

Abstract
The management of advanced renal cell carcinoma (RCC) has dramatically changed over the past decade. Therapies that target the vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) pathways have considerably expanded treatment options; however, most patients with advanced RCC still have limited overall survival. Increased understanding of the mechanisms of T cell-antigen recognition and function has led to the development of novel immunotherapies to treat cancer, chief among them inhibitors of checkpoint receptors — molecules whose function is to restrain the host immune response. In 2015, the FDA approved the first checkpoint inhibitor nivolumab for patients with advanced RCC following treatment with antiangiogenic therapy based on improved overall survival compared with the standard of care. Ongoing phase III trials are comparing checkpoint- inhibitor-based combination regimens with antiangiogenesis agents in the first-line setting. The field is evolving rapidly, with many clinical trials already testing several checkpoint inhibitors alone, in combination, or with other targeted therapies. In addition, different novel immune therapies are being investigated including vaccines, T-cell agonists, and chimeric antigen receptor T cells. Determining which patients will benefit from these therapies and which combination approaches will result in better response will be important as this field evolves.
Since Dr William Coley's observation in the late nineteenth century that activation of the immune system can result in tumour regression, scientists have been trying to harness the power of the immune system to treat cancer1. Renal cell carcinoma (RCC) is a natural target for testing novel immune therapies. Cytotoxic chemotherapy is generally ineffective in RCC, but cytokine-based immune therapies such as IL-2 and IFNα can be effective. For example, a small percentage of patients achieve durable remissions with high-dose IL-2 (REF. 2). Nevertheless, such treatment is not suitable for many patients owing to a substantial incidence of high-grade adverse events, including considerable cardiopulmonary toxicity3. For many years IL-2 and IFNα were the only approved cytokine-based therapies for advanced RCC, until improved understanding of the disease biology led to the development of molecularly targeted agents. With the serial approval of several compounds directed against vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) signalling, and registration trials proving these newer agents were superior to IFNα, cytokine therapies were largely replaced as standard therapies in this disease4–10.
A new generation of targeted immune therapies is now emerging as a powerful tool in oncology. In particular, checkpoint inhibitors — antibody therapies that counteract the molecular mechanisms by which tumour cells evade immune recognition — are demonstrating impressive activity across an increasing number of tumour types11. Given that immune-mediated therapies are known to be active in RCC, patients with renal cancer were included in the early checkpoint inhibitor trials, with very encouraging results12,13.
The field of cancer immunotherapy is evolving rapidly. In addition to checkpoint inhibitors, many other immune therapies for RCC are being investigated, including novel vaccines, T-cell agonists, and adoptive T-cell therapies (FIG. 1). This trend is reflected by a number of ongoing phase III trials comparing the current standard-of-care VEGF-targeted therapies for metastatic RCC with novel immune treatments. In this Review, we discuss the biological principles that underlie the mechanism of action of these new therapies, summarize completed and ongoing clinical trials of novel immunotherapies with a focus on checkpoint inhibition, and consider future directions for immune-directed therapies for RCC.
Figure 1. Selected immune therapies under investigation for renal cell carcinoma (RCC).
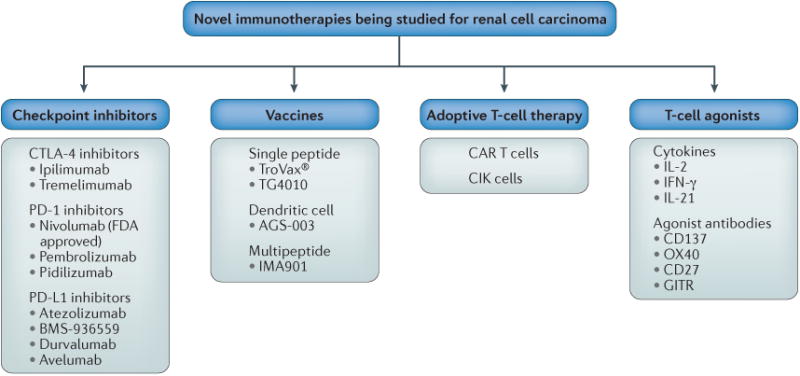
Checkpoint inhibitors under investigation include the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) inhibitors ipiliimumab and tremelimumab, the programmed cell death protein 1 (PD-1) inhibitors nivolumab (which is FDA approved), pembrolizumab and pidilizumab, and the programmed cell death 1 ligand 1 (PD-L1) inhibitors atezolizumab, BMS-936559, durvalumab, and avelumab. Vaccine strategies investigated in RCC include the single peptide vaccines TroVax® and TG4010, the dendritic cell vaccine AGS-003, and the multipeptide vaccine IMA901. Adoptive T-cell therapies such as chimeric antigen receptor (CAR) T cells and cytokine-induced killer (CIK) cells are also being investigated. Multiple T-cell agonists have been or are being studied, including the cytokines IL-2, IFNγ, and IL-21, as well as agonist antibodies to the co-stimulatory molecules CD137, OX40, CD27 and GITR.
Cancer immunology
Immunotherapy can induce long-lasting anticancer responses owing to the generation of antigen-specific immune memory, either through memory T cells or antibodies. Mellman and colleagues12 have outlined several crucial steps that are needed to mount an initial effective immune response against tumours. First, antigen-presenting cells (APCs), primarily dendritic cells, must encounter a tumour-associated antigen expressed on the tumour. Tumour-associated antigens can emerge via altered protein structure caused by acquired somatic mutations or differentially expressed proteins. The antigen expression also needs to be different enough from expression patterns on normal cells so that immune tolerance has not yet developed. Additionally, APCs require co-activating signals for maturation and function, such as factors released during tumour cell death. On encountering the tumour-associated antigen, APCs process it into peptide fragments, which then form a complex with major histocompatibility complex (MHC) class I and II molecules.
The first step in T-cell activation is recognition by the T-cell receptor (TCR) of the antigen presented on the MHC molecule. Full T-cell activation then requires a co-stimulatory signal, initiated by the CD28 receptors on the T cell binding to the B7 ligands (CD80 and CD86) on the APC14 (FIG. 2). CD3 and/or CD28 co- stimulatory molecules initiate a cascade of signals that results in increased cell metabolism and progression through the cell cycle15. CD8+ T cells are thought to be the main antitumour effectors, but the interplay between CD8+ and CD4+ T cells in cancer immunity is not fully under-stood16. Finally, the T cells need to travel to the tumour site and carry out their cytotoxic activity.
Figure 2. Site of action of checkpoint inhibitors and agonists being tested in advanced renal cell carcinoma (RCC).
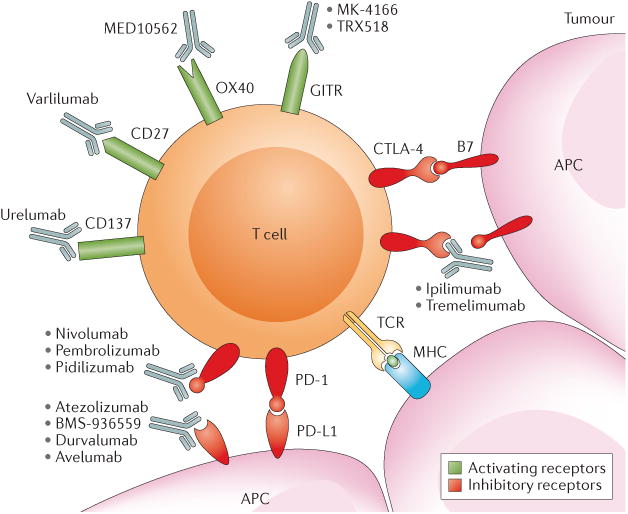
T-cell activation is regulated by various co-stimulatory and inhibitory checkpoints. Both agonistic antibodies to activating receptors and blocking antibodies to inhibitory receptors can stimulate T-cell activity and are being tested in advanced renal cell carcinoma and other solid tumours. Activation of T cells first requires an antigen-presenting cell (APC), such as a dendritic cell, to present an antigen. Here, an APC presents a tumour antigen complexed to major histocompatibility complex (MHC) class I to the T cell via the T cell receptor (TCR). Co-stimulatory signals are also needed at this time. At this point, B7 on an APC can bind to cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) creating an inhibitory signal, but ipilimumab or tremelimumab — CTLA-4 antibodies — can inhibit the inhibitory signal by binding to CTLA-4 and promote T-cell activation. Once the activated T cell is in the tumour environment it can recognize the antigen presented by an APC cell in the tumour. At this time, the programmed cell death protein 1 (PD-1) receptor can also send an inhibitory signal to the T cell when the receptor binds to programmed cell death 1 ligand 1 (PD-L1), which is often expressed on tumour cells. Inhibition of PD-L1 or PD-1 could block that signal. Several PD-1 inhibitors are under investigation for RCC, including pembrolizumab and pidilizumab, and nivolumab was recently FDA approved for patients with RCC who have failed prior antiangiogenic therapy. PD-L1 inhibitors under investigation include atezolizumab, BMS-936559, durvalumab and avelumab. In addition to inhibitory receptors, several activating receptors exist that stimulate T-cell activity, including CD137, CD27, OX40 and GITR. Similarly, several agonist antibodies target these receptors are under investigation for RCC. These include urelumab targeting CD137, varlilumab targeting CD27, MEDI10562 targeting OX40, and MK-4166 and TRX518 targeting GITR.
Multiple feedback mechanisms exert stimulatory or inhibitory effects on T cells, regulating immune function and preventing an excessive antigen response. These mechanisms include molecules on the surface of the T cells that act as checkpoints, and other immune cells such as regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSC), which can counteract the function of T cells17,18 (FIG. 3). Tumour cells in RCC and other diseases can take advantage of these mechanisms and prevent a potential anticancer immune response via cell– cell interactions and paracrine effects on the tumour microenvironment. Increased understanding of these processes has enabled the development of a number of targeted approaches.
Figure 3. Immunomodulatory effects of targeted therapies for renal cell carcinoma.
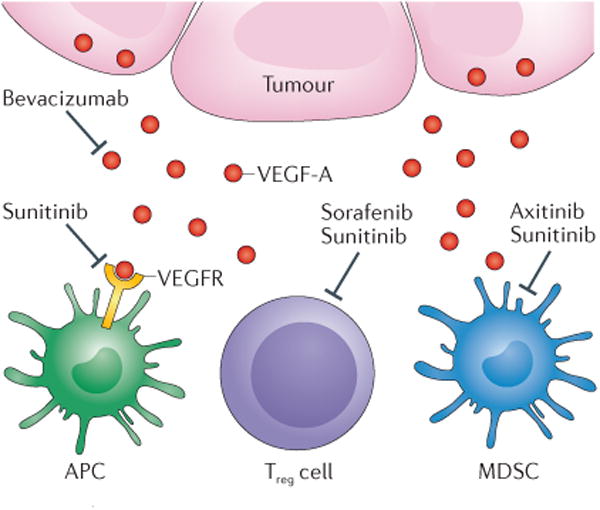
Tumour cells can secrete vascular endothelial growth factor-A (VEGF-A), which, when it binds to the VEGF receptor (VEGFR), can signal to halt antigen–presenting-cell (APC) maturation. Both bevacizumab — a monoclonal antibody against VEGF-A — and sunitinib — a small-molecule tyrosine kinase inhibitor — can block signalling through this pathway and, therefore, promote maturation of APCs. Both regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs) inhibit immune activation. VEGF-blockade with sunitinib or sorafenib can inhibit Treg cell function and treatment with sunitinib or axitinib has been found to inhibit function of MDSCs in preclinical and clinical models.
Checkpoint inhibitors
Immune-checkpoint receptors are negative immune regulators that limit proliferation and activity of T cells and other immune cells including macrophages and natural killer cells through signalling pathways. Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) are the best-studied inhibitory checkpoint receptors in the context of cancer immunotherapy, but several other surface receptors are also being explored as therapeutic targets, including the inhibitory receptors lymphocyte activation gene 3 protein (LAG-3), killer-cell immunoglobulin- like receptor (KIR), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and T cell immunoglobulin and mucin domain-containing 3 (TIM-3, also known as HAVCR2), as well as the tumour necrosis factor receptor superfamily member 4 (TNFRSF4 also known as OX40 or CD134), tumour necrosis factor receptor superfamily member 18 (TNFRSF18), CD27, and CD137 (REFS 19–21).
CTLA-4 and PD-1 are key regulators of adaptive immunity with complementary nonredundant functions. During early T-cell activation CTLA-4 is recruited to the plasma membrane, where it competes with the co- stimulatory receptor CD28 to bind to B7 ligands expressed on APCs22,23. CTLA-4 is thought to bind with both higher affinity and avidity than CD28 to B7 ligands24. CTLA-4 suppresses T-cell activation by competitively inhibiting CD28 binding to CD80 and/or CD86, and inducing downstream inhibitory signalling, which ultimately leads to decreased T-cell proliferation and IL-2 secretion23,25 (FIG. 2). Evidence also exists that CTLA-4 antibodies can deplete Treg cells, further contributing to activation of the immune system26. The importance of CTLA-4 regulation is highlighted by the fact that Ctla-4-knockout mice uniformly die at a young age owing to lymphoproliferative processes27.
Like CTLA-4, PD-1 is a transmembrane protein expressed in activated effector T cells, but not in resting T cells28. PD-1 has two known ligands, programmed cell death 1 ligand 1 (PD-L1) and PD-L2, which can be expressed on a variety of cells, including APCs, tumour cells and T cells themselves. When bound to its ligands, PD-1 also inhibits signalling pathways that normally lead to an effective T-cell response. In contrast to CTLA-4, which seems to mainly function in early activation of T cells, PD-1 has a role in limiting the activity of T cells in the periphery of the primary lymphoid organs during the inflammatory response29,30.
In the 1990s, data from preclinical studies showed that blockade of immune checkpoints bolsters the T-cell response and could result in tumour eradica-tion31. This observation led to the rapid development of many antibody therapies that block different steps in the immune-checkpoint pathway. The first checkpoint regulator to be clinically targeted was CTLA-4, but many others are currently in various stages of clinical development32 (FIG. 2).
Clinical experience
CTLA-4-directed antibodies
Ipilimumab — a monoclonal antibody against CTLA-4 — was the first checkpoint inhibitor to be tested in large-scale trials. It received FDA approval for the treatment of melanoma in 2011, after patients demonstrated superior overall survival in a phase III trial with either ipilimumab alone or in combination with the peptide gp100 vaccine compared with vaccine alone33. During its early clinical development, ipilimumab was evaluated for the treatment of advanced RCC. In a phase II trial, 61 patients were treated in one of two dosing cohorts (high or low) for up to 1 year, unless tumour progression or limiting toxicity was observed34. Response rates were 12.5% and 5% in the high-dose and low-dose cohorts, respectively. No complete responses or durable regressions were seen, in contrast to the experience in patients with melanoma. Higher toxicities were recorded for patients in the high-dose group than the low-dose group: 43% of these participants had grade 3, 4, or 5 toxicities compared with 18% for the low-dose group. Of note, a highly significant positive correlation between development of autoimmune toxicities and response was reported. The response rate for those who experienced toxicities was 30%, compared with 0% of those who did not experience toxicities (P <0.001). This correlation was only observed in a small cohort of patients, but could be important and should be further evaluated in larger phase III trials of checkpoint inhibitors in RCC. Some of the increased toxicity might be attributed to the dosing schedule used in this early study — nearly half of the patients who developed toxicities did so after five or more doses. In the phase II studies investigating ipilimumab for melanoma a total of four doses were used, which is now the widely practiced dosing regimen. Given the improved adverse-effect profile and efficacy of the newer PD-1 inhibitors, CTLA-4 antibody monotherapy in RCC has been largely abandoned in favour of combination trials with other checkpoint inhibitors35.
PD-1-directed antibodies
Nivolumab — a monoclonal antibody that blocks the PD-1 pathway — is the first checkpoint inhibitor to demonstrate a survival benefit in patients with metastatic RCC (TABLE 1). Motivated by the encouraging overall survival results of a dose-ranging phase II nivolumab trial, CheckMate 025 was designed as a phase III, open-label study comparing nivolumab with everolimus in patients with advanced RCC who had failed an anti-VEGF therapy, with overall survival as the primary end point36,37. A total of 821 patients were randomly assigned 1:1 to receive 3 mg/kg of nivolumab every 2 weeks (selected on the basis of its safety and efficacy profile in multiple tumour types) or 10 mg of everolimus daily.
Table 1. Results of selected checkpoint blockade trials in patients with renal cell carcinoma.
| Agent and trial | Phase | Population | Design | n | Response | Toxicities | Comments | Refs |
|---|---|---|---|---|---|---|---|---|
| Nivolumab (CheckMate 025) | III | Prior systemic therapies:
|
Open-label, 1:1 randomized trial | 821 | OS (months)
|
Grade 3–4 treatmentrelated AEs:
|
Hazard ratio for death with nivolumab = 0.73 (P = 0.002) | 37 |
| Nivolumab and ipilimumab | I |
|
Randomized trial of three dosing cohorts: Induction
|
100 | ORR:
|
Treatment in Arm C stopped owing to toxicity Grade 3–4 treatment related AEs:
|
Median OS not reached with median follow-up duration ranging from 46–90 weeks | 48 |
| Atezolizumab and bevacizumab | Ib | Various solid tumours allowed For RCC prior systemic therapies:
|
Two arms:
|
|
|
Grade 3–4 AEs regardless of attribution across all tumour types:
|
Atezolizumab and bevacizumab 15 mg/kg every 3 weeks chosen as the dose for a phase 2 trial | 62 |
AE, adverse event; MSKCC, Memorial Sloan Kettering Cancer Center; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; RCC, renal cell carcinoma.
The study was stopped after a prespecified interim analysis determined that the primary end point had been reached. The median overall survival was 25 months for nivolumab and 19.6 months for everolimus37. The overall survival benefit of nivolumab was present irrespective of Memorial Sloan Kettering Cancer Center risk group, number of previous therapies, or PD-L1 tumour expression level. The objective response rate (ORR) was also higher with nivolumab than everolimus (25% versus 5%), but complete responses were rare — only 1% for nivolumab and <1% for everolimus. Interestingly, the median progression-free survival was similar in both groups (4.6 months for nivolumab and 4.4 months for everolimus). An ad hoc sensitivity analysis was performed on data from patients who were alive without disease progression at 6 months. The median progression-free survival was 15.6 months in the nivolumab group versus 11.7 months in the everolimus group, suggesting a potential delayed benefit in progression-free survival with nivolumab37.
The toxicity profile was consistent with those observed in other studies on nivolumab: grade 3 or grade 4 treatment- related adverse events occurred in 19% of those treated with nivolumab and 37% of those treated with everolimus. Fatigue was the most common grade 3 or grade 4 adverse event reported for nivolumab, occurring in 2% of patients37.
Based on the results of CheckMate 025, nivolumab received approval from the FDA in November 2015 for the treatment of advanced RCC in patients who have received prior antiangiogenic therapy.
Nivolumab and other PD-1 inhibitors, such as pidili-zumab, are now being studied in various clinical settings, including in combination with other therapies and as adjuvant or neoadjuvant therapy38-40.
Anti-PD-L1 antibodies
Several anti-PD-L1 anti bodies including BMS-936559, durvalumab and atezolizumab (MPDL3280A) are being tested in clinical trials in various malignancies, including RCC. Anti-PD-L1 antibodies inhibit the PD-1-PD-L1 signalling pathway but spare PD-1-PD-L2 interaction. PD-L2 is thought to have less of a role in T-cell inhibition at tumour sites than PD-L1, but the full clinical implications of sparing PD-L2 are not completely understood41. PD-L2 binds to RGM domain family member B (RGMb), which is expressed in lung interstitial macrophages and alveolar epithelial cells, and regulates respiratory immunity42. Some authors have suggested that blockade of this interaction mediates the rare, but potentially fatal, pneumonitis that has been seen in trials of anti-PD-1 agents43.
In the phase I trial of BMS-936559, 17 patients with RCC were included and, of those, only two had an objective response, seven patients had stable disease, and progression-free survival at 6 months was 53%. The overall rate of grade 3 and grade 4 adverse events across all tumour types was 9%, mirroring the more benign adverse-effect profile of anti-PD1 therapies35.
Atezolizumab has also been tested in RCC as part of a phase I trial in multiple different types of solid tumours44. A total of 70 patients with either clear cell RCC (90%) or non-clear cell RCC (10%) were treated every 3 weeks for up to 1 year; dose escalation was performed up to 1,200 mg flat dose. Most of the patients had received previous treatment, including 57% who had received two or more lines of therapy. The ORR for patients with clear cell RCC was 15%, and median progression-free survival was 5.6 months (95% CI 3.9–8.2).%. The toxicity profile was again very similar to PD-1 inhibitors and no grade 3 or grade 4 pneumonitis was reported. Given promising preliminary data on combination checkpoint inhibitor and targeted therapies, atezolizumab is now being tested in combination with bevacizumab for RCC45. Another PD-L1 inhibitor, durvalumab, is being tested in combination with CTLA-4 inhibition in advanced RCC46.
Checkpoint inhibitor combinations
In an effort to improve on the single-agent activities of immune-based therapies, combinations with other immunotherapies, radiotherapy, chemotherapy, and targeted therapies are being studied in a broad range of malignancies. Preliminary clinical data indicates that some combinations can be synergistic, and the field is developing rapidly20 (TABLE 2).
Table 2. Ongoing combination checkpoint inhibitor and targeted therapy trials in RCC.
| Checkpoint inhibitor | Targeted therapy | Phase | Population | Identifier | Refs |
|---|---|---|---|---|---|
| Nivolumab |
|
I | Advanced RCC, prior cytokine therapy allowed | NCT01472081 (CheckMate 016) | 49 |
| Nivolumab | Bevacizumab | Neoadjuvant pilot | Metastatic clear cell RCC, prior therapy allowed | NCT02210117 | 38 |
| Nivolumab | Temsirolimus | Ib/II | Metastatic RCC, prior therapy allowed | NCT02423954 | 117 |
| Pembrolizumab | Pazopanib | I/II | Untreated, advanced clear cell RCC | NCT02014636 | 65 |
| Pembrolizumab | Axitinib | Ib | Untreated, advanced clear cell RCC | NCT02133742 | 67 |
| Pembrolizumab | Bevacizumab | Ib/II | Metastatic clear cell RCC treated with failure of at least one prior therapy | NCT02348008 | 66 |
| Pembrolizumab | Aflibercept | I | Metastatic RCC treated with at least one prior VEGF TKI | NCT02298959 | 118 |
| Avelumab | Axitinib | Ib | Untreated, advanced clear cell RCC | NCT02493751 | 119 |
| Atezolizumab | Bevacizumab | III | Untreated, advanced clear cell RCC | NCT02420821 | 45 |
RCC, renal cell carcinoma; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor.
One of the best-studied combinations is CTLA-4 and PD-1 blockade. These checkpoint inhibitors affect different stages of the tumour-directed T-cell response and early data suggests that their actions could be synergistic47. A randomized, double-blind, phase III trial in patients with melanoma comparing combination nivolumab and ipilimumab with ipilimumab alone or nivolumab alone showed an unprecedented 58% response rate for the combination, driving enthusiasm for testing the combination in other immunogenic malignancies48.
Results from combination immunotherapy trials for RCC have so far demonstrated manageable safety and promising antitumour activity. CheckMate 016 was a large phase I study on advanced RCC investigating nivolumab in combination with different agents — sunitinib, pazopanib, or ipilimumab49. The study included three dose cohorts of concurrent ipilimumab and nivolumab50. All patients received induction with the combination every 3 weeks for four doses, followed by a maintenance phase of nivolumab 3 mg/kg every 2 weeks. Three dosing regimens for the induction were used: ipilimumab 1 mg/kg and nivolumab 3 mg/kg, ipilimumab 3 mg/kg and nivolumab 1 mg/kg, and ipilimumab 3 mg/kg and nivolumab 3 mg/kg. The last dosing regimen was discontinued after six patients experienced excessive toxicity.
Analysis of an expansion cohort of the ipilimu-mab 1 mg/kg plus nivolumab 3 mg/kg and ipilimumab 3 mg/kg plus nivolumab 1 mg/kg arms (47 patients in each group) showed ORR of 38% and 43%, respec-tively51. The median duration of response was not reached in the patients receiving nivolumab 1 mg/kg plus ipilimumab 3 mg/kg and was 53.9 weeks in patients receiving nivolumab 3 mg/kg plus ipilimumab 1 mg/kg. Median overall survival had not been reached in either arm. Grade 3 or grade 4 treatment- related adverse events occurred in 34% of patients receiving ipilimumab 1 mg/kg plus nivolumab 3 mg/kg and 64% of patients receiving ipilimumab 3 mg/kg plus nivolumab 1 mg/kg. The most common adverse effects of any grade were fatigue, rash, pruritus, nausea, and diarrhoea.
These encouraging results motivated the design of CheckMate 214, an ongoing multinational, phase III trial comparing combination ipilimumab and nivolumab with sunitinib as first line treatment for metastatic RCC. This trial has completed accrual of >1,000 patients52. Given the adverse-event profiles seen in CheckMate 016, patients receive ipilimumab at 1 mg/kg and nivolumab 3 mg/kg every 3 weeks for four treatments, followed by maintenance nivolumab 3 mg/kg every 2 weeks. The primary endpoint for this trial is overall survival.
Checkpoint inhibitor and targeted therapy combinations
In RCC, immunotherapy in combination with approved targeted therapies are an interesting prospect as anti-angiogenic treatments and have been proposed to have immunomodulatory effects themselves53,54 (FIG. 3). The cytokine VEGF-A can have potent immuno-modulatory actions including promoting the proliferation of MDSCs and altering dendritic-cell maturation. Anti-VEGF agents like bevacizumab, sunitinib and axitinib have been shown to decrease the number and effectiveness of dendritic cells, Treg cells and MDSCs in various cancer models53,55–57. In a study analysing Treg cell response in patients with metastatic RCC before and after treatment with sunitinib, there was a reduction in Treg cells after 1–2 cycles of sunitinib, although this reduction did not reach statistical significance58. In another study of melanoma patients treated with ipili-mumab plus bevacizumab versus ipilimumab alone, those treated with the combination showed increased trafficking of CD8+ T cells and CD163+ dendritic macro-phages, as well as increased circulating memory T cells compared with immunotherapy alone59,60. These immune boosting actions could potentially synergize with the actions of checkpoint inhibitors.
The first trial to combine CTLA-4 blockade with an anti-VEGF agent showed that the combination was poorly tolerated. A phase I, dose-escalation trial tested the combination of sunitinib and tremelimumab — an anti-CTLA-4 antibody61. In total, 28 patients with metastatic disease who had received no more than one prior systemic therapy were enrolled. Dose-limiting toxicities (DLT) were experienced by two of five participants who received 50 mg of sunitinib and the lowest dose of tremelimumab 6 mg/kg, and this regimen was discontinued. The most common DLT was unexpected rapid- onset renal failure. Of the 21 evaluable patients, nine (43%) had a partial response.
Other trials have shown improved toxicity profiles for combination therapy. The combination that is furthest along in the clinical trial process is bevacizumab with atezolizumab — an anti-PD-L1 therapy. A phase I trial investigating two dosing regimens (2 week and 3 week) of this combination in patients with solid tumours, including 13 patients with RCC62. Of these participants, 12 were assigned to a dosing schedule of bevacizumab 15 mg/kg and atezolizumab 20 mg/kg every 3 weeks, and one assigned to receive bevacizumab 10 mg/kg and atezolizumab 15 mg/kg every 2 weeks. Treatment was well tolerated — in the entire solid-tumour cohort, grade 3 adverse events attributed to atezolizumab were experienced by 3% of patients in the 3-week dosing group and 17% in the 2-week dosing group. No grade 4 or grade 5 adverse events were attributed to atezolizumab. The ORR for RCC was 40%, including one complete remission. A three-arm phase II trial comparing this combination, atezolizumab monotherapy, or sunitinib in patients with untreated advanced disease has completed accrual63. Patients in the atezolizumab monotherapy arm are allowed to crossover to the combination upon progression. A phase III, open-label randomized trial is ongoing that compares this bevacizumab plus atezoli-zumab combination with sunitinib only, with a primary end point of progression-free survival45.
The phase I CheckMate 016 trial, in addition to testing ipilimumab plus nivolumab, investigated nivolumab at 2 mg/kg or 5 mg/kg every 21 days in combination with either pazopanib or sunitinib at standard doses64. Data from this trial demonstrated that combining PD-1-directed therapy with anti-VEGF can be challenging, owing to high toxicity. Treatment-related grade 3 and grade 4 adverse events were observed in >70% of patients enrolled in the study. Particularly, renal and hepatic toxicities were high: in the pazopanib arm, 20% of patients developed grade 3 or grade 4 hepatotoxicity and in the sunitinib arm, 3% of patients had grade 3 or grade 4 renal toxicity. Nevertheless, with an ORR of 52% and an additional 30% of participants having stable disease, the sunitinib plus nivolumab combination seems to be highly active. Similarly, the ORR of 45% for patients who received pazopanib plus nivolumab, all of whom had received previous anti-VEGF therapy, suggested synergism in this combination. Selecting anti-VEGF therapies without overlapping toxicities with checkpoint inhibitors will be important in ensuring improved safety profiles.
Other trials testing combinations are ongoing and include phase I trials of pembrolizumab in combination with axitinib, bevacizumab, or pazopanib65–67 (TABLE 2).
Other considerations
Assessing response
Traditional measures of assessing response to therapy, such as progression-free survival and ORR, might not be as predictive of the benefit of checkpoint inhibitors as they are for other forms of therapy. For example, the initial checkpoint inhibitor trials showed that some patients can have unique responses such as stable or even progressive disease initially and then a response. In the phase II nivolumab trial31, overall survival (range 18.2–25.5 months) was strikingly longer than expected from the progression-free survival (range 2.7–4.2 months), and in the phase III trial32, a significant overall survival benefit was observed in patients who received nivolumab compared with those who received everolimus (P = 0.002), but a progression-free survival benefit was not observed36,37. Additionally, long-term follow-up analysis of the patients involved in the phase I trial of nivolumab showed that many patients can have durable responses: 30% of responders had ongoing responses beyond 96 weeks68. One report detailed one RCC patient whose response evolved into a complete response >3 years after stopping anti-PD1 therapy69.
The relatively modest progression-free survival in these trials is probably the result of a few factors. First, infiltration of tumours by activated immune cells could mimic progression on imaging studies. This phenomenon has been termed pseudoprogression70. Second, tumour growth might initially outpace the activation of the immune system, showing initial progression followed by response. Finally, immune responses might be delayed compared with traditional targeted therapy responses. Immune-related Response Evaluation Criteria In Solid Tumours (RECIST) criteria have been proposed to address this phenomenom, but at the moment these criteria remain under investigation in RCC71.
Immune-related adverse events
Toxicities from checkpoint blockade therapy are different from those experienced with targeted therapy and are mostly attributable to a hyperactivated T-cell response causing inflammation in healthy tissues72, These toxicities are known as immune-related adverse events (irAEs). Commonly reported irAEs associated with CTLA, PD-1, and/or PD-L1 blockade include fatigue, skin rash, colitis, and asymptomatic hepatitis, but other rare toxicities have also been observed, such as hypophysitis, uveitis, and pneumonitis11,72.
Data from phase I trials studying checkpoint inhibitors for RCC suggest that both the frequency and grade of toxicities caused by CTLA blockade alone and in combination with PD-1 inhibitors are greater than those for PD-1- directed monotherapy. The rate of grade 3 or grade 4 toxicities with nivolumab monotherapy in the phase III trial was 19%73, which is consistent with the PD-1 inhibitor experience in other tumour types, in which follow-up duration is longer73. The most common adverse effects were fatigue, nausea, and pruritus, but rare hepatitis and endocrinopathies were also reported. In this trial, patients were monitored with comprehensive metabolic panels before each administration of the therapy and for thyroid function every other cycle. The rate of grade 3 or grade 4 toxicities in the phase I trial of ipilimumab and nivolumab were >30% for all dose levels tested, but phase III data for more definitive toxicity information are pending41,42.
Early recognition and withholding of the agent are thought to be key to managing toxicity, and systemic corticosteroids constitute the standard treatment for high-grade irAEs. In the phase II study of nivolumab, 15%, 19%, and 33% of patients required systemic corticosteroids in the 0.3 mg/kg, 2 mg/kg, and 10 mg/kg groups, respectively36. Additionally, immune-mediated endocrinopathies frequently require hormone replacement owing to organ dysfunction. When steroid- refractory symptoms occur, nonsteroidal immunosuppressants such as tumour necrosis factor inhibitors or mycophenolate mofetil can be used successfully; the choice of such agents depends on the affected organ. Most irAEs seem to be reversible with treatment discontinuation and immuno-suppression, but long-term follow-up data for some adverse effects, including endocrinopathies, CNS toxicity, and pneumonitis are lacking68.
The safety of these agents in people with an underlying autoimmune disease is unknown, as these patients were excluded from the trials. Emerging data in melanoma suggest that ipilimumab might be tolerated in some patients with an autoimmune disease, but data are still lacking for patients with RCC74. Extensive descriptions of irAEs and their treatment are reviewed elsewhere11,72,73,.
Predictive biomarkers
The precise mechanism of action of checkpoint blockade in RCC is still unknown and the search for biomarkers predictive of response is an ongoing area of research. Arguably, some tumours, especially melanoma and lung cancer tumours, respond to immunotherapy owing to their high overall burden of somatic mutations, facilitating host recognition and generating a stronger CD8+ T-cell response75. Interestingly, the somatic mutation association is low in RCC compared with other tumour types such as lung cancer or melanoma, raising the possibility that other biological aspects of RCC render this cancer susceptible to immuno-therapies75. In melanoma and non-small cell lung cancer, evidence suggests that response to checkpoint blockade correlates with CD8+ T-cell recognition of tumour neo-antigens that are thought to initiate a response mediated by MHC class I76,77, although such associations have not yet been observed in RCC.
PD-L1 expression levels in tumour cells and tumour-infiltrating lymphocytes and their correlation with response to blockade have been widely studied in RCC and other tumours. Early single-agent studies of nivolumab and atezolizumab demonstrated correlation between high PD-L1 tumour expression and ORRs, but lack of PD-L1 expression in a tumour does not exclude the possibility of the patient achieving an objective response37,78,79. In patients with >1% PD-L1 expression, median overall survival was 22 months with nivolumab and 19 months with everolimus (HR = 0.79; 95% CI, 0.53–1.17). Among patients with <1% PD-L1 expression, overall survival was 27 months with nivolumab and 21 months with everolimus (HR = 0.77; 95% CI, 0.60–0.97). These findings suggest that the benefit of nivolumab over everolimus is not dependent on PD-L1 expression status, but indicated that PD-L1 expression is likely to be a prognostic marker associated with worse outcomes in RCC, in line with previous data80,81. These analyses have many limitations including the heterogeneous expression of PD-L1 — both intratumoural and between primary and metastatic sites — and the fact that PD-L1 expression seems to be a dynamic process and, therefore, might be mis represented when tested on an archival tumour specimen. Another concern is that different trials have used different PD-L1 assays, targets (expression on tumour cells, lymphocytes, or both), and expression thresholds to define PD-L1 positivity, which are likely to vary substantially in their sensitivity and specificity. Generally, clinicians agree that, until further data is gathered, PD-L1 tumour expression should not be used to select patients for anti-PD-1 therapy.
Enthusiasm exists for investigating other bio markers predictive of immunotherapy response. Choueiri and colleagues78,79 led CheckMate 009, a large, dose-randomized phase II study of nivolumab in patients with advanced RCC designed to investigate tissue and serum biomarkers. In total, 67 patients who had received prior therapy received nivolumab at three different doses (0.3, 2.0, and 10.0 mg/kg every 3 weeks); 24 treatment-naive patients were treated at 10 mg/kg every 3 weeks. Fresh tumour biopsies and serum samples were acquired at baseline and after 4 weeks of therapy. Evidence of immunomodulatory effects was observed with treatment, including dynamic changes in CD3+ and CD8+ tumour-infiltrating cells and increases in chemokine CXCL9 and CXCL10 concentrations in peripheral blood79. Exploratory analyses suggested that benefit from nivolumab might correlate with post- treatment increases in T-cell counts in the tumour, increased expression of IFNγ-regulated genes, and decreases in T-cell receptor clonality in the blood. Expression profiling during treatment suggested feedback between different immune checkpoints, lending further support to the concept of combination checkpoint inhibitor therapy.
Non-clear cell renal cell carcinoma
Evidence exists for using anti-PD-1 inhibitors in non-clear cell RCC. Response to immunotherapy has been seen across epithelial tumours, with no clear predictive characteristic yet identified, other than mismatch repair status82. The FDA approved nivolumab for RCC without restriction to specific subtypes; however the experience in non-clear cell subtype is limited, as the phase II and phase III trials of this therapy did not include patients with this disease.
Choueiri et al.83 reported PD-L1 tumour expression in different RCC subtypes including chromophobe (5.6% PD-L1 expression), papillary (10% PD-L1 expression), Xp11.2 translocation (30% PD-L1 expression), and collecting duct (20% PD-L1 expression). However, PD-L1 expression status does not perfectly predict response to therapy with anti-PD-L1 and, therefore, should not limit patient selection in clinical trials.
The phase I study of atezolizumab did include 7 patients with non-clear cell histology, and of those, none had a response by conventional RECIST, but one patient had a response by immune-related response criteria that was ongoing at the cut-off point for data collection62. In addition, an anecdotal report demonstrated notable efficacy in a papillary tumour with sarcomatoid and rhabdoid features84. Clinical trials of checkpoint inhibitor therapy to assess efficacy are a priority, given the unmet need for effective systemic therapy for this heterogeneous group of tumours. In absence of more data, we recommend that patients with non-clear cell RCC be treated with immunotherapy in the setting of clinical trials whenever possible.
Vaccines
Antitumour vaccines constitute a strategy to stimulate the immune system to recognize and eliminate malignant cells. Historically, these vaccines had failed to show benefit in human subjects, but FDA approval in 2010 of the first therapeutic cancer vaccine sipuleucel-T for prostate cancer has resparked interest in the field.
Vaccine strategies in cancer immunology can differ in the antigen used, the manufacturing mode, and the adjuvant used85,86. Attempts at treatment with modified virus vaccines targeting a single tumour antigen have been disappointing. TG4010, a vaccine with a modified vaccinia virus expressing mucin-1 (MUC1), a glycoprotein that is overexpressed in RCC, in combination with adjuvant IL-2, showed an acceptable safety profile but no clinical activity in a phase II trial involving patients with advanced RCC87. MVA-5T4 (Tro Vax®, Oxford BioMedica, UK), an attenuated modified vaccinia virus vaccine encoding the tumour antigen 5T4 has also been studied. The antigen 5T4 is highly expressed on the cell surface of renal carcinomas, but rarely detected in normal adult tissues other than the placenta, making it an attractive target for vaccine development88. A large phase III trial showed that the addition of the vaccine to standard therapy was well tolerated, but added no overall survival benefit compared with standard therapy alone89.
Currently, only two vaccine therapies are under phase III trial investigation for advanced RCC. The first is AGS-003, an autologous dendritic-cell vaccine generated with use of patient-specific tumour-cell RNA isolated from fresh tumour tissue90. Dendritic cells are collected from the patient via leukapheresis, then exposed to tumour antigens and matured in vitro, and ultimately reinfused into the patient, with the aim of stimulating the immune response85. In a phase II trial, newly diagnosed patients with unfavourable prognosis received AGS-003 plus sunitinib91. Median progression-free survival was 11.2 months, and median overall survival was 30.2 months, exceeding historical controls for this subgroup of patients. Notably, no added toxicity from the vaccine was observed. The ADAPT study is an ongoing phase III trial, building on these early results, comparing AGS-003 plus sunitinib with sunitinib alone in newly diagnosed patients with metastatic clear cell RCC undergoing cytoreductive nephrectomy, with overall survival as the primary endpoint92.
A phase III trial of the second vaccine IMA901 (a multipeptide) has concluded93. IMA901 consists of ten tumour-associated peptides (TUMAPs) whose antigens are overexpressed in RCC, including MUC1, PLIN2, APOL1, CCND1, GUCY1A3, PRUNE2, MET, RGS5, HBV and MMP7. IMA901 has been developed in combination with granulocyte–macrophage colony- stimulating factor (GM–CSF), as well as cyclophosphamide, with the rationale that cyclophosphamide can inhibit Treg cells that could dampen the immune response. A phase I study compared IMA901 plus GM–CSF with and without cyclophosphamide94. Patients in both groups received 17 vaccinations over 9 months. An overall survival benefit was noted among those patients who had an immune response to multiple TUMAPs and also received cyclophosphamide. A phase III trial randomly assigned 339 patients in a 3:2 fashion to sunitnib plus IMA901, GM-CSF, and prevaccination cyclo phosphamide, or sunitinib alone. Results were presented at the European Society of Medical Oncology Annual Congress in 2015, showing that addition of the vaccine did not improve overall survival, the trial's primary end point95. No difference in progression-free survival was observed. Of interest, the patients in the control arm had higher rates of sunitinib exposure, which were significantly increased in the intermediate-risk group, than patients in the control arm, therefore, direct comparison of outcomes between the groups is not possible. Similar to the phase II data, adverse events were not significantly different between the treatment groups.
A natural next step of investigation is the combination of vaccine therapy and checkpoint inhibitor therapy. Vaccines can prime dendritic cells to present antigen, but the inhibitory effects in the immune milieu might limit their effect. Hence, checkpoint blockade therapy might enhance the effectiveness of vaccine therapy and vice versa. This concept is being investigated in an ongoing phase II trial evaluating PD-1 blockade alone or in combination with a dendritic cell vaccine, such as AGS-003 in advanced RCC96.
Future directions
Adoptive T-cell therapy
Adoptive T-cell transfer therapy refers to the autologous or allogeneic infusion of T cells. One such therapy involves the generation and infusion of chimeric antigen receptor (CAR) T cells — T cells that have been genetically modified to express a receptor specific to tumour epitopes independent of human leukocyte antigen (HLA). The promising efficacy of anti-CD19 CAR T cells in haematological malignancies has inspired further investigations in solid tumours97.
One of the key aspects of designing effective CAR T cells is finding a tumour-associated antigen that is uniformly expressed in tumour cells but not in normal tissue. Carbonic anhydrase 9 (CAIX) is an enzyme that is overexpressed in clear cell RCC but minimally expressed in normal tissue98. Early efforts in using CAIX as a tumour-associated antigen for CAR resulted in liver enzyme elevations that limited its use, likely owing to the therapy also targeting CAIX expressed in liver bile duct epithelium99. A group in the Netherlands gave patients a CAIX monoclonal antibody before infusion of CAR T cells to reduce this on-target-off-tumour toxicity100. This strategy was based on previous clinical trials using a radiolabelled CAIX monoclonal antibody in patients with RCC, in which low doses of antibody could saturate the liver in humans, but not saturate RCC tumour lines in vitro101. Compared with the eight patients without pretreated antibodies, of whom 50% had grade 2 or greater liver enzyme abnormalities after CAR T cell infusion, none of the patients who received pretreated antibodies exhibited grade 3 or greater liver enzyme abnormalities100. However, in the study, no clinical responses were observed, and the efficacy of CAIX CAR T cells has yet to be proven.
Another form of adoptive immune cell therapy tested in RCC is autologous cytokine-induced killer (CIK) cell immunotherapy. CIK cells are created in vitro by harvesting peripheral mononuclear cells in the blood using an anti-CD3 antibody, IL-1, IFNγ, and IL-2, and the resulting phenotype shares features of effector T cells and natural killer cells102. A phase II trial randomly assigned 148 patients with metastatic RCC to CIK cell immunotherapy or IL-2 combined with IFNα103. Progression-free survival and overall survival at 3 years in the CIK-cell therapy arm were 18% and 61%, respectively, compared with 12% and 23% in the IL-2 plus IFNα arm (P = 0.031 and P <0.001, respectively). This therapy is being further investigated in conjunction with dendritic cell vaccines and early results show that therapy is well tolerated and might have activity against RCC104.
T-cell agonists
Stimulatory molecules expressed on immune cells can also be targeted with agonist antibodies. CD137 is a co- stimulatory molecule for T cells that increases T-cell-effector activity and survival. Its use in combination with anti-DR5 and anti-CD40 antibodies in mouse models of RCC has shown to improve survival compared with control mice (P <0.001)105. The CD137 agonist PF-05082566 is currently being tested in combination with pembrolizumab in a phase I trial of advanced solid tumours, including RCC106. Varlilumab is an agonist antibody targeting CD27, and is also a co-stimulatory molecule that regulates T-cell activation, survival and memory response. In a phase I trial in solid tumours, including 11 patients with RCC, varlilumab was well tolerated and correlative studies showed immune activation consistent with CD27 co-stimulation. Of the six evaluable RCC patients, two had stable disease107. This antibody is currently being studied in combination with sunitinib and in combination with atezolizumab in phase I/II trials in RCC108.
The stimulatory cytokine IL-21 has also been tested as monotherapy and in combination with tyrosine kinase inhibitors in phase I trials in RCC109,110. In a phase I study of recombinant IL-21 in patients with melanoma or RCC, therapy was well tolerated; of the 19 RCC patients, four had partial responses and 13 had stable disease111. Combination with sunitinib resulted in dose-limiting thrombocytopenia and neutropenia, but combination with sorafenib was better tolerated, with a response rate of 21% and evidence of durable responses.
In addition to CD137 and CD27, other co-stimulatory molecules such as OX40 and GITR are also promising therapeutic targets. Trials of monotherapy with the OX40 agonist MEDI0562, and with the GITR agonists MK-4166 and TRX518, for example, are underway in solid tumour malignancies112–114. Like combination checkpoint blockade strategies, much enthusiasm exists for combined treatment strategies with other immunomodulatory agents115,116.
Conclusions
Novel immune therapies are emerging as an important addition to targeted therapies in the treatment of RCC. Many questions regarding their optimal use remain including timing, dose, length of treatment, and adjuvant or neoadjuvant use, among others. Additional investigations into predictive biomarkers are needed to optimize patient selection. As the field of cancer immuno therapy develops, the use rational approaches will be important to select and test optimal treatment combinations. To date, nivolumab has been approved in the second- line setting, and phase III, randomized trials with novel immuno therapy combinations are challenging the first-line standard of care in RCC — in the near future, immunotherapy will likely be a new standard therapy.
Key points.
Checkpoint inhibitors have shown promising efficacy and manageable safety profiles for patients with advanced renal cell carcinoma (RCC)
The programmed cell death protein 1 (PD-1) inhibitor nivolumab prolongs overall survival in patients with advanced RCC following therapy with an antiangiogenesis agent and has FDA approval in this setting
Promising combinations of checkpoint inhibitors with other checkpoint inhibitors or targeted therapies are being compared with antiangiogenic agents for patients with advanced RCC in clinical trials
Other novel immune therapies such as vaccines, cytokine-induced killer cells, and T-cell agonists for advanced RCC are being studied in clinical trials
Further studies should focus on how to best select patients who will benefit from immune therapies and on how to choose rational combination strategies
Footnotes
Competing interests statement: M.H.V. has been a consultant for Novartis, GlaxoSmithKline, Bayer, Calithera, and Natera and has received honoraria from Novartis and research funding from Brystol-Myers Squibb and Pfizer. R.J.M. has been a consultant for Pfizer, Novartis, and Eisai and has received research funding from Genentech, Brystol-Myers Squibb, Exelixis, Eisai, and GlaxoSmithKline. M.I.C. declares no competing interests.
Author contributions: All authors researched data for the article, contributed substantially to discussion of content, wrote and reviewed and edited the article before submission.
Review criteria: PubMed database searches were made using the following terms: “renal cell and CTLA-4”, “renal cell and PD-1”, “renal cell and PD-L1”, “renal cell and checkpoint”, “renal cell and vaccine”, and “renal cell and immunotherapy”. The ASCO and ESMO annual meeting website (http://meetinglibrary.asco.org/abstracts) was searched using similar terms.
References
- 1.Coley W. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;105:487–510. [PubMed] [Google Scholar]
- 2.McDermott DF. Immunotherapy of metastatic renal cell carcinoma. Cancer. 2009;115:2298–2305. doi: 10.1002/cncr.24236. [DOI] [PubMed] [Google Scholar]
- 3.Fyfe G, et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13:688–696. doi: 10.1200/JCO.1995.13.3.688. [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 5.Hudes G, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 7.Escudier B, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2015;370:2103–2111. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg CN, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 9.Rini BI, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 10.Escudier B, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:1125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 11.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brahmer JR, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 15.Appleman LJ, Chernova I, Li L, Boussiotis VA. CD28 costimulation mediates transcription of SKP2 and CKS1, the substrate recognition components of SCFSkp2 ubiquitin ligase that leads p27kip1 to degradation. Cell Cycle. 2006;5:2123–2129. doi: 10.4161/cc.5.18.3139. [DOI] [PubMed] [Google Scholar]
- 16.Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2:91–98. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- 17.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 21.Chauvin JM, et al. TIGIT and PD-1 impair tumor antigen – specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 23.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linsley PS, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano E, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 28.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fife BT, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. 2008;8:153–160. doi: 10.1038/nri2253. [DOI] [PubMed] [Google Scholar]
- 31.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 32.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang JC, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30:825–830. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robert C, Schachter J, Long GV. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:372–2521. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 36.Motzer RJ, et al. Nivolumab for metastatic renal cell carcinoma (mRCC): results of a randomized, dose-ranging phase II trial. J Clin Oncol. 2015;33:1430–1437. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motzer RJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02210117.
- 39.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02575222.
- 40.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/show/NCT01441765.
- 41.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Y, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211:943–959. doi: 10.1084/jem.20130790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015;7:85–96. doi: 10.1177/1758834014567470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDermott DF, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol. 2016;34:833–842. doi: 10.1200/JCO.2015.63.7421. [DOI] [PubMed] [Google Scholar]
- 45.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02420821.
- 46.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02762006.
- 47.Curran Ma, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT01472081.
- 50.Hammers HJ, et al. Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC) [abstract] J Clin Oncol. 2014;32(5s Suppl):4504. doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammers HJ, et al. Expanded cohort results from CheckMate 016: a phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC) [abstract] J Clin Oncol. 2015;33(Suppl):4516. [Google Scholar]
- 52.Hammers HJ, Plimack ER, Sternberg CN, McDermott DF, Larkin JMG. CheckMate 214: a phase III, randomized, open-label study of nivolumab combined with ipilimumab versus sunitinib monotherapy in patients with previously untreated metastatic renal cell carcinoma [abstract] J Clin Oncol. 2015;33(Suppl):TPS4578. [Google Scholar]
- 53.Terme M, et al. Modulation of immunity by antiangiogenic molecules in cancer. Clin Dev Immunol. 2012;2012:492920. doi: 10.1155/2012/492920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desar IME, et al. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129:507–512. doi: 10.1002/ijc.25674. [DOI] [PubMed] [Google Scholar]
- 56.Yuan H, et al. Axitinib augments antitumor activity in renal cell carcinoma via STAT3-dependent reversal of myeloid-derived suppressor cell accumulation. Biomed Pharmacother. 2014;68:751–756. doi: 10.1016/j.biopha.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 57.Du Four S, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology. 2015;4:e998107. doi: 10.1080/2162402X.2014.998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finke JH, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 59.Nishino M, Giobbie-Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer. 2014;2:40. doi: 10.1186/s40425-014-0040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hodi FS, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–642. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rini BI, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117:758–767. doi: 10.1002/cncr.25639. [DOI] [PubMed] [Google Scholar]
- 62.Lieu C, Bendell J, Powderly JD, Pishvaian M, Hochster S. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or chemotherapy (chemo) in patients (pts) with locally advanced or metastatic solid tumors. Ann Oncol. 2014;25(Suppl):iv361–iv372. [Google Scholar]
- 63.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT01984242.
- 64.Amin A, Plimack ER, Infante J, Ernstoff B, Rini BI. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC) [abstract] J Clin Oncol. 2014;32(5s Suppl):5010. [Google Scholar]
- 65.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02014636.
- 66.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02348008.
- 67.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02133742.
- 68.McDermott DF, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol. 2015;33:2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lipson EJ, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–468. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiou VL, Burotto M. Pseudoprogression and immune-related response in solid tumors. J Clin Oncol. 2015;33:3541–3543. doi: 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolchok JD, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2015;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 72.Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092–2099. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naidoo J, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26:2375–2391. doi: 10.1093/annonc/mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson DB, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol. 2016;2:234–240. doi: 10.1001/jamaoncol.2015.4368. [DOI] [PubMed] [Google Scholar]
- 75.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rizvi NA, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choueiri TK, Fishman M, Escudier B, Kim JJ, Kluger HM. Immunomodulatory activity of nivolumab in previously treated and untreated metastatic renal cell carcinoma (mRCC): biomarker-based results from a randomized clinical trial [abstract] J Clin Oncol. 2014;32(5s (suppl.)):5012. [Google Scholar]
- 79.Choueiri TK, Fishman M, Escudier B, McDermott DF, Kluger HM. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma (mRCC): association of biomarkers with clinical outcomes [abstract] J Clin Oncol. 2015;33(Suppl):4500. doi: 10.1158/1078-0432.CCR-15-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thompson RH, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA. 2004;101:17174–17179. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thompson RH, et al. Costimulatory molecule B7-H1 in primary and metastatic clear cell renal cell carcinoma. Cancer. 2005;104:2084–2091. doi: 10.1002/cncr.21470. [DOI] [PubMed] [Google Scholar]
- 82.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choueiri TK, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol. 2014;25:2178–2184. doi: 10.1093/annonc/mdu445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geynisman DM. Anti-programmed cell death protein 1 (PD-1) antibody nivolumab leads to a dramatic and rapid response in papillary renal cell carcinoma with sarcomatoid and rhabdoid features. Eur Urol. 2015;68:912–914. doi: 10.1016/j.eururo.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Melero I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 86.Pal SK, Hu A, Figlin RA. A new age for vaccine therapy in renal cell carcinoma. Cancer J. 2013;19:365–370. doi: 10.1097/PPO.0b013e31829d74b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oudard S, et al. A phase II study of the cancer vaccine TG4010 alone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: clinical and immunological findings. Cancer Immunol Immunother. 2011;60:261–271. doi: 10.1007/s00262-010-0935-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Southall PJ, et al. Immunohistological distribution of 5T4 antigen in normal and malignant tissues. Br J Cancer. 1990;61:89–95. doi: 10.1038/bjc.1990.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Amato RJ, et al. Vaccination of metastatic renal cancer patients with MVA-5T4: a randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16:5539–5547. doi: 10.1158/1078-0432.CCR-10-2082. [DOI] [PubMed] [Google Scholar]
- 90.Figlin RA. Personalized immunotherapy (AGS-003) when combined with sunitinib for the treatment of metastatic renal cell carcinoma. Expert Opin Biol Ther. 2015;15:1241–1248. doi: 10.1517/14712598.2015.1063610. [DOI] [PubMed] [Google Scholar]
- 91.Amin A, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): phase 2 study results. J Immunother Cancer. 2015;3:14. doi: 10.1186/s40425-015-0055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT01582672.
- 93.US National Library of Medicine. ClinicalTrials.gov. 2015 http://clinicaltrials.gov/show/NCT01265901.
- 94.Walter S, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 95.Rini B, et al. Results from an open-label, randomized, controlled Phase 3 study investigating IMA901 multipeptide cancer vaccine in patients receiving sunitnib as first-line therapy for advanced/metastatic RCC [abstract] Ann Oncol. 2015;26(Suppl.):17LBA. [Google Scholar]
- 96.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT01441765.
- 97.Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer J. 2014;20:151–155. doi: 10.1097/PPO.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bui MHT, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res. 2003;9:802–811. [PubMed] [Google Scholar]
- 99.Lamers CHJ, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 100.Lamers CH, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Steffens MG, et al. Targeting of renal cell carcinoma with iodine-131-labeled chimeric monoclonal antibody G250. J Clin Oncol. 1997;15:1529–1537. doi: 10.1200/JCO.1997.15.4.1529. [DOI] [PubMed] [Google Scholar]
- 102.Kim JS, et al. Preclinical and clinical studies on cytokine-induced killer cells for the treatment of renal cell carcinoma. Arch Pharm Res. 2014;37:559–566. doi: 10.1007/s12272-014-0381-x. [DOI] [PubMed] [Google Scholar]
- 103.Liu L, et al. Randomized study of autologous cytokine-induced killer cell immunotherapy in metastatic renal carcinoma. Clin Cancer Res. 2012;18:1751–1759. doi: 10.1158/1078-0432.CCR-11-2442. [DOI] [PubMed] [Google Scholar]
- 104.Wang D, et al. Clinical research of genetically modified dendritic cells in combination with cytokine-induced killer cell treatment in advanced renal cancer. BMC Cancer. 2014;14:251. doi: 10.1186/1471-2407-14-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Westwood JA, et al. Three agonist antibodies in combination with high-dose IL-2 eradicate orthotopic kidney cancer in mice. J Transl Med. 2010;8:42. doi: 10.1186/1479-5876-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02179918.
- 107.Infante J, Burris Ha, Ansell SM, Nemunaitis J, Weiss G. Immunologic activity of an activating anti-CD27 antibody (CDX-1127) in patients (pts) with solid tumors [abstract] J Clin Oncol. 2014;32(5s Suppl):3027. [Google Scholar]
- 108.US National Library of Medicine. ClinicalTrials.gov. 2016 http://clinicaltrials.gov/show/NCT02386111.
- 109.Grünwald V, et al. A phase I study of recombinant human interleukin-21 (rIL-21) in combination with sunitinib in patients with metastatic renal cell carcinoma (RCC) Acta Oncol. 2011;50:121–126. doi: 10.3109/0284186X.2010.509104. [DOI] [PubMed] [Google Scholar]
- 110.Bhatia S, et al. Recombinant interleukin-21 plus sorafenib for metastatic renal cell carcinoma: a phase 1/2 study. J Immunother Cancer. 2014;27:2. doi: 10.1186/2051-1426-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thompson JA, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol. 2008;26:2034–2039. doi: 10.1200/JCO.2007.14.5193. [DOI] [PubMed] [Google Scholar]
- 112.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02318394.
- 113.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02132754.
- 114.US National Library of Medicine. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/ct2/show/NCT02628574.
- 115.Sanmamed MF, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2016;42:640–655. doi: 10.1053/j.seminoncol.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 116.Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol. 2009;157:9–19. doi: 10.1111/j.1365-2249.2009.03912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.US National Library of Medicine. ClinicalTrials.gov. 2015 https://www.clinicaltrials.gov/ct2/show/NCT02423954.
- 118.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02298959.
- 119.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02493751.