

Abstract
Time-resolved electron transfer and electrogenic H+ translocation have been compared in a bd-type quinol oxidase from Escherichia coli and its E445A mutant. The high-spin heme b595 is found to be retained by the enzyme in contrast to the original proposal, but it is not reducible even by excess of dithionite. When preincubated with the reductants, both the WT (b5582+, b5952+, d2+) and E445A mutant oxidase (b5582+, b5953+, d2+) bind O2 rapidly, but formation of the oxoferryl state in the mutant is ≈100-fold slower than in the WT enzyme. At the same time, the E445A substitution does not affect intraprotein electron re-equilibration after the photolysis of CO bound to ferrous heme d in the one-electron-reduced enzyme (the so-called “electron backflow”). The backflow is coupled to membrane potential generation. Electron transfer between hemes d and b558 is electrogenic. In contrast, electron transfer between hemes d and b595 is not electrogenic, although heme b595 is the major electron acceptor for heme d during the backflow, and therefore is not likely to be accompanied by net H+ uptake or release. The E445A replacement does not alter electron distribution between hemes b595 and d in the one-electron reduced cytochrome bd [Em(d) > Em(b595), where Em is the midpoint redox potential]; however, it precludes reduction of heme b595, given heme d has been reduced already by the first electron. Presumably, E445 is one of the two redox-linked ionizable groups required for charge compensation of the di-heme oxygen-reducing site (b595, d) upon its full reduction by two electrons.
Keywords: oxidase, electron transfer reactions, respiratory chain
Cytochrome bd is a terminal respiratory oxidase required by many bacteria for survival and growth under conditions of low oxygen tension (1–3). Unlike the heme-copper oxidases (4, 5), cytochrome bd does not pump protons (6). However, the oxidation of ubiquinol by O2 catalyzed by cytochrome bd is linked to the generation of transmembrane electric potential difference (ΔΨ) (7–11) by virtue of the fact that the protons resulting from the oxidation of ubiquinol are released into the bacterial periplasm, whereas the protons used to convert O2 to H2O are taken from the bacterial cytoplasm. Cytochrome bd is a heterodimeric enzyme containing three hemes: b558, b595, and d (12–14) but no copper. Although the x-ray structure of cytochrome bd has not been determined, experimental studies support a topology with all of the three hemes being located near the periplasmic side of the membrane (15, 16). It is commonly accepted that the low-spin heme b558 is directly involved in oxidation of quinol, and that the high-spin heme d is the site where O2 is trapped and reduced to H2O (12). However the role of high-spin heme b595 is still a matter of debate. Together with heme d, heme b595 is proposed to form a binuclear site for the effective reduction of oxygen analogous to the high-spin heme/CuB binuclear center in the heme-copper oxidases (17–23). The electron transfer sequence in cytochrome bd is thought to be QH2 → b558 → [b595 → d] → O2 (24–26). It is not known whether heme b595 simply serves as a conduit to transfer electrons between heme b558 and heme d (25), or whether heme b595 has a more complex role in the oxygen chemistry.
Time-resolved electrometry and absorption spectroscopy have proven to be very useful tools for elucidation of the molecular mechanism of the heme-copper oxidases (27–34). These approaches have been successfully applied to the WT cytochrome bd oxidase to study its reaction with O2 (11). Oxidation of the three-electron-reduced WT cytochrome bd by O2 has been found to occur in two steps: (i) the electrically silent process of oxygen binding to heme d with formation of compound A (heme d oxy-complex, d2+-O2), followed by (ii) the monoexponential generation of ΔΨ with τ of ≈60 μs that corresponds to the conversion of compound A to compound F (ferryl-oxo heme d, Fe4+ = O2-) (11).
Recently, a mutant was reported that is catalytically inactive and selectively influences the properties of heme b595. This is the E445A mutant of subunit I (CydA) of cytochrome bd from Escherichia coli (35). Previous analysis concluded that heme b595 is either partially or completely absent from the E445A mutant (35). In the current work, it is shown that the E445A oxidase preparations actually still retain the heme, but this heme remains in the ferric state, even in the presence of a strong reductant (dithionite). This unique possibility to have two-electron-reduced enzyme was used to analyze the role of heme b595.
Materials and Methods
Materials. Sucrose monolaurate was from Anatrace (Maumee, OH), plant L-lecitin was from Avanti Polar Lipids, and sodium dithionite was from Merck. Other basic chemicals and biochemicals were from Sigma-Aldrich, Merck, Fluka, and Serva.
Sample Preparation. E. coli cells were obtained from GO105/pTK1 and GO105/pTK1/E445A strains (35). Both the WT and the E445A mutant cytochrome bd oxidases were purified as described (36), but the second (hydroxyapatite) chromatographic step was omitted as it can cause destabilization and substantial degradation of heme b595 in the E445A mutant enzyme (35). Besides, in this work for solubilization of the membranes sucrose monolaurate detergent was used instead of N-dodecyl-N,N-dimethylammonio-3-propane-sulfonate used in an earlier report (11). Change of detergent allowed us to isolate the enzyme containing bound quinone. Reconstitution of cytochromes bd into liposomes was performed as described (11) except that the final concentration of the enzyme was increased by four times.
Heme Analysis. The heme b contents of both WT and E445A mutant enzymes were measured by the pyridine hemochromogen assay, using a value of Δε556.5–540 of 23.98 mM-1·cm-1 (37). To determine the heme d content, a value of Δε628–670 of 25 mM-1·cm-1 from the absolute dithionite-reduced spectra was used. The conventional procedure of using the dithionite-reduced minus air-oxidized difference absorption spectrum appears to be error-prone because the mutant enzyme in the air-oxidized form has a much lower amount of the ferrous oxy species. Hence, the use of the Δε628–607 value of 10.8 mM-1·cm-1, which correctly quantifies the content of heme d in the WT enzyme (20), results in an incorrect value of heme d in the E445A mutant enzyme (35). The use of this traditional method results in overestimation of heme d in the mutant enzyme.
The lineshapes of the 630-nm absorption band of ferrous heme d in the WT and mutant oxidases are not identical. In the mutant, the 630-nm peak is broader on its blue side than that in the WT, whereas the lineshapes of the red side of the peak are virtually identical in the WT and mutant enzymes (A. M. Arutyunyan, V.B.B., R.B.G., and A.A.K., unpublished work). For this reason the content of heme d for the WT and mutant enzyme was determined from the ferrous heme d absorption spectra by using the wavelength pair 630-nm minus 670-nm (Δε628–670 of 25 mM-1·cm-1) instead of the frequently used pair 630-nm minus 607-nm (20). This extinction coefficient for measuring heme d gives the same result obtained by an independent method based on the intensity of the dithionite-reduced absolute CD spectrum in the 600- to 700-nm range (A. M. Arutyunyan, V.B.B., R.B.G., and A.A.K., unpublished work).
EPR Spectroscopy. An X-band EPR spectrometer ESP-300 (Bruker) was used. The field modulation frequency was 100 kHz. The temperature of the sample was controlled with an ESR 900 liquid helium cryostat and an ITC4 temperature controller (Oxford Instruments, Palo Alto, CA). All EPR data were corrected by subtracting the blank (EPR signal of tube with buffer).
Kinetic Measurements. Time-resolved electometric and transient absorbance measurements were made as reported (ref. 11 and references therein). matlab (Mathworks, South Natick, MA) was used for data manipulation and presentation.
Results
Heme b595 Is Present in the E445A Mutant of Cytochrome bd. Transient absorption changes in cytochrome bd were measured in the spectral region where the absorbance spectra of all three hemes are well defined. Fully reduced, unliganded WT cytochrome bd shows peaks at 629 nm (heme d2+), 595 nm (heme b5952+), 561 nm (superposition of the α-band of heme b5582+ and the β-band of heme b5952+), and 531 nm (the β-band of heme b5582+).
Fig. 1A compares the absorption changes after CO photolysis from heme d2+ in the dithionite-reduced WT (solid line) and E445A mutant (dotted line) enzymes. For the WT cytochrome bd, the CO recombination difference spectrum in the presence of 1% CO has a maximum at 642 nm, a minimum at 622 nm, and a bump ≈540 nm, which is consistent with CO rebinding to heme d2+ (38). In contrast, in the E445A mutant enzyme, the spectrum of the CO recombining phase (Fig. 1 A, dotted line) reveals not only CO binding to heme d2+, but also some internal electron redistribution consistent with electron transfer from ferrous heme b595 to ferric heme d. The latter is well illustrated by the difference between the CO recombination spectra of the WT and E445A enzymes (Fig. 1B). This difference spectrum has maxima at 595 and 562 nm and a minimum at 630 nm and is almost identical to the reported spectrum of the reduced minus oxidized spectrum of heme b595 (39, 40), with the addition of a trough at 630 nm, indicating the oxidation of heme d upon CO photolysis of the mutant. Although the amount of reduced heme b595 is small (≈20% of the maximum possible), the spectroscopic identification of reduced heme b595 is clear.
Fig. 1.

Absorption changes during CO recombination after flash photolysis of dithionite-reduced cytochrome bd. (A) Spectrum of CO photolysis of the E445A mutant enzyme (dotted line) was adjusted to match the amplitude of photolysis of the WT enzyme (solid line). The spectrum is a difference between spectra before and after the flash. The spectrum before the flash is constant, but the spectrum after the flash was obtained by extrapolation of the CO recombination kinetics at each wavelength to zero time. (B) Difference between CO recombination spectra of E445A mutant and WT. Conditions: cytochrome bd,25 μM (WT) and 12 μM (E445A mutant); 0.1% Tween 20; 100 mM Hepes-KOH, pH 7.5; sodium dithionite, 0.1 mM (WT) and 3 mM (E445A mutant); 1% CO; 1-cm light path; room temperature.
This result was unexpected because the E445A mutant enzyme was previously reported to be missing heme b595 (35). To resolve differences between previous and current results relating to the presence of heme b595, the pyridine hemochromogen analysis was repeated. Both in this work and ref. 35 the amount of heme B extracted from cytochrome bd has been compared with the specific content of heme d in the enzyme. However, in ref. 35, evaluation of heme d content was based on the reduced minus air-oxidized difference spectrum of cytochrome bd, whereas in the current work, the absolute spectra of the reduced enzyme (using the red side of the 630-nm peak) have been used to quantify the amount of heme d. This method allows us to avoid the problems associated with (i) interference of heme b595 at the wavelengths traditionally used to quantify heme d and (ii) the different yields of the oxyferrous species of heme d in the air-oxidized preparations of the WT and mutant cytochromes bd (see Materials and Methods for details). In addition, the heme d content of the WT and mutant oxidases, determined by using Δε628–670 for the dithionite-reduced absolute spectra, is equal to the content of heme b558 of each enzyme (Δε561–580 of 21.0 mM-1·cm-1) for the dithionite-reduced minus air-oxidized difference spectrum (19). Hence, for the WT and mutant enzymes, the heme b558/heme d ratio is 1.0, and the total heme b/heme d ratio is 2.0. Heme b595 is present in the E445A mutant oxidase.
The conclusion that heme b595 is present in the E445A mutant but remains high-spin ferric even in the presence of excess of reductant is directly supported by the EPR spectroscopy. After the addition of dithionite, no EPR signals are observed with the WT oxidase, because all of the hemes have been reduced to the EPR-silent ferrous forms. However, with the E445A mutant, there is a clear g ≈6 signal from a high-spin ferric heme despite the presence of dithionite (Fig. 2). The signal at g = 6 is very fast relaxing and does not show any saturation with the microwave power as high as 100 mW at 13 K. Even at 4 K at such power only slight saturation is observed.
Fig. 2.

EPR spectra of air-oxidized (dotted line) and dithionite-reduced (solid line) cytochrome bd from E445A mutant enzyme. EPR conditions: microwave power, 2 mW; microwave frequency, 9.429 GHz; modulation amplitude, 12 G; temperature, 12° K. Sample conditions: 26.6 μM enzyme; 20 mM Mops-KOH and 40 mM sodium phosphate, pH 7.6; 0.05% sarcosyl. Data indicated by the solid line were obtained in the presence of 5 mM sodium dithionite.
E445A Substitution Strongly Inhibits Charge Translocation Coupled to the Oxidation of Cytochrome bd by Oxygen. Electrometric measurements of ΔΨ generation associated with the reaction of both the WT and mutant enzymes with O2 were made under the same experimental conditions. Typical recordings are shown in Fig. 3. The kinetics of the electrometric response of the fully reduced WT enzyme (Fig. 3, trace 1) can be reasonably modeled by three sequential reactions. The first reaction is an electrically silent lag phase with a rate constant (k1) dependent on the concentration of O2 (11). Under the present experimental conditions k1 = 7.4 × 104 s-1 (τ1 ≈14 μs). This reaction reflects oxygen binding to the enzyme (R → A transition), where state A denotes the formation of the heme d2+-O2 adduct. The second phase with k2 of 1.1 × 104 s-1 (τ2 ≈90 μs) is electrogenic and very similar to the value reported previously (11) corresponding to the transition of the ferrous-oxy intermediate to the oxoferryl state (heme d4+ = O2-) (A → F transition). In previous studies of the WT cytochrome bd (11), the electrogenic events stopped at this point, as expected for the three-electron-reduced enzyme (11, 41). However, the preparation used in this work was modified (see Materials and Methods) so as to retain bound quinol, which was demonstrated by HPLC analysis (data not shown). Hence, there are reducing equivalents available to carry the reaction beyond state F. Indeed, there is an additional third kinetic phase with k3 of 1.65 × 103 s-1 (τ3 ≈600 μs) corresponding to the conversion of the oxoferryl state F to the oxidized enzyme O (heme d3+--OH) and/or to the A state.
Fig. 3.

Generation of a membrane potential during the reaction of the reduced cytochrome bd with O2. Trace 1, WT enzyme. Trace 2, E445A mutant enzyme. Conditions: 100 mM Mops-KOH (pH 7.0), 10 μM N,N,N′,N′-tetramethyl-1,4-phenylenediamine, 50 mM glucose, 0.5 mg/ml catalase, 1.5 mg/ml glucose oxidase, and 1% CO. Reaction was started by a laser flash after 400 ms from the beginning of the injection of 100 μl of oxygen-saturated buffer ([O2] = 1.2 mM). The fit of the presented experimental curves gives the following parameters for the phases: Trace 1, R → A, τ ≈13.5 μs with zero amplitude; A → F, τ ≈90.9 μs, amplitude of -3.2 mV; F → O, τ ≈606 μs, amplitude of -1.6 mV. Trace 2: R → A, τ ≈13.5 μs with zero amplitude; A → F1, τ≈1.3 ms, amplitude of -0.36 mV; A → F2, τ≈12.5 ms, amplitude of -1.7 mV. (Inset) Generation of ΔΨ during the reaction of the reduced E445A mutant cytochrome bd with O2 on the longer time scale.
The amplitude of the F → O transition (≈1.6 mV) is about half of that for the A → F transition (≈3.2 mV). This finding is likely caused by the loss of the bound quinone from a portion of the enzyme population during the vesicle reconstitution procedure. Enzyme-lacking bound quinone will not have enough reducing equivalents to proceed beyond the F state.
The reaction of the ascorbate (N,N,N′,N′-tetramethyl-1,4-phenylenediamine)-reduced E445A mutant with O2 does not reveal any measurable microsecond phase of ΔΨ generation that would be the counterpart of that in the WT enzyme. As shown in Fig. 3 (trace 2 and Inset), the initial nonelectrogenic binding of O2 to the E445A enzyme is followed by a minor electrogenic phase (≈0.36 mV) with the k value of 7.6 × 102 s-1 (τ ≈1.3 ms). After this small electrogenic phase, a large electrogenic event with the rate constant of ≈80 s-1 (τ ≈12.5 ms) and amplitude of ≈1.7 mV is observed. Both phases probably reflect the conversion of A to F in different subpopulations of the enzyme. The conclusion that the final product of this reaction is the F state was confirmed by the absolute absorption spectrum taken after completion of the reaction (2 s) and showing a peak at 680 nm, diagnostic of the heme d oxoferryl species (42).
E445A Replacement Does Not Inhibit Electron Backflow from Heme d2+ to Hemes b558 and b595 Induced by Flashing Off CO from Heme d2+ in the Singly Reduced Cytochrome bd. The WT and E445A cytochrome bd each readily forms the CO adduct of the one-electron-reduced enzyme (heme d2+-CO). Previous data (11) with the WT enzyme showed that photodissociation of CO results in electron redistribution from heme d to heme b558 and heme b595, referred to as backflow electron transfer. Here, the backflow electron transfer reactions were compared for the WT and E445A mutant enzymes. Remarkably, the results are very similar.
Recombination of CO with heme d after photolysis of the mixed-valence cytochrome bd includes (i) CO rebinding to the heme d after the flash and (ii) the return of the electrons from the transiently reduced hemes b558 and b595 to heme d (11). Therefore, the spectrum of the CO rebinding in the CO mixed-valence enzyme contains the spectral changes induced by the binding of CO to heme d as well as the changes caused by the concomitant electron redistribution. The spectral contribution caused by oxidoreduction of the hemes (Fig. 4) was obtained by subtracting CO-binding spectrum of fully reduced enzyme from such spectrums of the one-electron-reduced WT (solid line) and mutant (dotted line) enzymes. This spectrum shows the reduction of heme d and corresponding oxidation of hemes b558 and b595 as CO rebinds to heme d.
Fig. 4.

Spectra of electron backflow relaxation after CO photolysis from the one-electron-reduced WT (solid line) and E445A mutant (dotted line) enzymes. The spectra were obtained by subtraction of the spectrum of CO rebinding to heme d (solid line of Fig. 1) from the total spectra of the CO recombining phase, the latter is a sum of the two processes (CO rebinding to heme d and intraprotein electron redistribution). The amplitudes of CO recombination phases of mixed valence enzymes were adjusted to match the amplitude of CO photolysis of fully reduced WT enzyme. Conditions are as in Fig. 1, except that no dithionite was added.
Using the appropriate extinction coefficients for hemes d and b558 (see Materials and Methods), it is possible to quantify the extent to which hemes d and b558 are reduced and oxidized, respectively, concomitant with CO rebinding. Approximately 25% of the heme d undergoes a redox change upon photolysis. If the amount of heme d that becomes reduced as CO rebinds is taken as 100%, the data show that only ≈20% of this reducing equivalent can be accounted for by the oxidation of heme b558 in the WT and ≈18% in the mutant enzyme. The remaining source of electrons returning to heme d must be heme b595, because the bound quinone has midpoint redox potential value that is much lower than heme b595 (39). Hence, in both enzymes, the photolysis of CO from heme d2+ in the one-electron-reduced enzyme results in partial reduction of heme b558 and heme b595 with major fraction (≈80%) of the internally redistributed electron residing on heme b595.
E445A Replacement Does Not Affect Membrane Potential Generation Coupled to Intramolecular Electron Redistribution Between Hemes d2+ and b558. Electron redistribution is also coupled to movement of protons across the membrane, resulting in ΔΨ generation of opposite sign compared with that generated by the forward reaction of the oxidase with O2 (11). Analysis of the kinetic traces (Fig. 5) of ΔΨ generation after CO photodissociation from the one-electron-reduced WT enzyme (solid line) reveals two electrogenic phases with τ1 ≈110 μs and τ2 ≈380 μs. ΔΨ generation is reversed at longer times, concurrent with the reversal of electron backflow (data not shown), and this process corresponds to the bimolecular reaction of CO recombination with heme d (11). It has been shown by the H2O/D2O solvent isotope effect (11) that charge separation in cytochrome bd is caused by transmembrane movements of protons rather than electrons.
Fig. 5.

Normalized electrometric responses of cytochrome bd coupled to electron backflow after CO photolysis from the one-electron-reduced WT (solid line) and E445A mutant (dashed line) enzymes. The exponential fit of presented experimental curves gives the following time constants and relative amplitudes: solid line, τ1 = 112 μs, A1 = 66%; τ2 = 385 μs, A2 = 34%; and dashed line, τ1 = 83 μs, A1 = 51%; τ2 = 278 μs, A2 = 49%. Conditions are as in Fig. 3, except that no O2 and N,N,N′,N′-tetramethyl-1,4-phenylenediamine were added, the latter allowed the enzyme to avoid reduction.
The electrogenic backflow response of the E445A mutant enzyme (Fig. 5, dashed line) can be also fitted by two exponentials, yielding time constants τ1 ≈80 μs and τ2 ≈280 μs. The values of these rate constants are very similar to those for the WT enzyme. This finding is in contrast to the ΔΨ generation during the reaction of O2 with the reduced enzymes, in which case the electrogenic processes observed upon oxidation of the E445A mutant enzyme is markedly slower than that of the WT.
Discussion
Heme b595 Is Present in the E445A Mutant. One conclusion from the current work is that the E445A mutant of E. coli cytochrome bd contains heme b595. Although the enzyme contains heme b595, the heme remains in the ferric form even in the presence of dithionite as it can be seen by EPR spectroscopy. The dramatically perturbed redox properties of heme b595 correlate with the fact that the mutant enzyme does not support respiration (35). Remarkably, heme b595 can be reduced transiently by photolysis of the CO adduct of heme d2+, either in the one- or two-electron-reduced forms of the E445A mutant enzyme (Figs. 1 and 4). It is of interest to note cytochrome bd is not the only example of a protein that contains a heme not reducible by dithionite. Nonreducibility of a high-spin heme b by dithionite also has been observed with many catalases in which the proximal ligand of heme b is tyrosine rather than histidine (43).
Previous studies claiming that the E445A mutant enzyme has only one heme B per heme d were incorrect. This error in quantitation in previous work was caused by an error in quantifying the amount of heme d in the mutant enzyme. The contribution of heme b595 and the ferrous oxy species to the absorbance of the enzyme at the wavelengths used to measure heme d in the reduced minus oxidized difference spectrum are significant and are not the same for the WT and mutant enzymes. These differences are minimized in the current work by using the absolute spectra of the dithionite-reduced enzymes for quantifying heme d. In the current work, there is no doubt that heme b595 is present (albeit not reduced by dithionite). It is also clear that the E445A mutation has a very dramatic effect selectively on this heme component of the enzyme, as previously concluded.
The Role of Heme b595 in Catalytic Turnover. When heme b595 is locked into the ferric form, the enzyme is incapable of rapid catalysis. The reaction of O2 with the two-electron-reduced E445A mutant is ≈100-fold slower to form the F species. Formation of the oxoferryl species requires four electrons to split the O—O bond, along with at least one proton. Because heme b595 remains in the ferric form in reduced enzyme, there are not enough electrons present in the metal centers to rapidly catalyze the reaction. This reaction would normally use one electron each from heme b595 and heme b558 as well as two electrons from heme d. Because it is very likely that sufficient numbers of electrons are present in the form of reduced quinol, it can be concluded either that rapid catalysis with O2 specifically requires the ferrous form of heme b595 and/or that the reaction requires the proton from E445. Future work is needed to address this issue.
The Contribution of Heme b595 to the Optical and Electrogenic Measurements of Electron Backflow. The backflow optical experiments with the WT enzyme show that upon photolysis of the CO adduct to the one-electron-reduced enzyme the electron from heme d is redistributed predominantly to heme b595. If the amount of transiently oxidized heme d in the photolyzed one-electron-reduced enzyme is taken as 100%, then ≈80% of this reducing equivalent ends up on heme b595 and only ≈20% on heme b558. The same is observed with the one-electron-reduced E445A mutant, indicating that the midpoint potentials of the three hemes are not altered under these conditions by the mutation. Because, in the dithionite-reduced E445A mutant, heme b558 is reduced but heme b595 remains oxidized, the backflow experiments with this form of the mutant provide an opportunity to observe the electrogenic effects of electron transfer from heme d to heme b595 without the complication of concurrent backflow to heme b558. The results show that ΔΨ generation after CO photolysis from the one-electron-reduced E445A enzyme decreases proportionally with reduction of heme b558. In the two-electron-reduced state of the mutant, the amplitude of ΔΨ generation is close to zero although the electron backflow to heme b595 is still present (Fig. 1B). These data indicate that the ΔΨ after CO dissociation from heme d in the one-electron-reduced enzyme is caused entirely by electron redistribution between heme d and heme b558 and protonation events associated with this process. In contrast, redox equilibration between heme d and heme b595, which is the major event observed optically, is electrically silent. It is concluded that the two hemes are at the same electrical depth in the membrane and that electron transfer between heme d and heme b595 is not linked to electrogenic proton movement.
Why Is Heme b595 Not Reduced by Dithionite in the E445A Mutant? A Plausible Model of Electron and Proton Transfer Pathways in Cytochrome bd. The behavior of heme b595 in the E445A mutant is very peculiar. The heme remains in the oxidized (ferric) form even upon the addition of dithionite, which readily reduces the other two hemes of the oxidase (heme b558 and heme d). At the same time photolysis of the CO adduct does result in transient reduction of heme b595. The spectral changes match those previously reported for heme b595 in the WT enzyme (39, 40), implying that the heme itself is likely in a high-spin configuration, as in the WT enzyme, and that its ligation also is probably unchanged by the mutation. Hence, the data make it unlikely that perturbation of heme b595 is the result of the E445A mutation altering the axial ligand to the heme. The fact that part of heme b595 is transiently reduced upon photolysis of the heme d2+-CO adduct suggests that the mutation does not specifically block the reduction of heme b595 but, rather, prevents the complete simultaneous reduction of both heme d and heme b595.
A plausible model that is consistent with all of the data is shown schematically in Fig. 6. The proposed scheme of electron and proton transfer pathways in cytochrome bd can explain the two apparently conflicting observations of behavior of the E445A mutant: resistance of heme b595 toward reduction by dithionite and transient its reduction upon photolysis of CO from heme d in the mixed-valence enzyme.
Fig. 6.

Possible scheme of electron and proton transfer pathways in cytochrome bd oxidase. The mutation in the E445 residue prevents the complete two-electron reduction of the di-heme site by dithionite. The proton access to site Xp is pictured as being from the periplasmic side of the membrane (P-side). If this is the case, it is unlikely that this proton is used in the reaction catalyzed at the enzyme active site, but is re-released upon reoxidation of the hemes. N-side, negative side of the membrane.
It is proposed that there are two protonatable groups, denoted 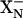 and
and  . The protonation states of both of these groups are influenced by the redox state of hemes d and b595 (Fig. 6). It is postulated that the pKa of
. The protonation states of both of these groups are influenced by the redox state of hemes d and b595 (Fig. 6). It is postulated that the pKa of 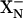 is higher than the pKa of
is higher than the pKa of 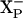 and that the
and that the  group is in protonic equilibrium with the bulk aqueous phase on the negative side of the membrane via a proposed proton-conducting channel. Proton transfer between the negative side of the membrane and the
group is in protonic equilibrium with the bulk aqueous phase on the negative side of the membrane via a proposed proton-conducting channel. Proton transfer between the negative side of the membrane and the  site through this channel, normal to the plane of the membrane, is proposed to result in generation of ΔΨ.
site through this channel, normal to the plane of the membrane, is proposed to result in generation of ΔΨ.
Upon reduction of both the WT and the mutant enzymes, the first electron transferred from heme b558 to the binuclear site is accompanied by the protonation of the 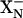 site (
site (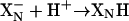 ). This electron is shared between heme d and heme b595 (80%/20%) and this distribution is the same in the E445A mutant and the WT oxidases. The second electron entering the binuclear site completes reduction of the di-heme site in the WT oxidase and is linked to the uptake of a second proton by another shared proton-accepting group,
). This electron is shared between heme d and heme b595 (80%/20%) and this distribution is the same in the E445A mutant and the WT oxidases. The second electron entering the binuclear site completes reduction of the di-heme site in the WT oxidase and is linked to the uptake of a second proton by another shared proton-accepting group,  . The E445A mutation is proposed to specifically block protonation of the
. The E445A mutation is proposed to specifically block protonation of the 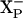 site.
site.
The consequence of preventing the protonation of the second postulated site (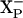 ) is that the reduction of the binuclear site in the E445A mutant enzyme by the second electron is not possible because the electron cannot be compensated by uptake of the second proton.
) is that the reduction of the binuclear site in the E445A mutant enzyme by the second electron is not possible because the electron cannot be compensated by uptake of the second proton.
There are no data to discriminate whether the proton taken up by  comes from the negative or periplasmic side of the membrane. In principle, the proton delivered to
comes from the negative or periplasmic side of the membrane. In principle, the proton delivered to 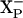 could come through the same channel as does the proton taken up by
could come through the same channel as does the proton taken up by 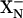 . In this case, the proton delivery would be electrogenic and, presumably the proton would be used by the chemistry of forming water at the active site. If E445 is embedded in the membrane, it is reasonable to postulate that E445 is the group
. In this case, the proton delivery would be electrogenic and, presumably the proton would be used by the chemistry of forming water at the active site. If E445 is embedded in the membrane, it is reasonable to postulate that E445 is the group 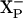 . On the other hand, it is possible that E445 is not
. On the other hand, it is possible that E445 is not 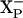 itself but is, rather, located on the surface and is required for the transfer of the proton to
itself but is, rather, located on the surface and is required for the transfer of the proton to 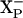 . In this case, the identity of the
. In this case, the identity of the 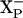 site also needs to be established.
site also needs to be established.
The midpoint redox potential values of hemes d and b595 are pH-dependent (44), consistent with the proposed model. Presumably, the protons taken up by one or both of these two groups are subsequently used to combine with O2 in the catalytic reaction to form H2O.
The addition of CO to the dithionite-reduced E445A mutant increases the midpoint redox potential of heme d so that the one electron equilibrated between heme d and heme b595 is now entirely found on heme d (>99%). Photolysis to remove CO results in the backflow of ≈20% of the reducing equivalent transiently from heme d to heme b595. When backflow between hemes d and b595 occurs, the redistributed electron is already compensated by the proton that was taken up by the 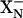 site. Hence this electron transfer is not associated with proton uptake and is nonelectrogenic.
site. Hence this electron transfer is not associated with proton uptake and is nonelectrogenic.
The results obtained upon photolysis of heme d2+-CO in the one-electron-reduced mutant enzyme are also explained by this model. For the E445A mutant, the situation with the one-electron-reduced enzyme is identical to that described above for the two-electron-reduced enzyme, except that heme b558 is also oxidized. With the WT enzyme, photolysis results in the redistribution of the electron that was initially on heme d to all three heme components. Of the 25% of heme d that is transiently oxidized, only approximately one-fifth of the transferred electron (5% of the single reducing equivalent present) ends up on heme b558. However, electron transfer from heme d to heme b558 is coupled to deprotonation of the group near heme d (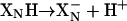 ), which makes this step electrogenic. The midpoint potential of heme b558 is pH-dependent. This finding indicates that the reduction of this heme is also accompanied by protonation. In our previous work (11) we showed that this proton uptake is electrogenic and there is a group (denoted
), which makes this step electrogenic. The midpoint potential of heme b558 is pH-dependent. This finding indicates that the reduction of this heme is also accompanied by protonation. In our previous work (11) we showed that this proton uptake is electrogenic and there is a group (denoted  in Fig. 6) at the periplasmic side of the membrane that picks up and releases a proton as heme b558 is reduced and oxidized.
in Fig. 6) at the periplasmic side of the membrane that picks up and releases a proton as heme b558 is reduced and oxidized.
In summary, these data point to a functional coupling between heme d and heme b595. Not only are these two hemes very close to each other, but their electrochemical properties are strongly linked to neighboring protonatable groups. These groups are critical to facilitating the chemistry of water formation and to coupling the chemical reaction to the generation of a membrane potential.
Acknowledgments
This work was supported by Biocentrum Helsinki, the Sigrid Juselius Foundation, and the Academy of Finland (M.I.V.); the Russian Foundation for Basic Research (V.B.B.), Howard Hughes Medical Institute International Scholar Award 55000320 (to A.A.K.), and National Institutes of Health Grant HL16101 (to R.B.G.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: ΔΨ, transmembrane electric potential difference.
References
- 1.Baughn, A. D. & Malamy, M. H. (2004) Nature 427, 441-444. [DOI] [PubMed] [Google Scholar]
- 2.Poole, R. K. & Cook, G. M. (2000) Adv. Microb. Physiol. 43, 165-224. [DOI] [PubMed] [Google Scholar]
- 3.Trumpower, B. L. & Gennis, R. B. (1994) Annu. Rev. Biochem. 63, 675-716. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson-Miller, S. & Babcock, G. T. (1996) Chem. Rev. 96, 2889-2908. [DOI] [PubMed] [Google Scholar]
- 5.Brzezinski, P. & Larsson, G. (2003) Biochim. Biophys. Acta 1605, 1-13. [DOI] [PubMed] [Google Scholar]
- 6.Puustinen, A., Finel, M., Haltia, T., Gennis, R. B. & Wikstrom, M. (1991) Biochemistry 30, 3936-3942. [DOI] [PubMed] [Google Scholar]
- 7.Kita, K., Konishi, K. & Anraku, Y. (1984) J. Biol. Chem. 259, 3375-3381. [PubMed] [Google Scholar]
- 8.Miller, M. J. & Gennis, R. B. (1985) J. Biol. Chem. 260, 14003-14008. [PubMed] [Google Scholar]
- 9.Kolonay, J. F., Jr., & Maier, R. J. (1997) J. Bacteriol. 179, 3813-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertsova, Y. V., Bogachev, A. V. & Skulachev, V. P. (1997) FEBS Lett. 414, 369-372. [DOI] [PubMed] [Google Scholar]
- 11.Jasaitis, A., Borisov, V. B., Belevich, N. P., Morgan, J. E., Konstantinov, A. A. & Verkhovsky, M. I. (2000) Biochemistry 39, 13800-13809. [DOI] [PubMed] [Google Scholar]
- 12.Junemann, S. (1997) Biochim. Biophys. Acta 1321, 107-127. [DOI] [PubMed] [Google Scholar]
- 13.Borisov, V. B. (1996) Biochemistry (Moscow) 61, 786-799. [PubMed] [Google Scholar]
- 14.Mogi, T., Tsubaki, M., Hori, H., Miyoshi, H., Nakamura, H. & Anraku, Y. (1998) J. Biochem. Mol. Biol. Biophys. 2, 79-110. [Google Scholar]
- 15.Osborne, J. P. & Gennis, R. B. (1999) Biochim. Biophys. Acta 1410, 32-50. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, J., Barquera, B. & Gennis, R. B. (2004) FEBS Lett. 561, 58-62. [DOI] [PubMed] [Google Scholar]
- 17.Hill, J. J., Alben, J. O. & Gennis, R. B. (1993) Proc. Natl. Acad. Sci. USA 90, 5863-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krasnoselskaya, I., Arutjunjan, A. M., Smirnova, I., Gennis, R. & Konstantinov, A. A. (1993) FEBS 327, 279-283. [DOI] [PubMed] [Google Scholar]
- 19.Tsubaki, M., Hori, H., Mogi, T. & Anraku, Y. (1995) J. Biol. Chem. 270, 28565-28569. [DOI] [PubMed] [Google Scholar]
- 20.Borisov, V., Arutyunyan, A. M., Osborne, J. P., Gennis, R. B. & Konstantinov, A. A. (1999) Biochemistry 38, 740-750. [DOI] [PubMed] [Google Scholar]
- 21.Vos, M. H., Borisov, V. B., Liebl, U., Martin, J.-L. & Konstantinov, A. A. (2000) Proc. Natl. Acad. Sci. USA 97, 1554-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borisov, V. B., Sedelnikova, S. E., Poole, R. K. & Konstantinov, A. A. (2001) J. Biol. Chem. 276, 22095-22099. [DOI] [PubMed] [Google Scholar]
- 23.Borisov, V. B., Liebl, U., Rappaport, F., Martin, J.-L., Zhang, J., Gennis, R. B., Konstantinov, A. A. & Vos, M. H. (2002) Biochemistry 41, 1654-1662. [DOI] [PubMed] [Google Scholar]
- 24.Hata-Tanaka, A., Matsuura, K., Itoh, S. & Anraku, Y. (1987) Biochim. Biophys. Acta 893, 289-295. [DOI] [PubMed] [Google Scholar]
- 25.Poole, R. K. & Williams, H. D. (1987) FEBS Lett. 217, 49-52. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi, K., Tagawa, S. & Mogi, T. (1999) Biochemistry 38, 5913-5917. [DOI] [PubMed] [Google Scholar]
- 27.Hill, B. H. (1994) J. Biol. Chem. 269, 2419-2425. [PubMed] [Google Scholar]
- 28.Konstantinov, A. A., Siletsky, S., Mitchell, D., Kaulen, A. & Gennis, R. B. (1997) Proc. Natl. Acad. Sci. USA 94, 9085-9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jasaitis, A., Verkhovsky, M. I., Morgan, J. E., Verkhovskaya, M. L. & Wikstrom, M. (1999) Biochemistry 38, 2697-2706. [DOI] [PubMed] [Google Scholar]
- 30.Siletsky, S., Kaulen, A. D. & Konstantinov, A. A. (1999) Biochemistry 38, 4853-4861. [DOI] [PubMed] [Google Scholar]
- 31.Morgan, J. E., Verkhovsky, M. I., Palmer, G. & Wikstrom, M. (2001) Biochemistry 40, 6882-6892. [DOI] [PubMed] [Google Scholar]
- 32.Einarsdottir, O., Szundi, I., Van Eps, N. & Sucheta, A. (2002) J. Inorg. Biochem. 91, 87-93. [DOI] [PubMed] [Google Scholar]
- 33.Ruitenberg, M., Kannt, A., Bamberg, E., Fendler, K. & Michel, H. (2002) Nature 417, 99-102. [DOI] [PubMed] [Google Scholar]
- 34.Bloch, D., Belevich, I., Jasaitis, A., Ribacka, C., Puustinen, A., Verkhovsky, M. I. & Wikstrom, M. (2004) Proc. Natl. Acad. Sci. USA 101, 529-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, J., Hellwig, P., Osborne, J. P., Huang, H. W., Moenne-Loccoz, P., Konstantinov, A. A. & Gennis, R. B. (2001) Biochemistry 40, 8548-8556. [DOI] [PubMed] [Google Scholar]
- 36.Miller, M. J. & Gennis, R. B. (1986) Methods Enzymol. 126, 87-94. [DOI] [PubMed] [Google Scholar]
- 37.Berry, E. A. & Trumpower, B. L. (1987) Anal. Biochem. 161, 1-15. [DOI] [PubMed] [Google Scholar]
- 38.Junemann, S., Wrigglesworth, J. M. & Rich, P. R. (1997) Biochemistry 36, 9323-9331. [DOI] [PubMed] [Google Scholar]
- 39.Koland, J. G., Miller, M. J. & Gennis, R. B. (1984) Biochemistry 23, 1051-1056. [DOI] [PubMed] [Google Scholar]
- 40.Lorence, R. M., Koland, J. G. & Gennis, R. B. (1986) Biochemistry 25, 2314-2321. [DOI] [PubMed] [Google Scholar]
- 41.Hill, B. C., Hill, J. J. & Gennis, R. B. (1994) Biochemistry 33, 15110-15115. [DOI] [PubMed] [Google Scholar]
- 42.Kahlow, M. A., Zuberi, T. M., Gennis, R. B. & Loehr, T. M. (1991) Biochemistry 30, 11485-11489. [DOI] [PubMed] [Google Scholar]
- 43.Deisseroth, A. & Dounce, A. L. (1970) Physiol. Rev. 50, 319-375. [DOI] [PubMed] [Google Scholar]
- 44.Lorence, R. M., Miller, M. J., Borochov, A., Faiman-Weinberg, R. & Gennis, R. B. (1984) Biochim. Biophys. Acta 790, 148-153. [DOI] [PubMed] [Google Scholar]