic atrophy. J Neurol Neurosurg Psychiatry 2014;85:486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takao M, Ghetti B, Yoshida H, et al. Early-onset dementia with Lewy bodies. Brain Pathol (Zurich, Switzerland) 2004;14:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takao M, Ghetti B, Hayakawa I, et al. A novel mutation (G217D) in the Presenilin 1 gene (PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta neuropathologica 2002;104:155–170. [DOI] [PubMed] [Google Scholar]
- 7.Takao M, Mori T, Orikasa H, et al. Postmortem diagnosis of Fabry disease with acromegaly and a unique vasculopathy. Virchows Arch 2007;451:721–727. [DOI] [PubMed] [Google Scholar]
- 8.Takao M, Aoyama M, Ishikawa K, et al. Spinocerebellar ataxia type 2 is associated with Parkinsonism and Lewy body pathology. BMJ case Rep 2011;2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higuchi Y, Hashiguchi A, Yuan J, et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 2016;79:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heidary G, Calderwood L, Cox GF, et al. Optic atrophy and a Leigh-like syndrome due to mutations in the c12orf65 gene: report of a novel mutation and review of the literature. J Neuroophthalmol 2014;34:39–43. [DOI] [PubMed] [Google Scholar]
- 13.Buchert R, Uebe S, Radwan F, et al. Mutations in the mitochondrial gene C12ORF65 lead to syndromic autosomal recessive intellectual disability and show genotype phenotype correlation. Eur J Med Genet 2013;56:599–602. [DOI] [PubMed] [Google Scholar]
- 14.Imagawa E, Fattal-Valevski A, Eyal O, et al. Homozygous p.V116* mutation in C12orf65 results in Leigh syndrome. J Neurol Neurosurg Psychiatry 2016;87:212–216. [DOI] [PubMed] [Google Scholar]
- 15.Montecchiani C, Pedace L, Lo Giudice T, et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 2016;139:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finsterer J. Mitochondriopathies. Eur J Neurol 2004;11:163–186. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Yang TL, Liu YZ, et al. Mitochondria-wide association study of common variants in osteoporosis. Ann Hum Genet 2011;75:569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CM, Huang CC. Gonadal dysfunction in mitochondrial encephalomyopathies. Eur Neurol 1995;35:281–286. [DOI] [PubMed] [Google Scholar]
- 19.Abramov AY, Smulders-Srinivasan TK, Kirby DM, et al. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain 2010;133:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paulus W, Straube A, Bauer W, Harding AE. Central nervous system involvement in Leber's optic neuropathy. J Neurol 1993;240:251–253. [DOI] [PubMed] [Google Scholar]
- 21.Boaretto F, Vettori A, Casarin A, et al. Severe CMT type 2 with fatal encephalopathy associated with a novel MFN2 splicing mutation. Neurology 2010;74:1919–1921. [DOI] [PubMed] [Google Scholar]