

Abstract
Background
A model-based analysis of oligonucleotide expression arrays we developed previously uses a probe-sensitivity index to capture the response characteristic of a specific probe pair and calculates model-based expression indexes (MBEI). MBEI has standard error attached to it as a measure of accuracy. Here we investigate the stability of the probe-sensitivity index across different tissue types, the reproducibility of results in replicate experiments, and the use of MBEI in perfect match (PM)-only arrays.
Results
Probe-sensitivity indexes are stable across tissue types. The target gene's presence in many arrays of an array set allows the probe-sensitivity index to be estimated accurately. We extended the model to obtain expression values for PM-only arrays, and found that the 20-probe PM-only model is comparable to the 10-probe PM/MM difference model, in terms of the expression correlations with the original 20-probe PM/MM difference model. MBEI method is able to extend the reliable detection limit of expression to a lower mRNA concentration. The standard errors of MBEI can be used to construct confidence intervals of fold changes, and the lower confidence bound of fold change is a better ranking statistic for filtering genes. We can assign reliability indexes for genes in a specific cluster of interest in hierarchical clustering by resampling clustering trees. A software dChip implementing many of these analysis methods is made available.
Conclusions
The model-based approach reduces the variability of low expression estimates, and provides a natural method of calculating expression values for PM-only arrays. The standard errors attached to expression values can be used to assess the reliability of downstream analysis.
Background
The statistical model proposed in [1] for one probe set in multiple oligonucleotide arrays has the form
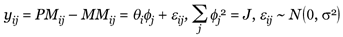 (1)
(1)
It states that the perfect match (PM)/mismatch (MM) difference in array i, probe j of this probe set is the product of model-based expression index (MBEI) in array i (θi) and probe-sensitivity index of probe j (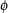 j) plus random error. Here J is the number of probe pairs in the probe set. Fitting the model, we can identify cross-hybridizing probes (j with large standard error (SE), which are excluded during iterative fitting) and arrays with image contamination at this probe set (θi with large SE), as well as single outliers (image spikes) which are replaced by the fitted values. In effect the estimated expression index θi is a weighted average of PM/MM differences:
j) plus random error. Here J is the number of probe pairs in the probe set. Fitting the model, we can identify cross-hybridizing probes (j with large standard error (SE), which are excluded during iterative fitting) and arrays with image contamination at this probe set (θi with large SE), as well as single outliers (image spikes) which are replaced by the fitted values. In effect the estimated expression index θi is a weighted average of PM/MM differences:
![]()
with larger weights given to probes with larger . The image of outliers (array and single outliers) identified through model-fitting can be used to assess the quality of an experiment and to identify unexpected problems such as a misaligned corner of a DAT file [1].
We have investigated several important properties of the model, including the reliability and stability of the fitted parameters MBEI (θ) and probe sensitivity indexes (), the performance of MBEI compared to the commonly used average difference (AD), and how the availability of SE facilitates downstream comparative and clustering analysis.
Results and discussion
Probe-sensitivity indexes are stable across tissue types
In practice, in an array experiment, a researcher hybridizes tissue or cell line samples, corresponding to different treatments or conditions, to a batch of arrays. Ideally, the probe-sensitivity index () should be independent of the tissue type. This condition, however, may not hold for those probes that have cross-hybridization affinity to non-target genes. Nevertheless, assuming that a non-target gene cross-hybridizes only to a few probes of a probe set, and its expression levels across arrays do not correlate with the target gene, the iterative probe-excluding procedure in [1] may be able to exclude cross-hybridizing probes, regardless of the tissue type hybridized. In addition, the relative probe-sensitivity indexes of the good probes called by the model are likely to be similar across sets of arrays hybridizing to different tissue samples.
The stability of the probe-sensitivity index is studied using 226 HU6800 arrays. We apply the model (equation 1) independently to six sets of HU6800 arrays (21 leukemia, lymphoma and mantle cell samples, 20 prostate cancer cell lines, 17 brain tumor samples, 55 cancer cell lines, 58 brain samples, and 55 lung tumor samples). Figure 1a shows the values fitted for probe set 6457 (used in Figure 1 and 2 of [1]) in the six array sets. The patterns resemble each other greatly, showing that the probe-sensitivity index is an inherent property of these non-cross-hybridizing probes and can be consistently identified from different sets of arrays. Figure 1b shows the patterns for another probe set. It is noteworthy that the probe 11 in array set 5 is likely to be cross-hybridizing, making its relative strength (here MM is consistently larger than PM and this leads to a negative ) dissimilar to the probe 11 in other array sets. The model identifies this probe as a 'probe-outlier' only for array set 5 and excludes it when calculating MBEI (0) for array set 5.
Figure 1.
values for probe sets. values estimated for probe sets (a) 6457, (b) 1248, and (c) 6571 in six array sets (shown in panels 1–6 from left to right for each probe set). values (constrained to have sum square equal to number of probes used in each array set) are on the y-axis, and probe pairs are labeled 1 to 20 on the x-axis. The title of each panel (for example, p = 0) indicates the proportion of arrays 'present' for the target gene in the array set. Large circles represent identified probe-outliers by negativity or large SE of .
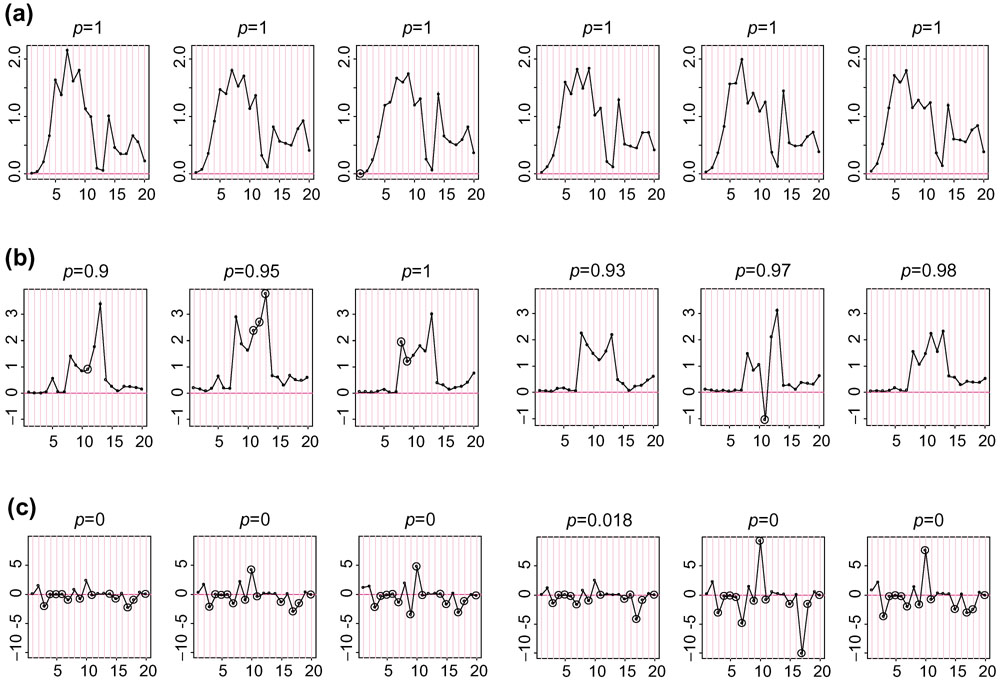
Figure 2.
Boxplots of average pairwise correlations of s between two array sets. They are stratified by average lower presence proportion in two array sets (the presence proportion of a probe set is the proportion of arrays in an array set where the target gene is called 'present' by GeneChip's algorithm). The average is taken over C(6, 2) = 15 pairwise comparison of two array sets for each probe set, and the correlation is calculated using probes that are not identified as an outlier in both array sets. The range of the average lower presence proportion for the six boxplots are: (0, 0.17), (0.17, 0.34), (0.34, 0.51), (0.51, 0.68), (0.68, 0.85), (0.85, 1). The title of each boxplot is the number of probe sets classified into this boxplot. Eleven probe sets with too few non-outlier probes to calculate correlations for all 15 comparisons are not included in the boxplots. The average lower presence proportion and average pairwise correlation for probe sets in Figure 1 are (a) 1, 0.95; (b), 0.93, 0.94; and (c) 0, 0.86.
In Figure 1a,b the target gene is present in most samples of all array sets. For a probe set whose target gene is mostly absent throughout samples (Figure 1c), many probes are identified as probe-outliers because of their negative indexes. Here, we cannot obtain correct probe-sensitivity indexes because of the absence of the target gene. Nevertheless, the PM-MM values for these probes are random fluctuations around zero, leading to a correct expression index close to zero. If the target gene becomes available for a future array set, the correct probe-sensitivity indexes will be recovered and these probes will be used for expression calculation.
Occasionally, a responsive probe set may give rise to very different estimates in two array sets. In Figure 1b, probes 8 and 13 have different relative responses in array set 1 and 4, leading to different probe-response patterns. This might be due to the possibility that the probes in this probe set are differentially cross-hybridized in different array sets, or that the same probe in different batches of arrays may systematically behave differently. Identification and flagging such probe sets is desirable and essential if we want to compare arrays hybridized to different tissue samples.
Figure 2 shows the boxplots of average pairwise correlations of values between two array sets, stratified by average lower presence proportion in the two sets. In general, when a gene is present in many samples of two array sets, the patterns estimated from the two sets are very similar. The target gene's presence in many arrays of an array set allows the probe-sensitivity index to be estimated accurately.
Model-based analysis for PM-only arrays
From Figure 1 of [1], one can see that some MM probes may respond poorly to the changes in the expression level of the target gene. This phenomenon raised questions on the efficiency of using MM probes, and led some investigators to design custom arrays that use PM probes exclusively (R. Abagyan and Yingyao Zhou, personal communication; B.R. Conklin, personal communication), and others to calculate fold changes using only PM probes (F. Naef, personal communication). This design greatly increases the number of genes that can be studied on one array. To investigate the relative performance of PM/MM versus PM-only designs, we exploited the model to estimate gene expression levels using only PM probes, and compared it to the MBEI using both PM and MM probes.
The full intensity model (equation 1 of [1]) specifies the relationship of PM probe responses and expression level θ:
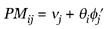 (2)
(2)
where vj is the baseline response of probe pair j due to nonspecific hybridization, and 'j is the sensitivity of PM probe of the probe pair j. The parameter estimates can be obtained by iteratively fitting θi and vj,'j, regarding the other set as known. The same outlier exclusion procedure in [1] is applied. The MM probe responses have a similar form as equation 2 except for different probe-sensitivity indexes. We fit a PM-only and an MM-only model to obtain expression values of all 20-probe probe sets using array set 1. For comparison, we also used half of the probe pairs (by alternatively picking one out of every two probes) in a 20-probe probe set to fit to the difference model (equation 1). For each probe set, these three sets of expression values were compared with the expression values of the original difference model using 20 probes, in terms of correlation of θs obtained by two methods across the 21 arrays. We assumed the 20-probe difference model provides the most accurate expression estimates. If, for a probe set, a simplified model (PM-only, MM-only or 10-probe difference model) performs reasonably well, we expect its θ estimates to correlate with that from the 20-probe difference model.
Figure 3 shows the histogram and Figure 4 the boxplot of correlations of θs estimated from the 20-probe difference model and θs estimated from the 10-probe difference model (a), the 20-probe PM-only model (b) and the 20-probe MM-only model (c). For probe sets with high presence proportion, both the 10-probe difference model and the PM-only model correlate well with the 20-probe difference model. The MM-only model yields noticeably lower correlations, however. We note that this comparison is intrinsically biased in favor of the 10-probe difference model because the 'truth' is constructed from PM-MM differences.
Figure 3.
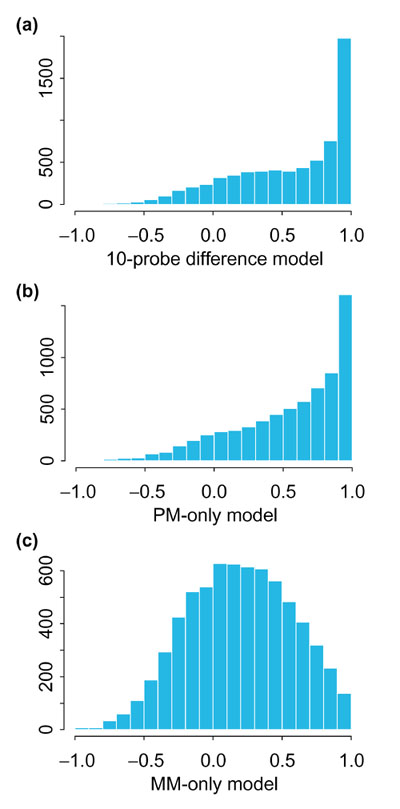
Histogram of correlations between model-based expression values estimated using the 20-probe difference model and those estimated using different models. (a) 10-probe difference model; (b) 20-probe PM-only model; (c) 20-probe MM-only model. All comparisons are across the 21 arrays in array set 1.
Figure 4.
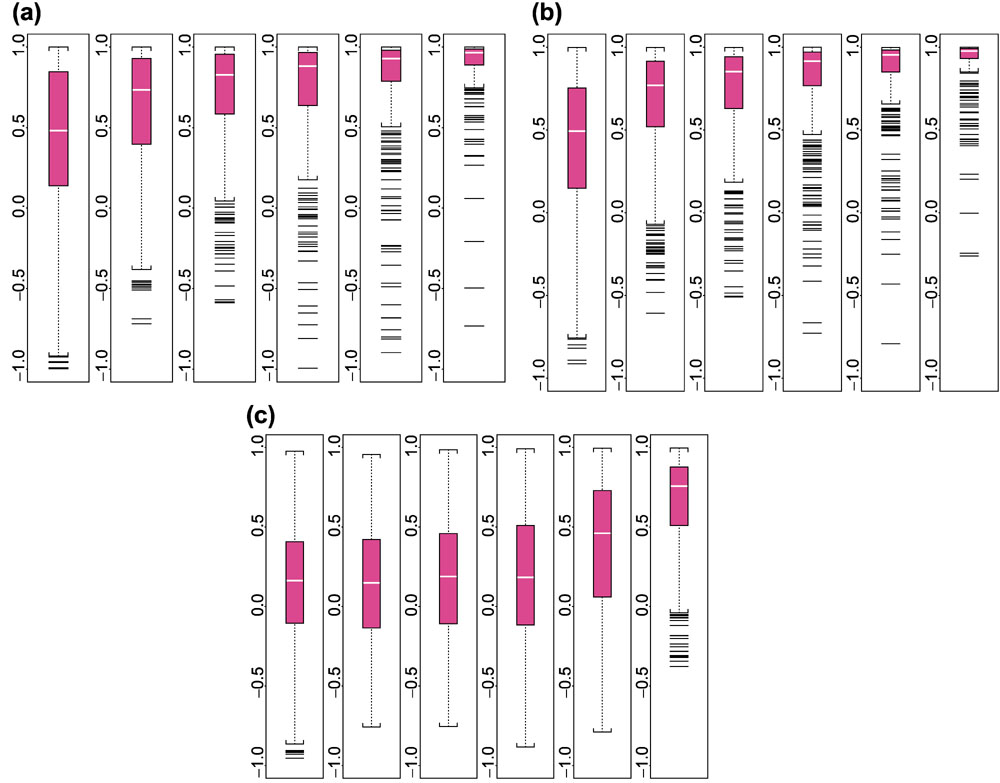
Boxplot of correlations between θ values estimated using the 20-probe difference model and θs estimated using different models, stratified by presence proportion. (a) 10-probe difference model; (b) 20-probe PM-only model; and (c) 20-probe MM-only model. The number of presence calls for a probe set in the 21 arrays and the subpopulation size for the six boxplots are: 0–3, 4,385; 4–7, 693; 8–11, 413; 12–15, 488; 16–19, 497; and 20–21, 323. Only 6,799 probe sets that have 20 probes are used.
This comparison corroborates the basic notion of the technology: the PM probes hybridize more strongly to the target signals than MM probes and contain most of the information. We stress that, whereas the above analysis illustrates the applicability of model-based analysis to PM-only arrays, the assessment presented here is only tentative because of the limited information provided by the HU6800 arrays on the comparisons. Definitive comparisons of the efficiency of the designs must await the availability of data from PM-only arrays.
MBEI reduces variability for low expression estimates
The array set 5 has 29 pairs of arrays [2]. Each pair consists of two arrays hybridizing to samples replicated at total mRNA level (the total mRNA sample is split and then amplified and labeled separately, and hybridized to two different arrays). The differences between the expression values of the two replicate arrays in a pair are due to the variation introduced in experimental steps after the split, the array manufacturing difference and analytical methods such as normalization and expression calculation. This difference provides a lower bound of biological variation that can be detected between two independently amplified samples, and serves as a good statistic for comparing different analytical methods.
The agreement of MBEI between two replicate arrays is shown in Figure 5a. For comparison, we also used the method in [3] to calculate ADs for all probe sets and plot them in Figure 5b (AD is based on normalized probe values, see Methods and materials section for the normalization method. Also note that GeneChip software excludes probes whose PM/MM difference is outside three standard deviations (SDs) of all probe differences in either of the two arrays in the comparison; here, as we are comparing multiple arrays at the same time, when calculating ADs a probe is excluded if its difference is an outlier in the above sense in any of the arrays, until a minimum of five probes is reached, where all five probes will be used). Both the MBEI and the AD method yielded some expression values differing by more than a factor of two, especially for genes at low expression level. This might be explained by the relatively larger amplification variation for weakly expressed genes, given a constant success rate of amplifying a sequence by a certain fold.
Figure 5.
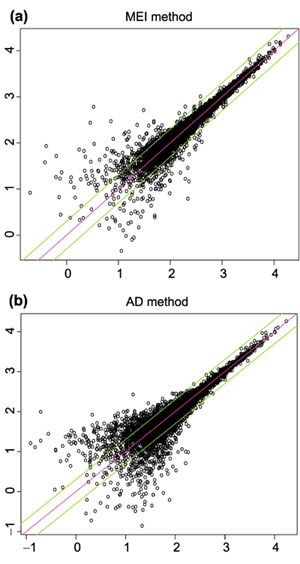
Log (base 10) expression indexes of a pair of replicate arrays (array 1 and 2 of array set 5) for different statistical methods. (a) MBEI method; (b) AD method. Only 6,695 (a) and 4,696 (b) probe sets with positive values in both arrays are used. The center line is y = x, and the flanking lines indicate the difference of a factor of two.
Researchers often use 'log ratio' between expression values of a gene in two arrays as the criterion for identifying differentially expressed genes. Between duplicate arrays, we expect these log ratios of expression values based on a good expression index (AD or MBEI) to be close to zero. Thus for every probe set we calculated its average absolute log (base 10) ratio of 29 pairs of duplicates as a statistic to compare the variation in expression levels between duplicates using the AD or the MBEI method. Figure 6 presents the results of the comparison. The average absolute log ratio distribution of the MBEI method is significantly lower than that of the AD method when expression level is low (and thus probe sets have a low proportion of detections of the target gene across arrays). As expression level becomes higher (when the target gene of a probe set is detected in more arrays), the AD method shows a rapid improvement in performance, approaching the level of the MBEI method. The same boxplots (Figure 7) for another set of 60 human U95A arrays consisting of 30 replicate pairs conveys similar information. These results suggest that the MBEI method is able to extend the reliable detection limit of expression to a lower mRNA concentration.
Figure 6.
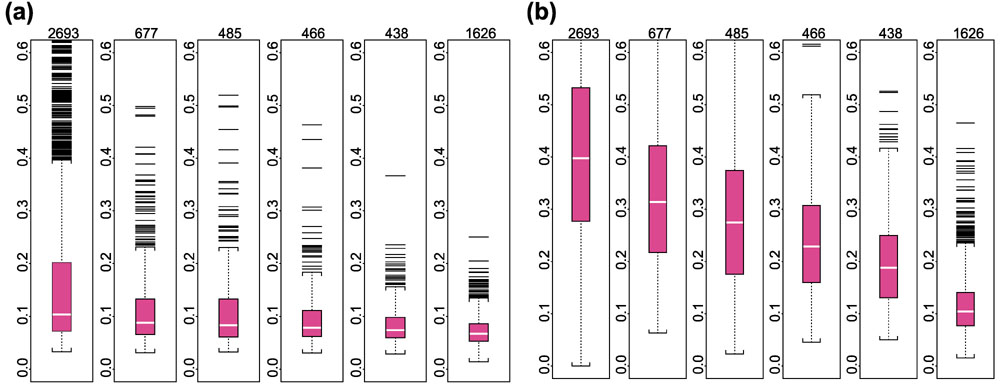
Boxplots of average absolute log (base 10) ratios between replicate arrays stratified by presence proportion for different statistical methods. (a) MBEI method; (b) AD method. The number of presence calls for a probe set in the 58 arrays for the six boxplots are: 0–9, 10–19, 20–29, 30–39, 40–49, 50–58. The title of each boxplot is the number of probe sets used for the boxplot. The average is taken over 29 replicate pairs. Log ratios are not calculated for negative expression values or expression values identified as 'array-outliers' by the MBEI method in either array of a replicate pair, and are not used to calculate the average. 744 probe sets are not included as their average absolute log ratios cannot be calculated for all the 29 pairs using either method.
Figure 7.
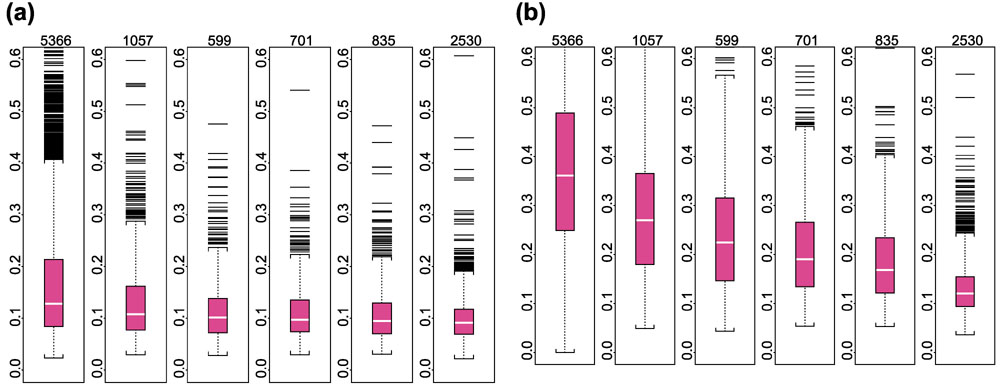
Similar plots as in Figure 6 for another set of 30 pairs of duplicated human U95A arrays. (a) MBEI method; (b) AD method.The number of presence calls for a probe set in the 60 arrays for the six boxplots are: 0–9, 10–19, 20–29, 30–39, 40–49, 50–60. The title of each boxplot is the number of probe sets used for the boxplot.
Confidence interval for fold change
After obtaining expression indexes using AD or MBEI, fold changes can be calculated between two arrays for every gene and used to identify differentially expressed genes. Usually, low or negative expressions are truncated to a small number before calculating fold changes, and GeneChip also cautions against using fold changes when the baseline expression is absent.
The availability of SEs for the model-based expression indexes allows us to obtain confidence intervals for fold changes. Suppose
![]()
where θ1 and θ2 are the real expression levels in the sample, and 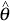 1 and 2 are the model-based estimates of expression levels. We substitute the model-based SEs for δ1 and δ2 Letting r = θ1/θ2 be the real fold change, then inference on r can be based on the quantity
1 and 2 are the model-based estimates of expression levels. We substitute the model-based SEs for δ1 and δ2 Letting r = θ1/θ2 be the real fold change, then inference on r can be based on the quantity
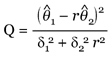
It can be shown that Q has a Χ2 distribution with 1 degree of freedom irrespective of the values of θ1 and θ2 [4]. Thus Q is a pivotal quantity involving r. We can use Q to construct fixed-level tests and to invert them to obtain confidence intervals (CI) for fold changes [5].
Table 1 presents the estimated expression indexes (with SEs) in two arrays and the 90% confidence intervals of the fold changes for 14 genes. Although all genes have similar estimated fold changes, the confidence intervals are very different. For example, gene 1 has fold change 2.47 and a tight confidence interval (2.06, 3.02). In contrast, gene 11 has a similar fold change of 2.48 but a much wider confidence interval (0.96, 18.18). Thus the fold change around 2.5 for gene 11 is not as trustworthy as that for gene 1. Further examination reveals that this is due to the large SEs relative to the expression indexes for gene 11. This agrees with the intuition that when one or both expression levels are close to zero for one gene, the fold change cannot be estimated with much accuracy. In addition, when image contamination results in unreliable expression values with large SEs, the fold changes calculated using these expression value are attached with wide CIs. In this manner, the measurement accuracy of expression values propagates to the estimation of fold changes.
Table 1.
Using expression levels and associated SEs to determine confidence intervals of fold changes
| Expression 1 | Std Error 1 | Expression 2 | Std Error 2 | Fold Change | Lower CB | Upper CB | |
| Gene 1 | 859.635 | 41.7808 | 347.57 | 36.0887 | 2.47327 | 2.06844 | 3.02672 |
| Gene 2 | 405.72 | 31.2305 | 164.014 | 44.2505 | 2.47369 | 1.66938 | 4.49127 |
| Gene 3 | 283.931 | 28.5281 | 114.705 | 18.4661 | 2.47531 | 1.83926 | 3.48466 |
| Gene 4 | 45.9821 | 64.2419 | 18.5727 | 84.5308 | 2.47579 | 0 | Infinity |
| Gene 5 | 225.178 | 57.489 | 90.9045 | 36.1766 | 2.47709 | 1.18104 | 7.48749 |
| Gene 6 | 247.002 | 50.6518 | 99.6642 | 19.5384 | 2.47834 | 1.51079 | 4.0211 |
| Gene 7 | 49.9739 | 21.5345 | 20.1514 | 23.5651 | 2.47992 | 0.487603 | Infinity |
| Gene 8 | 276.491 | 18.6883 | 111.373 | 36.1004 | 2.48256 | 1.59069 | 5.34635 |
| Gene 9 | 436.071 | 32.9779 | 175.384 | 21.0669 | 2.48638 | 1.98665 | 3.18811 |
| Gene 10 | 75.6914 | 17.7215 | 30.4395 | 17.9707 | 2.48662 | 1.07209 | 86.1656 |
| Gene 11 | 80.673 | 25.3085 | 32.4314 | 16.9626 | 2.4875 | 0.960787 | 18.1833 |
| Gene 12 | 181.528 | 42.4837 | 72.8751 | 28.1787 | 2.49094 | 1.24668 | 7.11945 |
| Gene 13 | 1122.28 | 99.2835 | 449.889 | 63.2821 | 2.49456 | 1.92075 | 3.35055 |
| Gene 14 | 168.234 | 40.629 | 67.4387 | 30.2982 | 2.49462 | 1.17639 | 9.81547 |
In practice, we find it useful to sort genes by the lower confidence bound ('Lower CB' in Table 1), which is a conservative estimate of the fold change. When an expression index is negative (as a result of taking PM/MM differences), we do not calculate the confidence intervals. In such a case, it is more helpful to filter genes by presence calls.
Standard errors help to assess clustering results
Cluster analysis is a popular method for analyzing the data of a series of microarrays [6, 7]. If two genes are co-regulated at the transcription level, their expression values across samples are likely to be correlated. Clustering algorithms use these correlations (or monotone transformation of correlations) to cluster co-regulated genes together. The correlation based on the estimated expression levels may, however, be different from that based on the real but unobserved expression levels. Also, the commonly used hierarchical clustering algorithm is an irreversible process: once two genes or nodes are merged, they will stay together, even if later on there is good reason to adjust previous clustering. Thus there is a need to assess the reliability of clusters.
A global way of using SE in hierarchical clustering is to resample or bootstrap [8] the whole 'gene by sample' data matrix and redo the clustering, then investigate the overall properties emerging from this repertoire of clustering trees. In Bittner et al. [9], the data matrix coming from cDNA microarray experiments is resampled using the estimated variation derived from the median SD of log ratios for a gene across samples. As we now have SEs for all data points, we can resample each expression value from a normal distribution with mean equal to the estimated expression value and SD equal to the attached SE.
Figure 8a shows a hierarchical clustering tree of 225 selected genes with presence proportion > 0.5 and coefficient of variation (SD/mean) > 0.7 across the 20 samples in array set 2. In trying to interpret this tree, we may be interested in the gene cluster colored in blue and the reliability of the gene members belonging to this cluster. The whole data matrix is resampled, and the clustering is performed again (Figure 8b). We notice that some blue genes (genes in the original cluster are colored blue) are clustered with other non-blue genes, and some non-blue genes are mixed into the main body of the blue genes. After each resampling, we identify a cluster that contains more than 80% of all the blue genes, but as few non-blue genes as possible (measured as a percentage of all genes in this cluster). This cluster is considered to be the cluster that corresponds to the original one in Figure 8a. In Figure 8b the root node of the 'corresponding cluster' is marked with small horizontal line intersecting the vertical line (representing the range of the cluster) on the right of the clustering picture. Then, for each of all the 225 genes, if it belongs to this 'corresponding cluster', we increase its 'in-cluster' count by 1. After resampling 30 times, the in-cluster counts are indicated in gray-scale on the left side of the original clustering (Figure 8c), with black representing 30 and white representing zero. A high 'in-cluster' count indicates a gene 'remains' in the original cluster in most of the resampled clustering trees.
Figure 8.
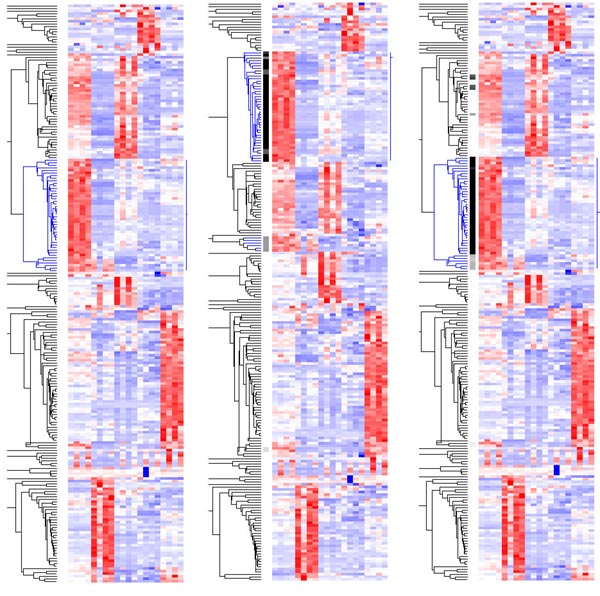
Gene clustering (a) 225 filtered genes are clustered based on their expression profiles across 20 samples. Each gene's expression values are standardized to have mean 0 and SD 1 across 20 samples. Dark blue represents low expression level and dark red high expression level. We might be particularly interested in the cluster colored in blue. (b) The clustering tree after a particular resampling. Although the original 'blue' genes are scattered to various places, we can still determine where the original cluster is, using the criteria described in the text. (c) After resampling 30 times, the reliability of the genes belonging to the original cluster is indicated by the vertical gray-scale bar on the left of the blue-red picture.
We can see from Figure 8c that most genes in the original cluster are reliable members, whereas a few genes at the bottom of the cluster are not (in fact they are merged into the original cluster last). Interestingly, some genes originally not in the original cluster group with the 'corresponding clusters' during resampling many times and have gray 'in-cluster' marks. These genes may be related to the original cluster in some way. In summary, this method can help us to distinguish reliable and unreliable gene members of a cluster, as well as draw our attention to related genes originally clustered somewhere else because of the accidental nature of hierarchical clustering.
Methods and materials
Software
We have developed a software package DNA-Chip Analyzer (dChip [10]) to perform invariant-set normalization (see below), calculation of MBEI [1], computation of confidence intervals of fold changes, and hierarchical clustering with resampling.
Our experience is that more than 10 arrays are appropriate for model training, outlier detection and MBEI calculation. Researchers with fewer than 10 arrays may seek arrays of the same chip type and hybridizing to similar tissue samples, and combine them in a single dChip analysis session. We are exploring model-based meta-analysis of many arrays of the same chip type but hybridizing to a heterogeneous set of tissues samples, and will present such analysis in future work.
Normalization of arrays based on an 'invariant set'
As array images usually have different overall image brightness (Figure 9a), especially when they are generated at different times and places, proper normalization is required before comparing the expression levels of genes between arrays. Model-based expression computation requires normalized probe-level data (from Affymetrix's DAT or CEL files). For a group of arrays, we normalize all arrays (except the baseline array) to a common baseline array having the median overall brightness (as measured by the median CEL intensity in an array).
Figure 9.
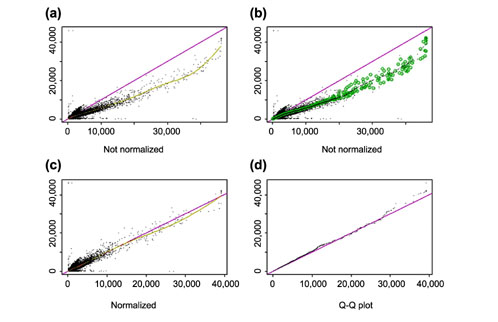
Normalization of gene expression levels between arrays. (a) The CEL intensities (see text) of a pair of replicate arrays (array 11 and 12 in array set 5) are plotted against each other. The baseline array 11 (shown on the y-axis) is not as bright as array 12 (shown on the x-axis). The smoothing spline (green curve) deviates from the diagonal line y = x (blue curve), indicating the need for normalization. (b) The same plot as (a) with superimposed circles representing the invariant set, on the basis of which a piecewise linear normalization relationship is determined (black dotted line, whose y-coordinate is the normalized value of array 12). The normalization curve is close to the smoothing spline curve in (a) as the two arrays are replicated arrays and all probes should be invariant. (c) After normalization (y-axis is the baseline array 11, and x-axis the normalized value of array 12), the scatterplot centers around the diagonal line and the array 12 is adjusted to have the similar overall brightness as array 11. The smoothing spline curve is also close to the diagonal line. (d) The Q-Q plot of probe intensities of array 11 and normalized array 12 shows the probes in the two sets have almost the same distribution.
A normalization relation can be understood as a curve in the scatterplot of two arrays with the baseline array drawn on the y-axis and the array to be normalized on the x-axis. A straight line running through the origin is a multiplicative normalization method (GeneChip's scaling method), and a smoothing spline through the scatterplot can also be used (Figure 9a, also see [11]).
We should base the normalization only on probe values that belong to non-differentially expressed genes, but generally we do not know which genes are non-differentially expressed (control or housekeeping genes may also be variable across arrays). Nevertheless, we expect that a probe of a non-differentially expressed gene in two arrays to have similar intensity ranks (ranks are calculated in two arrays separately). We use an iterative procedure to identify a set of probes (called the invariant set), which presumably consists of points from non-differentially expressed genes (Figure 9b). Specifically, we start with points of all PM probes (about 140,000 for HU6800 array). If a point's proportion rank difference (PRD, absolute rank difference in two arrays divided by n = 140,000) is small enough, it is kept for the new set. Here the threshold of being small is PRD < 0.003 when a points's average intensity ranks in the two arrays is small and PRD < 0.007 when it is large, accounting for fewer points at high-intensity range; and the threshold is interpolated in between. We chose these parameters empirically to make the selected points in the invariant set thin enough to naturally determine a normalization relation. In this way we may obtain a new set of 10,000 points, and the same procedure is applied to the new set iteratively, until the number of points in the new set does not decrease anymore. A piecewise linear running median line is then calculated and used as the normalization curve. After normalization, the two arrays have similar overall brightness. (Figure 9c). Figure 10 shows another pair of arrays where the normalization relationship is non-linear.
Figure 10.
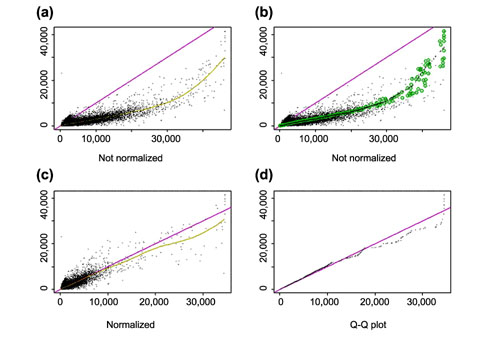
Similar plots as in Figure 9 for arrays hybridized to two different samples (array 24 and 36 of array set 5). (a) CEL intensities; (b) same plot as in (a) with superimposed circles representing the invariant set; (c) after renormalization; (d) Q-Q plot of normalized probe intensities. Note that the smoothing spline in (a) is affected by several points at the lower-right corner, which might belong to differentially expressed genes. The invariant set, on the other hand, does not include these points when determining the normalization curve, leading to a different normalization relationship at the high end.
Acknowledgments
Acknowledgements
We thank Sven de Vos, Dan Tang, Nik Brown, Stan Nelson, Jae K. Lee, Yaron Hakak, John Walker and Arindam Bhattacharjee for providing data, and the editor and referees who provided valuable suggestions. This work is supported in part by NIH grant 1 RO1 HG02341-01 and NSF grant DBI-9904701.
References
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodicka L, Dong H, Mittmann M, Ho M, Lockhart D. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- Wallace D. The Behrens-Fisher and Fieller-Creasy problems. In Lecture Notes in Statistics 1, RAFisher: An Appreciation Edited by Fienberg SE, Hinkley DV Springer-Verlag. 1988. pp. 119–147.
- Cox DR, Hinkley DV. Theoretical Statistics London: Chapman and Hall, 1974.
- Eisen M, Spellman P, Brown P, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E, Golub T. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. An Introduction to the Bootstrap New York: Chapman & Hall/CRC, 1993.
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Rad-macher M, Simon R, Yakhinik Z, Ben-Dork A, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- DNA-Chip Analyzer http://www.dchip.org
- Schadt EE, Li C, Su C, Wong WH. Analyzing high-density oligonucleotide gene expression array data. J Cell Biochem. 2001;80:192–202. [PubMed] [Google Scholar]