

Abstract
Ewing sarcoma/peripheral primitive neuroectodermal tumor (ES/pPNET) is a malignant small, round cell tumor arising from bone and soft tissue in children and young adults. It can occur at osseous and extraosseous sites. Its usual locations are diaphysis of long bones followed by pelvis, ribs, vertebrae, and rarely skull. We reviewed the literature and PubMed advanced search on ES/pPNET occurring at extraosseous sites, mainly involving the central nervous system (CNS). We reported a case of a 22-year-old male presenting with seizure finally diagnosed as a case of ES/pPNET. The challenges in management of this rare CNS tumor and its differential diagnosis are highlighted. We found that most cases of ES involving CNS represent secondary metastases from extracranial sites of ES/pPNET and there are rare case reports of primary intracranial ES-pPNET. Furthermore, among these intracranial tumors, most common tumors occupy an intraaxial location and only a handful of cases of dural-based or extraaxial tumors mimicking meningioma are reported. Differentiation of pPNET from central PNET (cPNET) is important as it has definitive therapeutic and prognostic implications. Awareness of this entity of ES/pPNET, its rare dural presentation, and differentiation from the more common cPNET is needed for appropriate patient management. Meningeal ES/pPNET has to be kept in mind in the differential diagnosis of meningeal tumors eroding bone.
Keywords: Central nervous system Ewing sarcoma/peripheral primitive neuroectodermal tumor, dural tumor, Ewing's sarcoma, intracranial primitive neuroectodermal tumor, meningioma, peripheral primitive neuroectodermal tumor, primary
Introduction
Ewing sarcoma/peripheral primitive neuroectodermal tumor (ES/pPNET) is a malignant small, round cell tumor arising from bone and soft tissue in children and young adults. It can occur at osseous and extraosseous sites.[1] Its usual locations are diaphysis of long bones followed by pelvis, ribs, vertebrae, and rarely skull. Intracranial ES/pPNET is usually metastases from extracranial sites of ES/pPNET. When primary ES/pPNET affects central nervous system (CNS), which is rare, it is usually intraparenchymal located supratentorially or in the spinal cord.[2] An extraaxial/dural presentation of ES/pPNET mimicking meningioma is extremely rare. These tumors pose a diagnostic challenge to neurosurgeons. Intraparenchymal cerebral tumors may be misdiagnosed as central PNET (cPNET) or other primary CNS tumors and the dural tumors very closely mimic meningioma, clinically and radiologically.[1,3] An early and accurate diagnosis of ES/pPNET is required so that multimodality treatment approach can be adopted as early as possible to prevent distant metastases which occurs quite early in this tumor. As the tumor presents in young age, long-term survival remains a challenge in management of ES/pPNET patients. We report a case of intracranial ES/pPNET presenting as meningioma in a 22-year-old male with complains of seizure and unconsciousness, an unusual presentation.
Case Report
A 22-year-old male presented with a single episode of generalized tonic-clonic seizure followed by unconsciousness. On examination, Glasgow coma score was E1 V1 M5. Papilledema was present with right-sided hemiparesis. Patient had no previous history of headache, vomiting, unconsciousness, and focal neurological deficit. There was no significant drug history or family history. Contrast-enhanced computed tomography(CECT) showed an extraaxial relatively defined hyperdense mass lesion measuring about 7 cm × 4.8 cm × 8.8 cm in size in left frontal region with internal foci of coarse calcifications which showed heterogeneous contrast enhancement. Significant mass effect in the form of contralateral shift of lateral ventricle and subfalcine and uncal herniation was noted [Figure 1]. Features suggested possibility of meningioma. Immediate fronto-parietal craniectomy was done. Intraoperatively, the tumor was seen as a soft reddish highly vascularized mass attached to the dura eroding the overlying bone in few areas. Gross total resection of the tumor with the attached dura was done. Histopathological examination of the resected bone and attached soft tissue pieces showed a richly vascular tumor with small round cells arranged in lobules, sheets, and cords [Figure 2]. The tumor was composed of monotonous small, round to oval cells with regular nuclear outline, vesicular chromatin, and scant amount of clear to pale eosinophilic cytoplasm [Figure 3] Cytoplasmic vacuolation was also noted. Some tumor cells were larger with prominent nucleoli and irregular nuclear contour. Homer wright rosettes were not seen. The cytoplasm of the tumor cells was stained positive with periodic acid-Schiff's staining. Necrosis was seen in few areas. There was involvement of dura and bony erosion [Figure 4]. Tumor did not invade the brain parenchyma. Immunohistochemistry showed positivity for CD99 and nonspecific enolase (NSE) [Figure 5]. It was negative for CD31, CD20, CD3, and glial fibrillary acidic protein. It was diagnosed as ES/pPNET. Postoperatively, patient was observed in surgical intensive unit. He showed recovery from hemiparesis and remained free from any neurologic deficits. Extensive workup was done including CECT scans of the thorax, abdomen, and pelvis and a bone scan with technetium Tc-99 m, to search for extracranial primary sites or metastatic deposits, which were all negative. Final diagnosis was primary ES/pPNET of dura. Chemotherapy (CT) and radiotherapy (RT) were given. Patient has no complaints and he is under routine follow-up for past 7 months.
Figure 1.
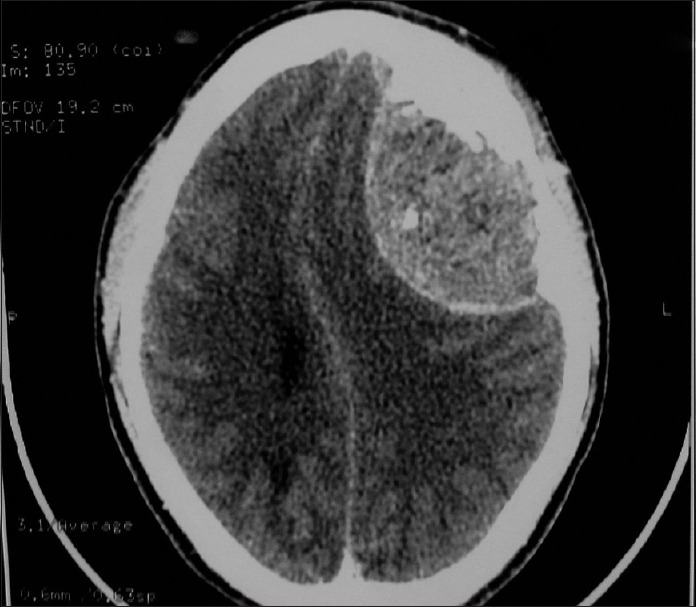
Axial CECT showing a relatively well-defined dural-based tumor showing contrast enhancement
Figure 2.
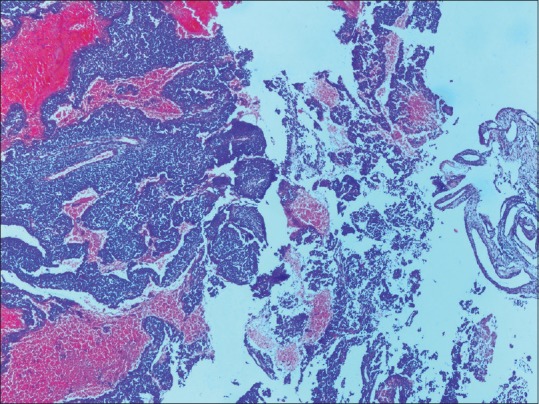
Photomicrograph showing a richly vascular small round cell tumor adjacent the dura (H and E, ×100)
Figure 3.
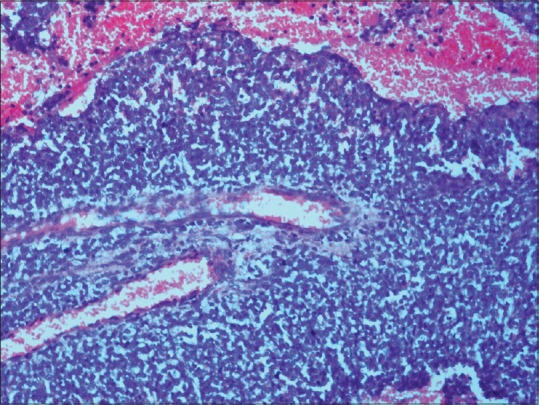
Photomicrograph showing tumor composed of sheets of uniform small round cells with scant cytoplasm an area of necrosis and several mitotic figures (H and E, ×200)
Figure 4.
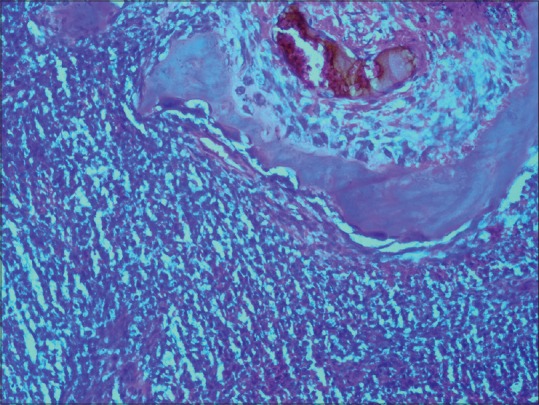
Photomicrograph showing the small round cells adjacent to the overlying bone (H and E, ×200)
Figure 5.
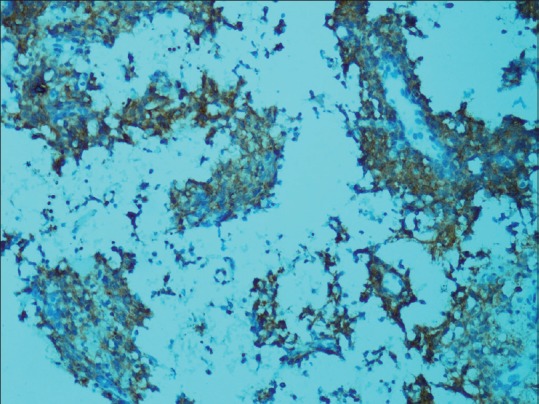
Tumor cells showing membranous expression of CD99 immunohistochemistry (×200)
Discussion and Review of Literature
Primary intracranial ES/pPNET is a recently recognized entity of CNS PNET. There are < 15 case reports of this entity in the literature.[4] A study conducted by Paulus et al. showed that out of total 2500 cases of brain tumors, only 9 were sarcomas. Of these, only one case was reported as ES.[5] A recent study by Krishnamani et al. showed that of 332 cases of ES diagnosed over an 11 years period, seven cases were of primary ES skull.[6] These tumors clinically present with signs of increased intracranial pressure, headache, vomiting, or with seizures and disturbances of consciousness. Motor deficit or uncommonly visual disturbances and endocrine abnormalities can occur depending on location. Ever since the first description of this tumor by Ewing as “diffuse endothelioma” controversy has persisted about the histogenesis of ES. The present case is discussed to clarify and stress upon two different aspects: First, the confusing terminology of ES, pPNEt, and cPNET and second is the unusual presentation of this tumor as a meningioma. ES and PNET are defined as round cell sarcomas showing varying degrees of neuroectodermal differentiation. The term “ES” is used for tumors with absent or limited neuroectodermal differentiation whereas “PNET” is employed for tumors demonstrating definite neuroectodermal features. “ES/pPNET” best describes this overlapping entity. The cPNET is defined as an embryonal tumor composed of undifferentiated or poorly differentiated neuroepithelial cells displaying divergent differentiation along neuronal, astrocytic, muscular, or melanocytic lines occurring within any region of the CNS other than cerebellum.[7] This group includes CNS-PNET not otherwise specified (NOS), neuroblastoma, ganglioneuroblastoma, medulloepithelioma, ependymoblastoma, and embryonal tumor with abundant neutrophils and true rosette.
Both cPNETs and pPNETs are aggressive tumors, but they differ in their cell of origin. The cPNETs arise from a precursor cell of the subependymal matrix of the CNS or external granular layer of the cerebellum, pinealocytes, and subependymal cells of the ventricles whereas pPNETs derive from the neural crest located outside the CNS. Cases of pPNET in meninges of cranial vault and epidural space of the spinal canal have been reported.[8] It is clinically important to differentiate the two as their clinical presentation, treatment, and prognosis differ variably.
Table 1 shows the important differences of the two entities. Compared to cPNET, ES/pPNET is relatively well-circumscribed tumors with broad implantation on the dura which allows gross total resection of the lesion.[9] pPNETs remain largely localized to the CNS and rarely metastases elsewhere while cPNETs involve the cerebrospinal fluid in 10–30% of cases at the time of diagnosis. The present case also showed a localized tumor attached to dura and bone with no distant metastasis or CSF involvement. All these factors together with sensitivity to CT probably contribute for the relatively better prognosis of pPNET. The long-term disease-free survival is reported in ES/pPNET cases which is uncommon among patients with intracranial cPNET patient.[10] Recent advances have made the distinction between cPNET and pPNET more clear. Immunohistochemistry for CD99 (MIC-2 gene product) shows strong membranous positivity in 97% of ES/pPNET cases while cPNETs are reported to be negative for CD99. CD99 is also detected in other small, blue, round cell tumors such as neuroblastoma, lymphoma, and rhabdomyosarcomas where cells show cytoplasmic positivity. Neuroectodermal differentiation is seen by immunopositivity for NSE, synaptophysin, and electron microscopy showing the presence of neurosecretory granules and microtubules. The immunopositivity for CD99 confirmed the diagnosis in the present case. Ewing family of tumors is characterized by a recurrent t(11;22) (q24;q12) chromosomal translocation, detectable in approximately 85% cases and t(21;22) (q22;q12) translocation in the remaining 10–15% of cases.[7,11]
Table 1.
Differences between peripheral primitive neuroectodermal tumor and central primitive neuroectodermal tumor
ES/pPNET is rarely arising as a primary dural-based neoplasm radiologically mimicking meningioma. The clinical features of these mimickers are further misleading, supporting a preoperative diagnosis of meningiomas as also seen in the present case. In general, patients with dural-based tumors do not present with a neurosurgical emergency. Neurological signs and symptoms usually arise when the tumor is large, compressing or invading the brain parenchyma.[12] Meningiomas are the most common dural-based tumor accounting for approximately 20–30% of all primary intracranial tumor. They are mostly benign cured by surgical resection alone. Only atypical meningioma or anaplastic meningioma with or without bony erosion may need further treatment. On the other hand, pPNET is a rare intracranial aggressive tumor which needs multimodality treatment combining surgery, CT, and RT. Distinction of both carries great prognostic and therapeutic significance and an incorrect diagnosis leads to undue delay in appropriate therapy under the mistaken impression of meningioma, causing deleterious effects on patient survival. Awareness of unusual presentations of ES/pPNET is important for early diagnosis of the tumor and having a high clinical suspicion for this infrequent tumor.[7,13] Even radiology may not help in such cases as both the tumors may present as well-defined dural masses showing contrast enhancement and can even show bony erosion.
Table 2 shows various cases of ES/pPNET presenting as extraaxial/dural-based masses reported in literature.
Table 2.
Present and previous reported cases of Intracranial extraaxial Ewing sarcoma/peripheral primitive neuroectodermal tumor tumors
It is clearly seen that the tumor presents in a wide age range from 5 to 56 years. These are contrast enhancing masses. In most cases, gross total resection is possible and only in one tabulated case subtotal resection was done. Most cases of intracranial pPNET show a favorable prognosis without any extracranial metastates after complete surgical resection and CT. One case however showed an aggressive course with local recurrence and metastases. Review of literature reveals that pPNET shows two different patterns of meningeal involvement. First is a diffuse involvement of the cranial and spinal leptomeninges without any primary intraparenchymal or meningeal tumor. The second is as a localized dural-based mass, mimicking meningioma, as encountered in the present case. A number of other dural and leptomeningeal nonmeningothelial neoplasm may radiologically and clinically simulate meningioma besides pPNET. These include hemangiopericytoma, solitary fibrous tumor, smooth muscle tumors (leiomyoma and leiomyosarcoma), epithelioid hemangioendothelioma, intradural chordoma, leptomeningeal medulloblastoma, melanocytic tumors, and hemangioblastoma.[24,25] Dural metastases from systemic cancer such as breast, prostate, and lung cancers can also mimic meningioma. Brain parenchyma is the most common site involved by tumor metastases, but dural compartment may also be affected in about 9–10% of all patients with systemic cancer.[26,27] Being aware can lead to early treatment. Immunohistochemistry for cytokeratins, vimentin, epithelial membrane antigen, CD45, endothelial markers CD31, CD34, and other specific markers for different metastatic tumor may help in differentiating these tumors.
There is no certain therapeutic protocol for management of patients with ES/pPNET affecting the CNS. However, studies show that a multimodality treatment approach is necessary and CT forms the backbone of treatment of localized ES. Surgical treatment is necessary to achieve diagnosis and decompression, which is usually followed by improvement of symptoms.[13,28,29] Our case involved a tumor presenting as a dural mass. This may have implications for survival as a well-circumscribed dural mass is more amenable to surgical resection than a tumor with extensive spread. Irrespective of the location, the primary surgical goal is to achieve total excision as this is an important factor for long-term survival. Removal of the involved bone is to be done.[8,30] The primary treatment consists of neoadjuvant systemic CT, with a combination of vincristine, doxorubicin, and cyclophosphamide, alternating with ifosfamide and etoposide, given for 12–24 weeks. This is followed by definitive local treatment of surgery or radiation, or surgery and postoperative radiation directed at the primary site. The treatment is completed with administering CT with a similar combination for a total of 36–49 weeks.[31,32] The multi-agent CT may also include adriamycin and actinomycin-D. Although these are commonly used for localized ES/pPNET, there is no standard regime for recurrent and metastatic disease, especially for intracranial ES/pPNET. Making the distinction between cPNETs and pPNETs is essential as already discussed. cPNETs are treated with intrathecal methotrexate as well as systemic CT, including platinum agents, etoposide, lomustine, and vincristine. The tumor, ES/pPNET is also radiosensitive and dose of 1.5–2 gy/days for 5 days/weeks is recommended followed by four drug CT regime. In contrast to pPNET, entire neuraxis radiation is used in children with cPNET. Isolated RT is indicated in inoperable cases.[12,33,34]
Because of the small number of patients, the prognosis of Es/pPNET is not clearly known. The largest series of primary cranial ES was reported by Desai et al. in 2000.[35] In this series, the 5 years survival was found to be more than 57%. Long-term survival (7.5 years) of 45–60% was noted for pPNET patients while cPNET patient survival was much shorter, with a 3 years survival of only 33% in one review which included 33 cases. Other studies showed 5 years survival of 77% in pPNET, compared with 44% in cPNET. However, a similar 5 years survival of 50–70% in pPNETs and cPNETs is seen in case of localized disease with similar treatment of adequate surgical resection followed by aggressive CT. Primary ES is reported to have a better prognosis as compared to ES elsewhere.[36,37,38]
Conclusion
This case comes as a reminder to neurosurgeons as well as radiologists and pathologists for careful consideration of rare tumors in the differential diagnosis of common tumors like meningioma. Awareness of this entity of ES/pPNET, its rare dural presentation, and differentiation from the more common cPNET is needed for appropriate patient management. Meningeal ES/pPNET has to be kept in mind in the differential diagnosis of meningeal tumors eroding bone.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Choudhury KB, Sharma S, Kothari R, Majumder A. Primary extraosseous intracranial Ewing's sarcoma: Case report and literature review. Indian J Med Paediatr Oncol. 2011;32:118–21. doi: 10.4103/0971-5851.89798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dedeurwaerdere F, Giannini C, Sciot R, Rubin BP, Perilongo G, Borghi L, et al. Primary peripheral PNET/Ewing's sarcoma of the Dura: A clinicopathologic entity distinct from central PNET. Mod Pathol. 2002;15:673–8. doi: 10.1038/modpathol.3880585. [DOI] [PubMed] [Google Scholar]
- 3.Choi HY, Kim YH, Kim JH, Kim IA, Choe G, Kim CY. Ewing's sarcoma/Peripheral primitive neuroectodermal tumor in the cerebellopontine angle: Diagnosis and treatment. J Korean Neurosurg Soc. 2011;49:359–62. doi: 10.3340/jkns.2011.49.6.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amita R, Sandhyamani S, Nair S, Kapilamoorthy TR. Intracranial Ewings sarcoma/peripheral primitive neuroectodermal tumor. Neurol India. 2014;62:432–3. doi: 10.4103/0028-3886.141300. [DOI] [PubMed] [Google Scholar]
- 5.Paulus W, Slowik F, Jellinger K. Primary intracranial sarcomas: Histopathological features of 19 cases. Histopathology. 1991;18:395–402. doi: 10.1111/j.1365-2559.1991.tb00869.x. [DOI] [PubMed] [Google Scholar]
- 6.Krishnamani K, Kumar TN, Gandhi LV, Raghunadharao D, Sadashivudu G, Megha U. Primary Ewing's sarcoma of the cranium: Case series and review of literature. J Cancer Res Ther. 2014;10:377–80. doi: 10.4103/0973-1482.136663. [DOI] [PubMed] [Google Scholar]
- 7.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC; 2007. pp. 141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole M, Parajuli S, Laske D, Goldstein L, Morrison T, Mukherjee A, et al. Peripheral primitive neuroectodermal tumor of the dura in a 51-year-old woman following intensive treatment for breast cancer. Am J Case Rep. 2014;15:294–9. doi: 10.12659/AJCR.890656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt D, Herrmann C, Jürgens H, Harms D. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing's sarcoma. A report from the kiel pediatric tumor registry. Cancer. 1991;68:2251–9. doi: 10.1002/1097-0142(19911115)68:10<2251::aid-cncr2820681025>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 10.Antonelli M, Caltabiano R, Chiappetta C, Oliva MA, Giangaspero F, Lanzafame S. Primary peripheral PNET/Ewing's sarcoma arising in the meninges, confirmed by the presence of the rare translocation t(21;22) (q22;q12) Neuropathology. 2011;31:549–55. doi: 10.1111/j.1440-1789.2010.01196.x. [DOI] [PubMed] [Google Scholar]
- 11.Sandberg AA, Bridge JA. Updates on cytogenetics and molecular genetics of bone and soft tissue tumors: Ewing sarcoma and peripheral primitive neuroectodermal tumors. Cancer Genet Cytogenet. 2000;123:1–26. doi: 10.1016/s0165-4608(00)00295-8. [DOI] [PubMed] [Google Scholar]
- 12.Gadani S, Mody RP, Solanki RN, Mahajan A. Primary Ewing sarcoma of skull vault in a child. Indian J Radiol Imaging. 2003;13:303–5. [Google Scholar]
- 13.Barresi V, Caffo M, Branca G, Caltabiano R, Tuccari G. Meningeal tumors histologically mimicking meningioma. Pathol Res Pract. 2012;208:567–77. doi: 10.1016/j.prp.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Katayama Y, Kimura S, Watanabe T, Yoshino A, Koshinaga M. Peripheral-type primitive neuroectodermal tumor arising in the tentorium. Case report. J Neurosurg. 1999;90:141–4. doi: 10.3171/jns.1999.90.1.0141. [DOI] [PubMed] [Google Scholar]
- 15.Antunes NL, Lellouch-Tubiana A, Kalifa C, Delattre O, Pierre-Kahn A, Rosenblum MK. Intracranial Ewing sarcoma/‘peripheral’ primitive neuroectodermal tumor of dural origin with molecular genetic confirmation. J Neurooncol. 2001;51:51–6. doi: 10.1023/a:1006432919281. [DOI] [PubMed] [Google Scholar]
- 16.Pekala JS, Gururangan S, Provenzale JM, Mukundan S., Jr Central nervous system extraosseous Ewing sarcoma: Radiologic manifestations of this newly defined pathologic entity. AJNR Am J Neuroradiol. 2006;27:580–3. [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta A, Bansal S, Chaturvedi S. Primary Ewing's sarcoma of frontoparietal bone with major soft tissue extension: An unusual presentation and review of the literature. Case Rep Pathol 2012. 2012:713836. doi: 10.1155/2012/713836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mobley BC, Roulston D, Shah GV, Bijwaard KE, McKeever PE. Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: Case reports and review. Hum Pathol. 2006;37:845–53. doi: 10.1016/j.humpath.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Tanboon J, Sitthinamsuwan B, Paruang T, Marrano P, Thorner PS. Primary intracranial Ewing sarcoma with an unusually aggressive course: A case report and review of the literature. Neuropathology. 2012;32:293–300. doi: 10.1111/j.1440-1789.2011.01258.x. [DOI] [PubMed] [Google Scholar]
- 20.Stechschulte SU, Kepes JJ, Holladay FP, McKittrick RJ. Primary meningeal extraosseous Ewing's sarcoma: Case report. Neurosurgery. 1994;35:143–7. doi: 10.1227/00006123-199407000-00023. [DOI] [PubMed] [Google Scholar]
- 21.Papotti M, Abbona G, Pagani A, Monga G, Bussolati G. Primitive neuroectodermal tumor of the meninges: An histological, immuno-histochemical, ultrastructural, and cytogenetic study. Endocr Pathol. 1998;9:275–80. doi: 10.1007/BF02739968. [DOI] [PubMed] [Google Scholar]
- 22.D’Antonio A, Caleo A, Garcia JF, Marsilia GM, De Dominicis G, Boscaino A. Primary peripheral PNET/Ewing's sarcoma of the Dura with FISH analysis. Histopathology. 2004;45:651–4. doi: 10.1111/j.1365-2559.2004.01961.x. [DOI] [PubMed] [Google Scholar]
- 23.Mellai M, Caldera V, Comino A, Fortunato M, Bernucci C, Schiffer D. PNET/ESFT of the cranial vault: A case report. Clin Neuropathol. 2010;29:372–7. doi: 10.5414/npp29372. [DOI] [PubMed] [Google Scholar]
- 24.Kleinschmidt-DeMasters BK, Mierau GW, Sze CI, Breeze RE, Greffe B, Lillehei KO, et al. Unusual dural and skull-based mesenchymal neoplasms: A report of four cases. Hum Pathol. 1998;29:240–5. doi: 10.1016/s0046-8177(98)90042-9. [DOI] [PubMed] [Google Scholar]
- 25.Ali AE, Fazl M, Bilbao JM. Primary intracranial leiomyoma: A case report and literature review. Virchows Arch. 2006;449:382–4. doi: 10.1007/s00428-006-0252-z. [DOI] [PubMed] [Google Scholar]
- 26.Nayak L, Abrey LE, Iwamoto FM. Intracranial dural metastases. Cancer. 2009;115:1947–53. doi: 10.1002/cncr.24203. [DOI] [PubMed] [Google Scholar]
- 27.Savage NM, Alleyne CH, Vender JR, Figueroa R, Zhang H, Samuel TA, et al. Dural-based metastatic carcinomas mimicking primary CNS neoplasia: Report of 7 cases emphasizing the role of timely surgery and accurate pathologic evaluation. Int J Clin Exp Pathol. 2011;4:530–40. [PMC free article] [PubMed] [Google Scholar]
- 28.Furuno Y, Nishimura S, Kamiyama H, Numagami Y, Saito A, Kaimori M, et al. Intracranial peripheral-type primitive neuroectodermal tumor. Neurol Med Chir (Tokyo) 2008;48:72–6. doi: 10.2176/nmc.48.72. [DOI] [PubMed] [Google Scholar]
- 29.Ludwig JA. Ewing sarcoma: Historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol. 2008;20:412–8. doi: 10.1097/CCO.0b013e328303ba1d. [DOI] [PubMed] [Google Scholar]
- 30.Bricha M, Jroundi L, Boujida N, El Hassani MR, Chakir N, Jiddane M. Primary Ewing sarcoma of the skull vault. J Radiol. 2007;88:1899–901. doi: 10.1016/s0221-0363(07)78370-1. [DOI] [PubMed] [Google Scholar]
- 31.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 32.Hadfield MG, Quezado MM, Williams RL, Luo VY. Ewing's family of tumors involving structures related to the central nervous system: A review. Pediatr Dev Pathol. 2000;3:203–10. doi: 10.1007/s100249910026. [DOI] [PubMed] [Google Scholar]
- 33.Saeedinia S, Nouri M, Alimohammadi M, Moradi H, Amirjamshidi A. Primary spinal extradural Ewing's sarcoma (primitive neuroectodermal tumor): Report of a case and meta-analysis of the reported cases in the literature. Surg Neurol Int. 2012;3:55. doi: 10.4103/2152-7806.96154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thacker MM, Temple HT, Scully SP. Current treatment for Ewing's sarcoma. Expert Rev Anticancer Ther. 2005;5:319–31. doi: 10.1586/14737140.5.2.319. [DOI] [PubMed] [Google Scholar]
- 35.Desai KI, Nadkarni TD, Goel A, Muzumdar DP, Naresh KN, Nair CN. Primary Ewing's sarcoma of the cranium. Neurosurgery. 2000;46:62–8. [PubMed] [Google Scholar]
- 36.Cugati G, Singh M, Pande A, Symss NP, Chakravarthy VM, Ramamurthi R. Isolated skull base primary Ewing's sarcoma: An extremely rare location. J Cancer Res Ther. 2013;9:741–2. doi: 10.4103/0973-1482.126479. [DOI] [PubMed] [Google Scholar]
- 37.Agrawal A, Dulani R, Mahadevan A, Vagaha SJ, Vagha J, Shankar SK. Primary Ewing's sarcoma of the frontal bone with intracranial extension. J Cancer Res Ther. 2009;5:208–9. doi: 10.4103/0973-1482.57129. [DOI] [PubMed] [Google Scholar]
- 38.Bano S, Yadav SN, Garga UC. Case Report: Intracranial peripheral primitive neuroectodermal tumor – Ewing's sarcoma of Dura with transcalvarial-subgaleal extension: An unusual radiological presentation. Indian J Radiol Imaging. 2009;19:305–7. doi: 10.4103/0971-3026.57215. [DOI] [PMC free article] [PubMed] [Google Scholar]