

Abstract
Rosai-Dorfman disease (RDD) is a rare, idiopathic, benign histioproliferative disorder. Extranodal involvement is seen in around 25–40% of patients. Central nervous system manifestation of RDD is uncommon and suprasellar location of the lesion is a distinct rarity. Surgery is the cornerstone of management of intracranial RDD. However, tumor recurrence or regrowth is a potential problem. Hence, low dose conformal radiotherapy (RT) should be considered in patients undergoing sub-total resection or having unresectable recurrent disease. Though cranial RT usually leads to satisfactory improvement of symptoms and long-term disease stabilization or regression, in few patients there may be an eventual progression of disease for which systemic chemotherapy may be considered. We have highlighted the salient features of this enigmatic disease by citing a case of a 50-year-old male patient with suprasellar RDD treated by maximal safe surgery and deferred radiation therapy on progression.
Keywords: Intracranial, Rosai-Dorfman disease, suprasellar
Introduction
Rosai-Dorfman disease (RDD) is a benign disorder of unknown etiology characterized by an excessive proliferation of histiocytes.[1] About 25–40% of patients have extranodal involvement including involvement of the central nervous system (CNS).[1,2] CNS involvement in RDD is rare, accounting for only 4% of all reported cases.[3] Common intracranial locations of RDD include the cerebral convexities, parasagittal and petroclival region. Involvement of suprasellar region is extremely rare, and less than ten cases of suprasellar Rosai-Dorfman syndrome have been reported until date.[4] We herein report a case of a 50-year-old male patient with suprasellar Rosai-Dorfman syndrome treated by maximal safe resection and deferred radiation therapy on progression.
Case Report
A 50-year-old male patient presented with complaints of gradually progressive loss of vision in his right eye over 3 months. There was no history of fever, headache, vomiting, seizure, loss of consciousness, photophobia, and weakness of body. On examination, the patient was alert, orientated, and afebrile. There was no evidence of peripheral lymphadenopathy. There was a loss of perception of light in the right eye. Rest of the neurological examination did not reveal any abnormality. A contrast-enhanced magnetic resonance imaging (MRI) of the brain revealed an intensely enhancing solid mass in an extradural location in the suprasellar cistern, close to the pituitary stalk. Anteriorly, the mass extended to the right orbital apex [Figure 1a]. The possible differential diagnoses were meningioma and RDD. Endocrinological evaluation did not reveal any abnormality. He underwent maximal safe resection (optic nerve decompression) via fronto-temporal craniotomy approach. Intraoperative findings included a soft to firm, moderately vascular tumor, lifting and surrounding the optic nerve more on the right side. It was extending into carotico-optic space partially encasing the internal carotid artery (ICA). Postoperative MRI of the brain suggested complete removal of the lesion, with minimal dural enhancement along the right temporal lobe [Figure 1b]. Postoperative histopathological report showed a tumor consisting of variable numbers of histiocytes intermixed with plasma cells, eosinophills and lymphocytes. Histiocytes showed features of emperipolesis. On immunohistochemistry, the histiocytes showed immunopositivity for S-100 protein suggesting a diagnosis of intracranial RDD [Figure 2].
Figure 1.

(a) Axial T1-weighted turbo spin echo postcontrast MRI image reveals an intensely enhancing solid mass (arrow) in extradural location in the suprasellar cistern, close to the pituitary stalk. Anteriorly the mass extends till the right orbital apex; (b) axial T1-weighted turbo spin echo postcontrast MRI image after surgery reveals complete removal of the lesion, with minimal dural enhancement along the right temporal lobe; (c-g) recurrence of the central nervous system lesions 1 year after surgery. Axial T2-weighted turbo spin echo magnetic resonance image (c) shows a hypointense extradural mass (arrow) in the right suprasellar cistern, abutting the right temporal lobe. Axial T1-weighted turbo spin echo postcontrast MRI image (d) shows intense contrast enhancement in the mass as well as extension to the contralateral side. Coronal T1-weighted turbo spin echo postcontrast images (e and f) reveal the extension of mass in the right cavernous sinus and encasement of the intracavernous segment of the right internal carotid artery (arrow in e); and extension anteriorly along the anterior cranial fossa dura (arrows in f). Axial T1-weighted turbo spin echo postcontrast image through the C-P angle cistern (g) reveals contrast enhancement and thickening involving the left vestibulo-cochlear and facial nerves, extending into the left internal auditory canal; (h-j) progression of the central nervous system lesion 20 months after completion of cranial radiotherapy. T1-weighted turbo spin echo postcontrast axial (h), coronal (i) and axial (j) images show a 5 cm × 4.9 cm × 4 cm intensely enhancing mass lesion in suprasellar region involving bilateral cavernous sinus and internal carotid artery (right > left), compressing optic chiasm and reaching up to left superior orbital fissure without obvious intra-orbital extension. Another 2.6 cm × 1.6 cm enhancing lesion is seen in the right occipito-parietal lobe (arrow in h). Similarly, enhancing lesion is also noted in the left cerebello-pontine angle involving the intracranial segment of left VII and VIII cranial nerve (arrow in j)
Figure 2.
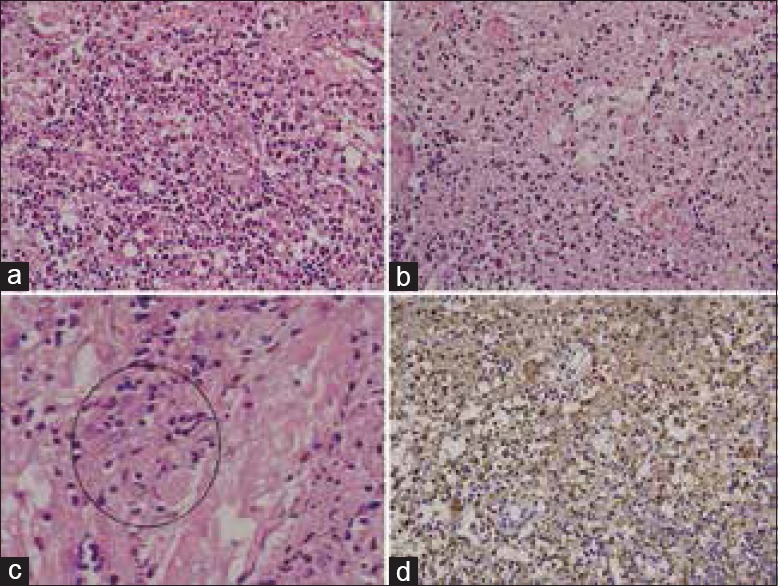
(a and b) The infiltrates are composed of variable numbers of histiocytes intermixed with plasma cells, eosinophills and lymphocytes (H and E, ×20), (c) histiocytes showing emperipolesis (H and E, ×40c), (d) Histiocytes are immune-positive for S-100 protein (Immunohistochemistry, ×20)
He was symptomatically stable for 1 year. Thereafter, he presented with gradually progressive diminution of hearing in his left ear over 3 months and acute worsening of vision in his left eye over 3 weeks. On examination, visual acuity had decreased to finger counting at 2 feet on the left and there was loss of perception of light in the right eye. Pure tone audiometry revealed complete loss of hearing in his left ear although hearing was intact in his right ear. A repeat MRI of the brain showed an intensely contrast enhancing mass lesion in the right suprasellar cistern, extending into the right cavernous sinus, encasing the intracavernous segment of ICA and abutting the right temporal lobe. There was also contrast enhancement and thickening involving the left vestibulo-cochlear and facial nerves, extending into the left internal auditory canal [Figure 1c–g]. He was given a trial of oral corticosteroids, but there was symptomatic progression after 2 weeks. Hence, local radiation was delivered to the suprasellar mass and the lesion involving the left vestibulo-cochlear and facial nerves to a total dose of 20 Gy in 10 fractions over 2 weeks by three-dimensional conformal radiotherapy (3D-CRT). He was evaluated 1 month after completion of radiation. Pure tone audiometry showed an improvement in hearing in his left ear though ophthalmological evaluation did not show any improvement in visual acuity. A brain MRI, done 3 months after completion of radiation therapy showed radiologically stable disease. Twenty months after completion of radiotherapy, he complained of decrease in vision in the left eye and brain MRI revealed a 5 cm × 4.9 cm × 4 cm intensely enhancing mass lesion in suprasellar region involving bilateral cavernous sinuses and ICAs (right > left), compressing optic chiasm and reaching up to left superior orbital fissure without obvious intra-orbital extension. Similarly, enhancing lesions were noted in bilateral tentorium, left cerebello-pontine angle, intracranial segment of left VII and VIII cranial nerve. A 2.6 cm × 1.6 cm enhancing lesion was also noted in the right occipito-parietal lobe along tentorium. The entire dura mater was thickened with nodular enhancements suggestive of diffuse leptomeningeal disease [Figure 1h–j]. In view of progressive, diffuse leptomeningeal disease, the patient is being considered for systemic chemotherapy.
Discussion
Rosia-Dorfman syndrome is a benign histiocytic proliferative disorder, first described by Rosai and Dorfman in 1969 as a triad of massive cervical lymphadenopathy, expanded lymph node sinus and characteristic histiocytes showing emperipolesis (sinus histiocytosis with massive lymphadenopathy).[5] Intracranial RDD predominantly affects males in the fourth to fifth decade.[2,6] The symptoms may vary depending on the site of the lesion. The common presenting symptoms are focal neurological deficit, seizures and headache owing to mass effect.[2,7] Symptoms of hypothalamic-pituitary axis dysfunction such as diabetes insipidus have also been reported.[8] Though 90% of patients with RDD present with massive, painless, bilateral cervical lymphadenopathy often associated with a history of fever and weight loss, such presentation is unusual in patients with the intracranial disease.[7]
Majority of intracranial RDD have dural attachment and some of them may cause adjacent bone erosion. Brain MRI usually shows a homogeneously enhancing meningeal-based mass on T1-weighted postcontrast images with a variable amount of peri-lesional edema on T2-weighted/fluid-attenuated inversion recovery images, mimicking meningioma. However, low signal intensity on T2-weighted MRI is the key feature that differentiates RDD from meningioma.[4,9] Sze and Zimmerman opined that the low signal intensity on T2-weighted MRI may be due to the presence of free radicals produced by macrophages during active phagocytosis.[10] Intraparenchymal lesions of RDD mimicking CNS lymphoma or tubercular granuloma have also been reported.[7] Though intracranial RDD is usually solitary, multiple lesions have also been described in the literature.[11,12,13]
Histopathological examination along with immunohistochemistry study is the gold standard to establish the diagnosis of intracranial RDD. Histologically, RDD is characterized by a polymorphous infiltrate of lymphoplasmacytic cells and histiocytes of varying size. The histiocytes typically show features of emperipolesis and are usually immune-positive for S-100 and CD-68, while immune-negative for CD-1a, which is the marker of Langerhans cell histiocytosis.[2,7,14]
Surgery in the form of maximal safe resection plays the most crucial role in the management of isolated CNS RDD.[2] However, recurrence after surgery is a common problem and is usually difficult to manage. In a comprehensive review of intracranial RDD by Petzold et al., tumor recurrence or regrowth has been reported in 14% of patients.[2] In a more updated analysis of 49 previous case reports of intracranial RDD by Symss et al., complete excision of lesions from sites such as dural convexity, parasagittal region, cerebello-pontine angle and the sphenoid wing has been associated with prolonged progression-free survival.[7] Complete resection is technically difficult in tumors involving the cavernous sinus, suprasellar region, petroclival region and multiple intracranial sites.[7] Subtotal resection is often done in these difficult sites and is associated with a higher rate of recurrence (20–25%).[7] However, the role of postoperative treatment is not well-defined because of the rarity of the disease.
Spontaneous resolution of the residual lesion after surgery has been reported in few cases. Regression of residual intracranial RDD following administration of postoperative corticosteroids has been reported by some authors.[11,12,15]
Radiation therapy is a useful therapeutic option in patients with intracranial RDD. Trudelin 1984 reported a case of RDD involving the petrous bone, where the use of postoperative radiation (15 Gy) after near-total excision of the lesion led to complete response, sustained for 14 months.[16] Petzold et al. and Symss et al. have advocated the use of postoperative low dose radiotherapy (RT) (20 Gy in 10 fractions over 2 weeks) in the case of subtotal resection.[2,7] Gamma knife radio-surgery (8–12 Gy depending on the site of lesion) has also been successfully applied in the management of residual intracranial RDD after subtotal resection.[13,17]
Systemic chemotherapy has also been used in a few reports with modest efficacy. Horneff et al. postulated the use of a combination of low dose methotrexate and 6 mercaptopurine for 4 weeks to ensure remission followed by maintenance therapy with 6 mercaptopurine for 2 years.[18] With this approach, there was a durable complete response (for 7 years) in a 3-year-old girl with steroid-resistant RDD.
In our patient, the primary site of the lesion was the suprasellar cistern, which is a very rare location for intracranial RDD. One year after maximal safe resection, the patient had multiple recurrent lesions involving the suprasellar cistern and the left VII and VIII cranial nerves, which were unresectable because of the proximity to critical vascular (ICA) and neural (II, VII and VIII cranial nerves) structures. Hence, low dose conformal RT was considered in his case. Use of RT led to prolonged radiological disease stabilization for 20 months and worthwhile functional improvement without any significant treatment-related toxicity. Unfortunately, thereafter, the patient had progressive diffuse leptomeningeal disease for which he is being considered for systemic chemotherapy.
Conclusion
Suprasellar RDD is a rare histiocytic proliferative disorder. Surgery is the main-stay of treatment. However, tumor recurrence or regrowth is a potential problem. Recurrent tumors are often not resectable due to their proximity to vital neurovascular structures. Low-dose external beam conformal RT is a safe and effective treatment modality in patients with recurrent disease where surgery is not feasible. RT usually leads to prolonged stabilization or regression of RDD and amelioration of symptoms. However, even after administration of cranial radiotherapy, there may be an eventual progression of disease in some patients. Although there is not enough data regarding the efficacy of systemic corticosteroids and chemotherapy, they may be considered for patients who experience disease progression despite adjuvant radiotherapy.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990;7:19–73. [PubMed] [Google Scholar]
- 2.Petzold A, Thom M, Powell M, Plant GT. Relapsing intracranial Rosai-Dorfman disease. J Neurol Neurosurg Psychiatry. 2001;71:538–41. doi: 10.1136/jnnp.71.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M, Provias J, Bernstein M. Rosai-Dorfman disease mimicking multiple meningioma: Case report. Neurosurgery. 1995;36:1185–7. doi: 10.1227/00006123-199506000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Lou X, Chen ZY, Wang FL, Ma L. MR findings of Rosai-Dorfman disease in sellar and suprasellar region. Eur J Radiol. 2012;81:1231–7. doi: 10.1016/j.ejrad.2011.02.059. [DOI] [PubMed] [Google Scholar]
- 5.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63–70. [PubMed] [Google Scholar]
- 6.Wan S, Teng X, Zhan R, Yu J, Gu J, Zhang K. Isolated intracranial Rosai-Dorfman disease mimicking suprasellar meningioma: Case report with review of the literature. J Int Med Res. 2008;36:1134–9. doi: 10.1177/147323000803600535. [DOI] [PubMed] [Google Scholar]
- 7.Symss NP, Cugati G, Vasudevan MC, Ramamurthi R, Pande A. Intracranial Rosai Dorfman disease: Report of three cases and literature review. Asian J Neurosurg. 2010;5:19–30. [PMC free article] [PubMed] [Google Scholar]
- 8.Ng HK, Poon WS. Sinus histiocytosis with massive lymphadenopathy localized to the sella. Br J Neurosurg. 1995;9:551–5. doi: 10.1080/02688699550041223. [DOI] [PubMed] [Google Scholar]
- 9.Udono H, Fukuyama K, Okamoto H, Tabuchi K. Rosai-Dorfman disease presenting multiple intracranial lesions with unique findings on magnetic resonance imaging. Case report. J Neurosurg. 1999;91:335–9. doi: 10.3171/jns.1999.91.2.0335. [DOI] [PubMed] [Google Scholar]
- 10.Sze G, Zimmerman RD. The magnetic resonance imaging of infections and inflammatory diseases. Radiol Clin North Am. 1988;26:839–59. [PubMed] [Google Scholar]
- 11.Camp SJ, Roncaroli F, Apostolopoulos V, Weatherall M, Lim S, Nandi D. Intracerebral multifocal Rosai-Dorfman disease. J Clin Neurosci. 2012;19:1308–10. doi: 10.1016/j.jocn.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 12.McPherson CM, Brown J, Kim AW, DeMonte F. Regression of intracranial Rosai-Dorfman disease following corticosteroid therapy. Case report. J Neurosurg. 2006;104:840–4. doi: 10.3171/jns.2006.104.5.840. [DOI] [PubMed] [Google Scholar]
- 13.Sato A, Sakurada K, Sonoda Y, Saito S, Kayama T, Jokura H, et al. Rosai-Dorfman disease presenting with multiple intracranial and intraspinal masses: A case report. No Shinkei Geka. 2003;31:1199–204. [PubMed] [Google Scholar]
- 14.Juskevicius R, Finley JL. Rosai-Dorfman disease of the parotid gland: Cytologic and histopathologic findings with immunohistochemical correlation. Arch Pathol Lab Med. 2001;125:1348–50. doi: 10.5858/2001-125-1348-RDDOTP. [DOI] [PubMed] [Google Scholar]
- 15.Bing F, Brion JP, Grand S, Pasquier B, Lebas JF. Tumor arising in the periventricular region. Neuropathology. 2009;29:101–3. doi: 10.1111/j.1440-1789.2008.00950.x. [DOI] [PubMed] [Google Scholar]
- 16.Trudel M. Dural involvement in sinus histiocytosis with massive lymphadenopathy. Case report. J Neurosurg. 1984;60:850–2. doi: 10.3171/jns.1984.60.4.0850. [DOI] [PubMed] [Google Scholar]
- 17.Hadjipanayis CG, Bejjani G, Wiley C, Hasegawa T, Maddock M, Kondziolka D. Intracranial Rosai-Dorfman disease treated with microsurgical resection and stereotactic radiosurgery. Case report. J Neurosurg. 2003;98:165–8. doi: 10.3171/jns.2003.98.1.0165. [DOI] [PubMed] [Google Scholar]
- 18.Horneff G, Jürgens H, Hort W, Karitzky D, Göbel U. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Response to methotrexate and mercaptopurine. Med Pediatr Oncol. 1996;27:187–92. doi: 10.1002/(SICI)1096-911X(199609)27:3<187::AID-MPO10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]