

Abstract
Background
The arteriovenous fistula (AVF) is the preferred form of hemodialysis access for patients with chronic kidney disease. However, AVFs are associated with significant problems including high incidence of both early and late failures, usually attributed to inadequate venous arterialization and neointimal hyperplasia, respectively. Understanding the cellular basis of venous remodeling in the setting of AVF could provide targets for improving AVF patency rates.
Methods and Results
A novel vascular smooth muscle cell (VSMC) lineage tracing reporter mouse, Myh11‐Cre/ERT2‐mTmG, was used to track mature VSMCs in a clinically relevant AVF mouse model created by a jugular vein branch end to carotid artery side anastomosis. Prior to AVF surgery, differentiated medial layer VSMCs were labeled with membrane green fluorescent protein (GFP) following tamoxifen induction. Four weeks after AVF surgery, we observed medial VSMC layer thickening in the middle region of the arterialized vein branch. This thickened medial VSMC layer was solely composed of differentiated VSMCs that were GFP+/MYH11+/Ki67−. Extensive neointimal hyperplasia occurred in the AVF region proximal to the anastomosis site. Dedifferentiated VSMCs (GFP+/MYH11−) were a major cellular component of the neointima. Examination of failed human AVF samples revealed that the processes of VSMC phenotypic modulation and intimal hyperplasia, as well as medial VSMC layer thickening, also occurred in human AVFs.
Conclusions
We demonstrated a dual function for mature VSMCs in AVF remodeling, with differentiated VSMCs contributing to medial wall thickening towards venous maturation and dedifferentiated VSMCs contributing to neointimal hyperplasia. These results provide valuable insights into the mechanisms underlying venous adaptations during AVF remodeling.
Keywords: arteriovenous fistula, neointima, remodeling, vascular smooth muscle differentiation, vein, venous maturation
Subject Categories: Stenosis, Vascular Disease
Introduction
Hemodialysis vascular access failure is the primary cause of morbidity in patients with end‐stage renal disease and contributes to the economic burden of this patient population. Vascular access complications account for nearly 30% of the hospitalization of hemodialysis patients in the United States, with an annual cost of $1 billion.1 The native arteriovenous fistula (AVF) is the preferred form of dialysis access but is associated with a high early failure rate. Some studies have emphasized that up to 60% of AVFs do not mature and of the remainder that do mature, 35% fail after 2 years.1, 2
The adaptive wall remodeling in response to increased blood flow in the venous segment of AVFs is associated with venous wall dilation and thickening. This arterialization of venous segments—also called maturation—is the desired clinical outcome.3, 4 The conditions and mechanisms that may lead to adequate maturation are poorly understood. It is thought to require the partial loss of the internal elastic lamina to facilitate medial reorganization and deposition of new layers of vascular smooth muscle cells (VSMCs) and extracellular matrix with the resultant characteristic outward remodeling.4 More recent studies have focused on the characterization of animal models and molecular pathways of arterial versus venous specification that may recapitulate some of the characteristic features associated with outward vein remodeling in humans.5, 6 However, the accumulated data are clearly limited.
In addition to lack of maturation, a major cause of primary and secondary AVF failure is the development of a hyperplastic response leading to a dramatic increase in neointimal cell deposition.7 Most commonly, neointimal hyperplasia after AVF creation is found near the anastomotic site but can also be observed in other segments of the draining vein.8, 9 In contrast to restenosis associated with preocclusive atherosclerotic arteries after angioplasty and stenting, the vessels involved are in many cases free from atherosclerotic plaques.10 Histologically, the neointimal lesions in AVFs consist of proliferating and migrating vascular cells that are presumed to be of many different origins including mature VSMCs, adventitial progenitor cells, or bone marrow stem cells.11, 12, 13 In a study using transgenic rats constitutively overexpressing the green fluorescent protein (GFP), it was suggested that neointimal cells originate from the local resident cells in the venous limb of the fistula.14 In other cases, a role for adventitial fibroblasts and cells derived from the arterial side of the artery has been suggested.15, 16 Overall, the contribution of specific cell types including VSMCs through specific cell lineage tracing has not been documented. The significance of such studies is that the determination of the different cell types associated with AVF may suggest specific cell targeting for future therapeutic approaches.
In the present study, we evaluated the contribution of mature VSMCs to AVF maturation and failure using a recently described murine model of AVF with a similar anatomical configuration (vein end to artery side) as typically used in humans. We crossed a transgenic mouse line expressing CreERT2 recombinase under the control of the promoter of Myh11, the most smooth muscle cell (SMC)–specific gene,17 with mTomato/GFP reporter mouse to achieve Myh11‐Cre/ERT2‐mTmG reporter strain. In this novel reporter mouse, only mature differentiated VSMCs possess Cre recombinase activity following tamoxifen (TMX) induction, thereby are labeled with membrane GFP, whereas other cell types remain labeled with mT. We then surgically created an AVF in these transgenic mice and analyzed the anastomosed vein (AVF) using fluorescence microscopy. Our results indicate both outward remodeling and neointimal hyperplasia with a significant contribution of VSMCs to maturation and neointimal hyperplasia, a finding that was supported by immunohistochemical analysis in human tissue sections of failed AVFs.
Methods
Generation of Myh11‐Cre/ERT2‐mTmG Reporter Mice and TMX Induction of Cre Recombinase
Protocols of the related animal breeding and mouse AVF model were approved by the Institutional Animal Care and Use Committee. In order to fate map mature VSMCs during AVF placement and at failure, mice carrying Myh11‐Cre/ERT2 (+/−)18 were bred with ROSAmT/mG mice (Jackson Laboratory, Bar Harbor, ME) to generate Myh11‐Cre/ERT2‐mTmG mice. TMX injection (33 μg/g body weight in 100 μL sunflower oil) was performed intraperitoneally to specifically activate Cre recombinase in mature VSMCs. The reporter mice were viable and born at the expected mendelian ratio. Mice at 10 to 12 weeks of age were used for AVF surgery 3 weeks after TMX injection.
Mouse AVF Model
The AVF mouse model in which the end of the jugular vein branch was ligated to the side of the carotid artery recapitulates the radiocephalic or brachiocephalic AVF performed in the clinic19 (Figure 1A and 1B). A total of 39 mice (C57/BL6 and mTmG reporter mice) were used in this study, 26 of which (66%) underwent successful AVF surgery and were subjected to further pathological evaluation. Briefly, mice were anesthetized with 1.5% isoflurane followed by the creation of a skin incision on the ventral midline of the neck under the microscope. The dorsomedial distal branch of the right external jugular vein was dissected with a vascular clamp placed proximally and transected distally. The right common carotid artery was dissected and clamped both proximally and distally and an incision (1 mm) was created longitudinally in the carotid artery. The end of the dissected vein was anastomosed to the incision of carotid artery. Immediately after AVF construction, mice were injected with heparin (0.2 IU/g body weight). Success of AVF construction was initially determined by visualization of a sudden increased blood flow in the ligated vein from the artery side and further confirmed by histopathological examination of the outward remodeling and neointima formation. Neointima formation in the proximal region of AVF were quantitated using ImageJ software as previously described,20 and the unit for the absolute area of the neointima and lumen is expressed as pixel square.
Figure 1.
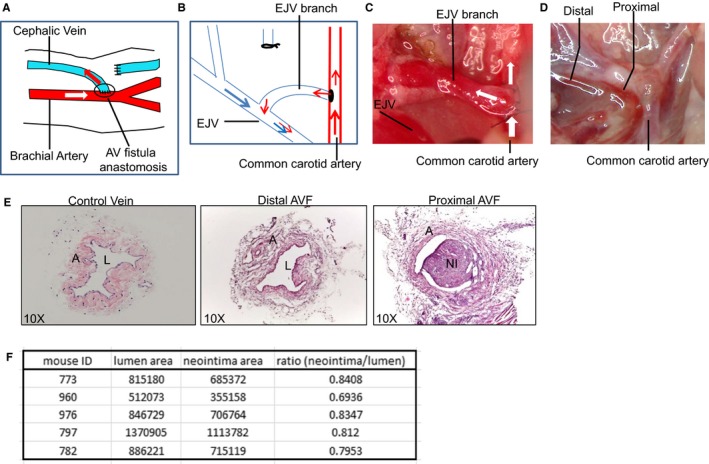
Arteriovenous (AV) fistula (AVF) model construction and histological characterization. A, Schematic illustrating the creation of a brachiocephalic AVF in humans. Most commonly, an AVF is created through anastomosis of the end of a cephalic vein branch to the side of the brachial artery. Arrows indicate the direction of blood flow. B, Schematic for mouse AVF construction, which recapitulates human AVF as shown in (A). C, Mouse AVF is constructed by anastomosing the end of one branch of the external jugular vein to the side of the common carotid. White arrows indicate blood flow direction. D, Mouse AVF after saline perfusion 4 weeks after AVF surgery. The indicated distal and proximal regions in the anastomosed vein were selected for histological examination. E, Representative hematoxylin and eosin (HE) staining for the control unligated jugular vein branch and the AVF samples (distal and proximal regions), which were isolated as depicted in (D). n=5. F, Quantitation of neointima formation at the proximal AVF region as depicted in (E). n=5. Unit for absolute area is expressed as pixel square. A indicates adventia; EJV, external jugular vein; L, lumen; NI, neointima.
Mouse Tissue Harvesting and Processing
AVF samples at 1, 2, and 4 weeks after AVF surgery were initially isolated to optimize the time point for observing adequate venous outward remodeling and neointima formation. We found that this was best achieved 4 weeks after AVF placement and most lineage tracing studies were performed at this time point. Tissues were fixed in 4% paraformaldehyde at 4°C for 4 hours before embedded in either optimal cutting temperature compound or paraffin for histology. Samples were first immersed in specimen processing HistoGel (HG‐4000‐012, Thermo Fisher Scientific, Waltham, MA) to maintain the shape of soft veins before paraffin processing and embedment. Eight micrometer cross sections were prepared continuously from the distal region of the ligated vein to the anastomosis site.
Immunofluorescence Staining
The cryosections were dried at room temperature for 30 minutes, baked at 55°C for 20 minutes, cooled down at room temperature for 10 minutes, and finally immersed in PBS Tween (PBST) for 5 minutes to remove optimal cutting temperature compound. To retain the mTmG fluorescence, trypsin‐mediated antigen retrieval (REF TA‐015–TR; Lab Vision Corporation, Fremont, CA) was employed. Sections were incubated with 1x trypsin at 37°C for 10 minutes for antigen retrieval, and a cool stopping buffer was used to stop the reaction. Sections were then permeabilized by 0.5% Triton X‐100, blocked with DAKO blocking buffer (Carpinteria, CA), and probed with primary antibody at 4°C overnight. After 3 washes with 0.1% PBST, sections were finally incubated with Alex Fluor conjugated secondary antibody (1:500; Life Technologies, goat anti‐rabbit 647, A21245 for MYH11 and CNN1; donkey anti‐mouse 647, A31571 for ACTA2; goat anti‐rabbit 488, A11034 for Ki67) for 1 hour at room temperature. After 3 washes with PBST, sections were mounted with anti‐fade mounting medium, which contains 4′,6‐diamidino‐2‐phenylindole (DAPI; H‐1200, VECTASHIELD, Vector Laboratories, Burlingame, CA). Fluorescent signal was captured using a confocal microscope (DMI 4000B; Leica Microsystems, Wetzlar, German) and processed using ImageJ under the same conditions. Information of all the primary antibodies used were as follows: MYH11 (1/200, ALFA AESAR Co, #J64817); ACTA2 (1/500, #M0851; DAKO); CNN1 (1/300, #13938‐AP; Proteintech); Ki67 (1/500, #Ab15580; Abcam); and CD45 (1/20 per instruction, #550539; BD Pharmingen).
For paraffin‐embedded sections, antigen retrieval was performed using a pressure cooker with high pressure for 10 minutes in 1x target retrieval solution (pH=6; Dako‐S1699). The remaining procedure for staining was performed as described above.
Determination of VSMC Layer Thickness, Membrane GFP (+), MYH11 (+), and Ki67 (+) Cells in Neointima
Images containing neointima proximal to the anastomosis site were captured by confocal microscopy and analyzed using ImageJ software. The neointima area was defined as the area inside of the internal elastic lamina. Total cell number in the neointima for each image was counted according to the number of DAPI‐labeled nuclei. Membrane GFP (+), MYH11 (+), and Ki67 (+) cells were quantified automatically using ImageJ and the percentage of each individual cell type in the neointima was calculated. Media thickness measurements were based on GFP (+)/MYH11 (+) staining in both ligated and nonligated control veins. At least 10 images from 4 animals per group were examined.
Human Sample Harvesting, Tissue Processing and Histology, and Immunofluorescence Staining
Human AVF samples were deidentified discarded segments from patients undergoing surgical revision of failed AVFs at Albany Medical College (institutional review board protocol IRB# 3733). AVF samples were dissected of any adherent fibrous or fatty acid tissue before immediate preparation for histological studies. Tissue sections (7–10 μm) were cut from prepared optimal cutting temperature blocks using a Leica CM1850 cryostat and transferred to Colorfrost Plus Microscope slides (Thermo Fisher Scientific) and stored at −80°C until use. For hematoxylin and eosin staining, slides were air‐dried briefly, fixed in 10% formalin, then stained with a progressive hematoxylin and eosin staining kit (Cardinal Health, Dublin, OH). For elastin/collagen staining, tissues were stained with Thermo Scientific Richard‐Allen Scientific Chromaview–Advanced Testing Elastic Stain kit according to the manufacturer's instructions. Briefly, the tissues were fixed in 10% formalin for 30 minutes, immersed in prepared working elastic stain solution for 30 minutes, rinsed briefly in running tap water, then decolorized in working differentiating solution. The tissues were monitored under a microscope to prevent overdifferentiation. Sections were rinsed in water and immersed in sodium thiosulfate solution for 1 minute, rinsed again in water, and then immersed in van Gieson stain solution for 1 minute. Tissues were dehydrated with two changes of anhydrous alcohol, and cleared with 3 changes of xylene. Both hematoxylin and eosin and elastin slides were coverslipped with VectaMount permanent mounting media (Vector Laboratories). For immunofluorescence, the sections were air‐dried, fixed with ice‐cold acetone for 10 minutes then outlined with a liquid blocker super PAP pen (Cardinal Health). Slides were rinsed with PBS/0.1% Triton X‐100 (PBST), blocked with 0.5% Fish gelatin (Sigma) for 30 minutes, incubated with primary antibody overnight at 4°C, washed in PBST, incubated with secondary antibody for 1 hour at room temperature, washed again, and then coverslipped with VectaShield Antifade Mounting Medium (Vector Laboratories) with DAPI. The coverslips were sealed with clear nail polish and the slides were stored at 4°C until viewing. Primary antibodies included MYH11 as above, ACTA2 (Proteintech, Rosemont, IL), and CNN1 (Sigma‐Aldrich, St Louis, IL). Secondary antibodies were Alexa Fluor goat anti‐rabbit 488 and chicken anti‐mouse 594 (Invitrogen, Eugene, OR).
Statistical Analysis
Statistical analysis was performed using the statistical package included in GraphPad. Differences between groups were evaluated with Student unpaired t test (2‐tailed). Results shown are means±SD. P values of <0.05 were considered significant.
Results
AVF Model Construction and Histological Characterization
The mouse AVF used in the present study was created through anastomosis of the end of a jugular vein branch with the side of the carotid artery4 (Figure 1B), an anatomical configuration similar to the human brachiocephalic AVF (Figure 1A). Patency of the AVF can be initially determined by visualization of the increased blood flow from the artery side to the ligated vein branch immediately after surgery (Figure 1C). Four weeks after AVF creation, a pulsatile blood flow, which was presumably arterial blood flow from the carotid artery, was clearly observed flowing through the ligated vein (data not shown), indicating the successful AVF construction. At this time point, we frequently observed a neointima‐like structure extending from the anastomosis site to the middle region of the ligated vein after left ventricle saline perfusion (Figure 1D).
Hematoxylin and eosin staining of sections derived from the distal and proximal regions of the mouse AVF was performed for initial histological and morphological characterization. The control jugular vein branch was soft with much fewer cells sporadically distributed all around the vessel wall compared with the anastomosed vein (Figure 1E). Evident outward remodeling characterized by medial and adventitial thickening occurred in the proximal and distal region of the AVF, with a neointima in the proximal region but none in the distal (Figure 1E and 1F). Compared with control veins, hematoxylin and eosin staining also revealed marked accumulation of matrix deposition and a considerably increased cell population in the ligated vein both distally and proximally to the anastomosis site, indicating that venous cells are undergoing phenotypic transition to a “synthetic “and “proliferative” state towards arterialization, in response to the arterial homodynamic environment.
Characterization of VSMC Differentiation Program in Mouse AVF Model
Because extensive neointima formation and outward remodeling occurred 4 weeks after surgery in the proximal region of the mouse AVF, we examined VSMC differentiation program in this region by immunofluorescence staining of contractile proteins. We found that adventitial cells did not express any VSMC contractile proteins such as ACTA2, CNN1, or MYH1117, 21, 22 (Figure 2A through 2D), suggesting that adventitial remodeling in the setting of AVF may not involve fibroblasts to myofibroblasts or VSMCs transition. Neointimal cells expressed relatively low levels of ACTA2 and CNN1, two early but less specific VSMC markers, compared with control veins (Figure 2A and 2B). We observed little to no expression of MYH11, the most specific VSMC marker, in the neointima17 (Figure 2C). Furthermore, most of the neointimal cells were positive for Ki‐67, indicating that these cells were proliferative (Figure 2D). Together, these results suggest that—similar to arterial remodeling—VSMC phenotypic switching from the quiescent/differentiated state to a dedifferentiated/proliferative mode may also be the underlying mechanism for neointima formation in the context of AVF.
Figure 2.
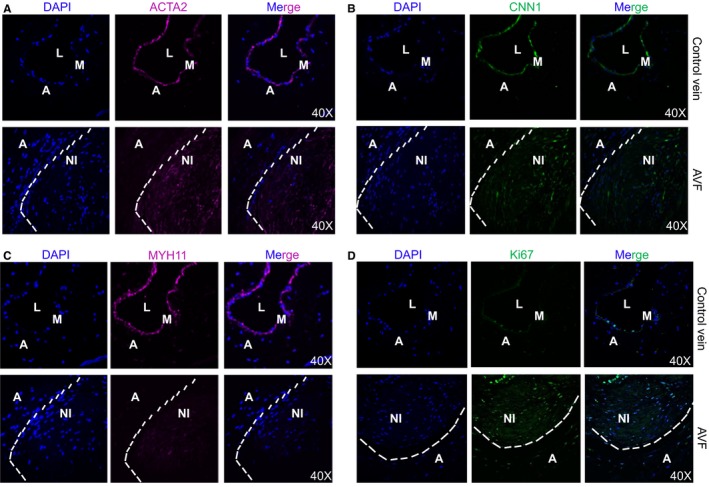
Characterization of vascular smooth muscle cell (VSMC) differentiation program at 4 weeks of arteriovenous fistula (AVF) construction. Immunofluorescent staining for the indicated VSMC contractile proteins including ACTA2 (A), CNN1 (B), and MYH11 (C) and cell proliferation marker Ki‐67 (D) in the control unligated jugular vein branch and the proximal AVF samples. Representative images from 4 independent animals are shown. Note: Compared with the control vein, there is reduced VSMC contractile protein expression in the neointima (NI) cells of AVF proximal to the anastomosis site and the NI cells are proliferative. A indicates adventitia; DAPI, 4′,6‐diamidino‐2‐phenylindole; L, lumen; M, medial smooth muscle layer.
Lineage Tracing of Mature VSMCs Using Myh11‐Cre/ERT2‐mTmG Reporter Mouse
To faithfully track the fate of mature VSMCs during AVF maturation and failure, we sought to utilize a novel reporter mouse strain carrying Myh11‐Cre‐ERT2/mTmG. This is a dual fluorescence labeling system driven by a chimeric cytomegalovirus and β‐actin promoter (pCAG). Before TMX induction, all cells were labeled with membrane localized tomato (mT). Upon TMX induction, only differentiated mature VSMCs were labeled with membrane localized GFP (mG) whereas all other cells remained mT (Figure 3A). We isolated the branch of external jugular vein and carotid artery (which we utilized for AVF construction), aorta, and bladder to validate the specific labeling for mature SMCs. As expected, confocal microscopy confirmed that SMCs in all these tissues showed mT‐labeled red fluorescence prior to TMX induction (Figure 3B, –TMX). After TMX induction, only SMCs in these tissues were labeled with mG (Figure 3B, +TMX).
Figure 3.
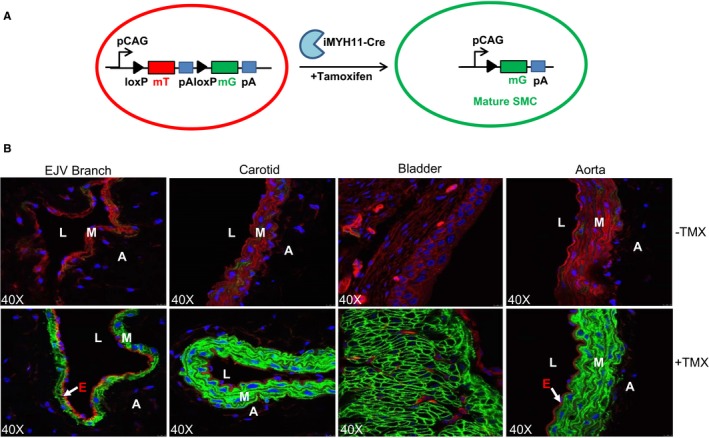
Lineage tracing mature vascular smooth muscle cells (SMCs) using Myh11‐Cre/ERT2‐mTmG reporter mouse. A, ROSA mT/mG mice were bred with Myh11‐Cre/ERT2 (+/−) to generate Myh11‐Cre/ERT2‐mTmG mice. ROSA mT/mG mice carry a single copy of the gene cassette comprised of cytomegalovirus enhancer and β‐actin chimeric promoter (pCAG) and a floxed membrane localized Tomato followed by a membrane localized green fluorescent protein (GFP) in ROSA 26 gene locus. Following tamoxifen (TMX) activation, Myh11‐Cre mediates the excision of the mTomato transgene, and thus the pCAG directs the expression of membrane GFP, which exclusively occurs in mature differentiated SMCs in Myh11‐Cre/ERT2‐mTmG mice. B, Myh11‐Cre/ERT2‐mTmG mice were injected with TMX or oil control for 5 consecutive days. Confocal microscopy for mT and mG fluorescence was performed in the indicated tissues (containing mature differentiated smooth muscle cells), which were isolated 3 weeks after TMX induction (n=3). All cells in the isolated tissues are labeled by mT(Red) before TMX induction, and only mature SMCs are labeled by mG(Green) after TMX induction. A indicates adventitia; E, endothelium; EJV, external jugular vein; L, lumen; M, medial smooth muscle layer; mG, membrane GFP; mT, membrane tomato; pA, polyadenylation.
Contribution of Mature VSMCs to Venous Maturation
We applied the system described above to fate map mature VSMCs in AVF remodeling. As described earlier in Figure 2, we found that the optimal time for the anastomosed vein to achieve outward remodeling and neointima formation was 4 weeks after AVF surgery, and sections from the distal, middle, and proximal regions were prepared at this time point as described in Figure 4A. Control jugular veins were characterized by a single medial VSMC layer that was GFP+ after TMX induction (Figure 4B, left panel). Four weeks after AVF surgery, we observed a thickening of the VSMC layer in the middle region of AVFs, with 2 to 3 layers of VSMCs labeled with membrane GFP (Figure 4B, right panel), which was not seen in the distal AVF region (data not shown). Quantitative analysis showed a 4‐fold increase in the thickness of the medial VSMC layer in AVFs compared with the control vein (Figure 4C). The majority of the medial GFP+ cells (≈99%) in the middle region of AVFs exhibited comparable protein levels of MYH11 to those in control veins (Figure 4D and 4E). Similar to control vein VSMCs, the GFP+ cells in the middle region of AVFs were negative for Ki67 staining (data not shown). All together, we demonstrated that maturation of the AVF in this mouse model was associated with medial wall thickening. This medial layer was solely composed of differentiated/quiescent VSMCs, which originated from previously differentiated mature VSMCs.
Figure 4.
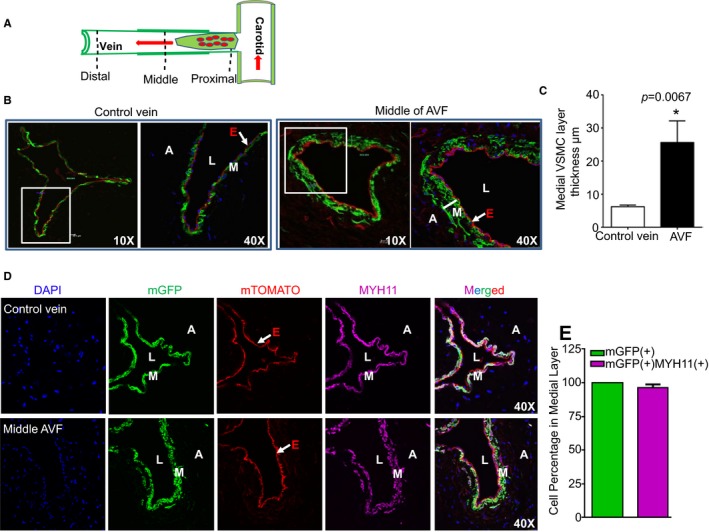
Mature vascular smooth muscle cells (VSMCs) contribute to arteriovenous fistula (AVF) maturation. A, Schematic illustration of the AVF regions used for immunofluorescence staining and histological examination in Myh11‐Cre/ERT2‐mTmG reporter mice at 4 weeks after AVF surgery. B, Representative images for mT‐ or mG‐labeled vessel wall of the control jugular vein branch and the middle AVF region in tamoxifen‐induced Myh11‐Cre/ERT2‐mTmG mice at 4 weeks after AVF surgery. C, Quantitative analysis of the thickness of the medial VSMC layer in the control jugular vein branch and the middle AVF region as described in (B) by software provided by the confocal microscope machine (n=4). There is medial VSMC layer thickening compared with control unligated vein branch at this time point. D, Immunofluorescence staining for MYH11in samples, as described in (A). Representative images from 4 different animals are shown. E, Quantitative analysis for the percentage of MYH11 (+) green fluorescent protein (+) cells in the medial VSMC layer of the middle AVF region by ImageJ software (n=4). The thickened medial layer is comprised of contractile VSMCs derived from previously differentiated mature VSMCs. A indicates adventitia; DAPI, 4′,6‐diamidino‐2‐phenylindole; E, endothelium; L, lumen; M, medial smooth muscle layer; mGFP, membrane green fluorescent protein.
Contribution of Mature VSMC to Neointima Formation
Extensive neointimal hyperplasia was frequently seen in the region adjacent to the anastomosis site, a phenomenon recapitulating pathological characteristics of failed human AVF8, 9 (Figure 5A, bottom panel). A large portion of neointimal cells were GPF+, indicating that mature VSMCs were a primary source of the cells populating the neointima. Consistent with Figure 2, the majority of neointimal cells exhibited barely detectable levels of MYH11 (Figure 5A). Quantitative analysis showed that 50% were GFP+ and 15% of neoinitmal cells expressed MYH11 protein but with much decreased levels. Interestingly, among all of the MYH11+ cells (15%), only 8.5% were labeled with GFP (Figure 5B). Neointimal cells expressed decreased CNN1 protein compared with control vein (Figure 5C). Quantitative analysis revealed that 42% of neointimal cells were CNN1+ (Figure 5D). Similarly, 24.5% (of 42% CNN1+) of cells were positive for both GFP and CNN1 (Figure 5D). This suggested that, during the course of neointima formation, some cells of unknown origin (such as resident progenitor cells) may transdifferentiate to VSMC‐like cells. Ki67 staining for cell proliferation revealed that 12% of neointima cells were positive, with 4.7% (of 12%) of the cells positive for both GFP and Ki67, indicating that beyond proliferative VSMCs, neointima formation might also involve non‐SMCs such as infiltrated inflammatory cells (Figure 5E and 5F). Indeed, we found there were CD45 (+) GFP (−) cells present in the neointima region (data not shown). Taken together, we demonstrated that VSMCs phenotypically switching from a differentiated to a dedifferentiated state was one of the underlying mechanisms for neointima formation in this AVF mouse model.
Figure 5.
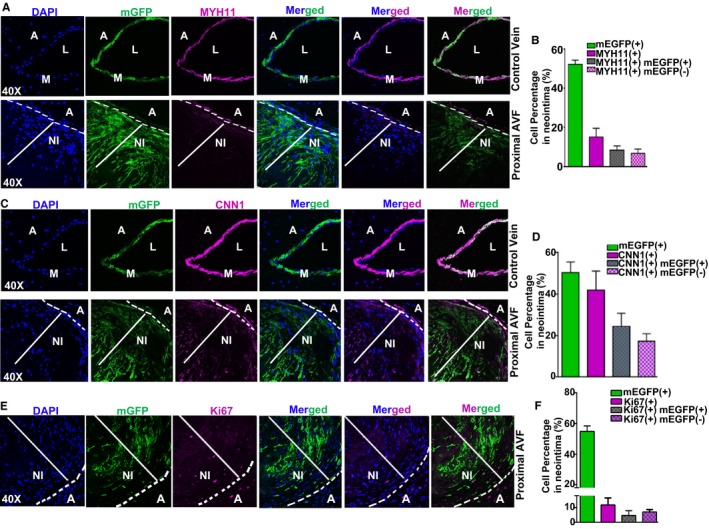
Mature vascular smooth muscle cells (VSMCs) contribute to neointima (NI) formation in arteriovenous fistula (AVF). Confocal microscopy for membrane green fluorescent protein (mGFP) and immunofluorescence staining for MYH11 (A) and CNN1 (C) in the control unligated vein and proximal AVF region, and Ki67 (E) in the proximal AVF region of Myh11‐Cre/ERT2‐mTmG mice at 4 weeks after AVF surgery. Quantitation of the percentage for each indicated cell category was analyzed using ImageJ and results are shown in B, D, and F, respectively (n=4). Extensive NI formation frequently occurs at the proximal region to the anastomosis site. A large portion of NI cells are GFP+ and with decreased levels of contractile proteins including MYH11 and CNN1. A indicates adventitia; DAPI, 4′,6‐diamidino‐2‐phenylindole; L, lumen; M, medial smooth muscle layer.
Characterization of VSMC Differentiation in Human AVF Samples
To evaluate whether our findings in the mouse AVF model could be extended to human AVFs, we examined deidentified human AVF samples collected from hemodialysis patients undergoing AVF placement or revision. Verhoeff‐van Gieson staining showed black staining for the elastin fibers and clearly marked the internal elastic lamella in the samples (Figure 6A). Consistent with a normal vein, the medial region of 10 of the 12 placement vessels consisted of 3 to 4 cell layers, with only two placement vessels showing higher numbers. In contrast, all of the revision vessels analyzed displayed an increase in medial cell layers relative to the placement vessels. Quantitative analysis showed a significant increase in the thickness of the medial layer in revision samples compared with placement veins (Figure 6B). Examination of contractile proteins by immunofluorescence staining demonstrated that the 3 contractile proteins CNN1, MYH11, and ACTA2, were abundantly expressed in the medial SMC layer in placement veins (Figure 7). Significantly, the thicker medial cell layer in the revision veins had comparable levels of contractile proteins to those found in the placement samples, suggesting that medial VSMCs‐mediated venous maturation is also operative in human AVF. All revision AVF samples showed extensive hyperplastic neointima in addition to venous maturation. Some neointimal cells in the revision AVF samples displayed some levels of VSMC contractile protein expression, albeit at lower levels than those in the medial layer. In conclusion, similar to our findings in the mouse AVF model, both medial thickening (venous maturation or arterialization) and VSMC‐derived neointima formation were also found to be operative in human AVF.
Figure 6.
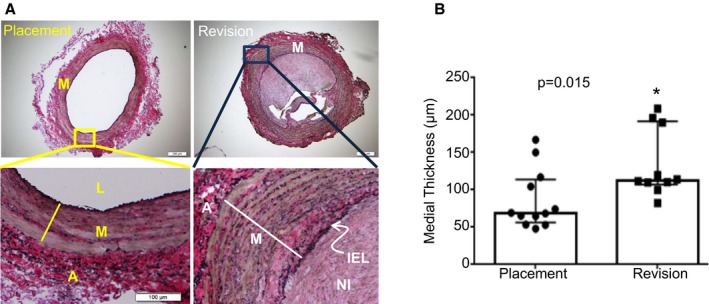
Morphometric analysis of human venous samples. A, Representative images of Verhoeff‐van Gieson stainings for cross sections of tissue samples obtained from patients undergoing arteriovenous fistula (AVF) placement or revision. The Verhoeff‐van Gieson staining marked elastin fibers in black, collagen in red‐pink, and the cell‐rich medial layer in brown. The bottom images are a zoomed‐in version of the upper panel. B, The medial‐intimal thickness of placement and revision vessels was measured, as described in the Materials and Methods section. There was a statistically significant increase in the thickness of the medial layer in the revision AVF samples compared with the placement venous vessels. The bottom and top boundaries of the bars represent the 25th and 75th percentiles, respectively. Columns denote medians and filled symbols depict individual values. *Paired Student t test, change is statistically significant at P<0.05. A indicates adventitia; IEL, internal elastic lamella; L, lumen; M, medial smooth muscle layer; NI, neointima.
Figure 7.
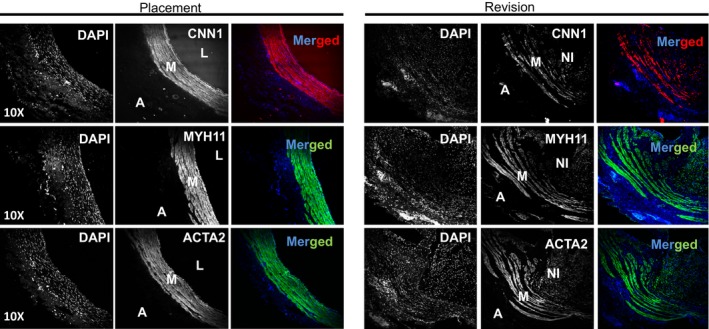
Characterization of vascular smooth muscle cell (VSMC) differentiation program in human venous samples. Venous samples obtained from patients undergoing arteriovenous fistula (AVF) placement or at revision of a failed AVF were subjected to immunostaining for VSMC contractile proteins including CNN1, MYH11, and ACTA2. Representative images for each protein staining in the consecutive cross sections are shown. Strong staining of the three VSMC markers (CNN1, MYH11, and ACTA2) was seen in the medial layer (M) of the placement vessels. However, there was little to no signal in the adventitia (A). Strong staining of the 3 VSMC markers was also observed in the thickened medial layer of the failed revision AVF samples, but there was decreased intensity seen in the neointima (NI) of revision samples. L indicates the lumen of the vessel. The image is representative of 4 placement and 4 revision veins, each from different individuals (8 individuals in total). DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Discussion
Patients with end‐stage renal disease rely on hemodialysis, which requires a functional high‐flow vascular access preferably achieved through an AVF conduit.2 However, inadequate venous maturation and neointimal hyperplasia leading to occlusion are frequently found in AVF constructed in patients.23 The underlying mechanism of venous maturation and neointima formation in AVF is poorly understood. In this study, we used a novel VSMC lineage tracing reporter mouse model to faithfully track mature VSMCs in an AVF mouse model with a vessel configuration similar to human AVF. We show that the VSMC phenotypic switch classically associated with arterial remodeling is also operative in the course of venous remodeling and that dedifferentiated VSMCs are a major cellular component of the neointima in AVF. Most significantly, we demonstrate for the first time a profound medial VSMC layer thickening in the draining vein of mouse AVF 4 weeks after surgery, indicating maturation of the vessel. This thickened medial SMC layer is solely composed of GFP‐labeled VSMCs, which are also positive for MYH11, a rigorous marker of the VSMC contractile phenotype. When examining human samples derived from patients undergoing revision of failed AVF, we confirmed that both medial VSMC layer thickening and VSMC phenotypic modulation–mediated neointima formation also occur in humans. Therefore, we demonstrated a dual function of mature VSMCs in the setting of AVF, with differentiated VSMC contributing to medial wall thickening towards beneficial venous maturation and dedifferentiated VSMC contributing to detrimental neointimal hyperplasia.
Consistent with previous results by Wong et al,19 we observed prominent neointimal hyperplasia at the anastomosis site of the mouse AVF. It has long been believed that intima hyperplasia, stenosis, and the consequential thrombosis are the major pathological causes of AVF failure.23 In the context of arterial remodeling, detailed studies have demonstrated that several cell populations, including medial contractile VSMCs phenotypically switching to a dedifferentiated/synthetic phenotype, resident multipotent progenitor cells transdifferentiating to VSMC‐like lineage, and inflammatory cells all contribute to neointima formation.24, 25, 26 However, in the context of AVF, most of the related studies have been limited to polymerase chain reaction analysis and immunostaining of nonspecific marker genes (such as vimentin, ACTA2, and inflammatory cytokines).13, 27 Polymerase chain reaction analysis of the whole vessel is rather ambiguous for determining cell lineage because of the heterogeneous RNA samples. In addition, gene programs during vascular remodeling are dynamic and mutually interactive. Accordingly, immunostaining of even the most definitive marker genes cannot define cellular origins. Because the influence of gene programs and pathways on vascular pathology is cell‐context dependent, precise characterization of cellular contribution to venous remodeling is of high interest for an effective therapeutic strategy for AVF failure.
Genetically engineered mouse models offer a superior approach to map a particular cell lineage in a given disease context. The reporter mouse Myh11‐Cre‐ERT2/mTmG is a TMX‐inducible VSMC‐specific Cre‐mediated dual fluorescence labeling system in which all mature VSMCs are labeled with membrane GFP at the time of TMX induction. Since Cre‐mediated genome engineering is permanent and transmissible to progeny cells, all mature VSMCs are permanently labeled with membrane GFP following TMX induction regardless of the varied gene expression in these cells under different pathophysiological conditions. This system has been successfully utilized to track mature VSMCs in arterial disease models of atherosclerosis and vascular injury, and has defined a pivotal role of mature VSMCs in these settings.28, 29 In contrast, the role of mature VSMCs in AVF remodeling has never been precisely characterized. Following AVF construction, the surgical trauma, sudden increased blood flow, high shear stress, exacerbated inflammatory response, and redox stress might all contribute to endothelial dysfunction in a fashion similar to some pathophysiologies associated with arteries.4 A distinct feature of AVF remodeling is the adaptation of the draining vein to the arterial hemodynamic environment, which results in outward remodeling, a process usually referred to as venous maturation or arterialization.4, 23 With this distinction in mind, we would like to speculate that different and maybe more complex changes related to mature VSMC functions might occur in AVF remodeling. We found that the neointima surrounding the anastomosis site is mainly composed of previously differentiated VSMCs labeled with GFP but negative for MYH11. This suggests that VSMC phenotypic adaptation operates during neointima formation in the setting of AVF. Interestingly, we observed a small portion of GFP (−) neointimal cells but positive for MYH11, which could possibly be derived from alternative cell lineages such as resident progenitor cells.
Our findings together with those from a previous report indicating that migratory arterial VSMCs could contribute to neointimal hyperplasia in an end‐to‐end AVF mouse model demonstrate that VSMC phenotypic modulation is critical for neointima formation in AVF.16 This is in contrast to a previous study using a vein graft mouse model in which endothelial‐to‐mesenchymal transition was shown to play an important role in venous remodeling.12 The construction of an AVF creates a high flow and high shear stress arterial environment, whereas vein grafting results in moderate flow and moderate shear stress.4 It is likely that high flow and shear stress causes more severe endothelium damage and that in the absence of endothelium, neointima formation in AVF is dominated by a VSMC response. This suggests that therapeutic strategies to limit neointimal hyperplasia in AVF and vein grafting might need to reflect their pathophysiological differences.
Outward remodeling, a process characterized by the thickening of the vessel wall outside the internal elastic lamella, is generally considered a beneficial adaption of veins to the arterial hemodynamic environment. Our mechanistic understanding of venous arterialization is somewhat limited. Related to vein graft remodeling, recent studies have focused on molecular mechanisms of venous versus arterial identity.16, 30 An important finding of the present study is the demonstration of outward remodeling or, more specifically, medial VSMC layer thickening in our mouse model of AVF. The intact mouse jugular vein consists of only one single SMC layer. Four weeks after surgery, multiple VSMC layers (2–3) appeared in the draining vein. Although there was comparable adventitia layer thickening, virtually no GFP‐labeled cells were present in the adventitia except for cells in the vasa vasorum (data not shown). This suggests that the adventitia remodeling in AVF involves different cellular origin(s). It is noteworthy that the thickened VSMC layer is solely composed of cells that are GFP+/MYH11+/Ki67−, indicative of an authentic VSMC contractile phenotype. This raises the question of the mechanism by which mature VSMCs contribute to medial SMC layer thickening. One possible answer is that it is a unique response of the local venous SMCs to the arterial environment. Venous SMCs could proliferate to produce more SMCs and at the same time still retain contractile features due to the beneficial arterial hemodynamic environment. Alternatively, the thickened VSMC layer could be populated by migrating arterial VSMCs, whereby arterial VSMCs initially dedifferentiate, migrate into the venous segment, and differentiate back to a contractile phenotype. This would be an important, yet difficult, question to answer due to the lack of a Cre‐driver to distinguish venous from arterial VSMCs currently.
Evidence for a direct morphological and biochemical characterization of mature vessels associated with clinical maturation (ie, adequate access flow) in humans is difficult to establish because the failed AVF is usually left ligated in situ in the patient. In the present study, we provided direct evidence that the contribution of mature VSMCs in venous maturation and neointima formation extends to human AVF. Most significantly, we observed medial VSMC layer thickening in the failed revision AVF samples. The expanded VSMCs in the thickened medial layer of the human revision AVF displayed equivalent levels of contractile proteins (MYH11, CNN1, and ACTA2) to those of placement vein samples. We also found that the neointimal cells in the revision veins showed some expression of contractile proteins but at reduced levels compared with medial VSMCs. This would suggest that VSMC phenotypic modulation is also operative for neointima formation in human AVF. We speculate that promoting VSMC differentiation after AVF construction could simultaneously promote beneficial venous maturation and prohibit the detrimental neointimal hyperplastic response. This could be a novel and effective “two birds with one stone” therapeutic strategy to treat AVF failure.
Our work provides new and important results on the maturation and adaptation of venous segments to the arterial environment. Our results suggest a dual role for mature VSMCs in venous maturation and neointimal hyperplasia during AVF remodeling. This delineates an important foundation for more detailed studies aimed at optimizing venous adaptation to the arterial environment during postsurgical processes. As previously established in other human vessels, examination of epigenetic histone markers in the promoter regions of MYH11 will facilitate the precise characterization of VSMC lineage in human AVF remodeling and in other vein graft conduits.31
Conclusions
By using a novel VSMC lineage tracing reporter mouse, Myh11‐Cre/ERT2‐mTmG in a clinically relevant AVF mouse model, we have shown that contractile VSMCs derived from previously differentiated VSMCs are the sole cell components in the thickened medial wall of the arterialized vein branch. Similar to arterial remodeling, previously differentiated VSMCs also undergo phenotypic transition to dedifferentiated VSMCs, contributing to neointimal hyperplasia. Importantly, we confirmed that both medial VSMC layer thickening and VSMC phenotypic modulation also occur in AVF constructed in patients. The present studies have uncovered a dual function for mature VSMCs during AVF remodeling. These results provide novel and valuable insights into the cellular mechanisms underlying both beneficial venous maturation and detrimental neointima formation following AVF construction.
Sources of Funding
This work was supported by the Dialysis Clinic, Inc. (DCI) Paul Teschan Research Fund (PTRF grant to Asif and Singer; PTRF grant No. 2015_04 to Long); DCI Research Fund Project grant No. C‐3804 (Asif and Singer); National Institutes of Health R01HL122686 (Long) and R01HL49426 (Singer); and American Heart Association 16GRNT31280002 (Jourd'heuil).
Disclosures
None.
(J Am Heart Assoc. 2017;6:e004891 DOI: 10.1161/JAHA.116.004891.)28360226
Contributor Information
David Jourd'heuil, Email: jourdhd@mail.amc.edu.
Xiaochun Long, Email: longx@mail.amc.edu.
References
- 1. Schwab SJ, Harrington JT, Singh A, Roher R, Shohaib SA, Perrone RD, Meyer K, Beasley D. Vascular access for hemodialysis. Kidney Int. 1999;55:2078–2090. [DOI] [PubMed] [Google Scholar]
- 2. Dember LM, Beck GJ, Allon M, Delmez JA, Dixon BS, Greenberg A, Himmelfarb J, Vazquez MA, Gassman JJ, Greene T, Radeva MK, Braden GL, Ikizler TA, Rocco MV, Davidson IJ, Kaufman JS, Meyers CM, Kusek JW, Feldman HI; Dialysis Access Consortium Study Group . Effect of clopidogrel on early failure of arteriovenous fistulas for hemodialysis: a randomized controlled trial. JAMA. 2008;299:2164–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riella MC, Roy‐Chaudhury P. Vascular access in haemodialysis: strengthening the Achilles' heel. Nat Rev Nephrol. 2013;9:348–357. [DOI] [PubMed] [Google Scholar]
- 4. Lu DY, Chen EY, Wong DJ, Yamamoto K, Protack CD, Williams WT, Assi R, Hall MR, Sadaghianloo N, Dardik A. Vein graft adaptation and fistula maturation in the arterial environment. J Surg Res. 2014;188:162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muto A, Yi T, Harrison KD, Davalos A, Fancher TT, Ziegler KR, Feigel A, Kondo Y, Nishibe T, Sessa WC, Dardik A. Eph‐B4 prevents venous adaptive remodeling in the adult arterial environment. J Exp Med. 2011;208:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang M, Collins MJ, Foster TR, Bai H, Hashimoto T, Santana JM, Shu C, Dardik A. Eph‐B4 mediates vein graft adaptation by regulation of endothelial nitric oxide synthase. J Vasc Surg. 2017;65:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roy‐Chaudhury P, Arend L, Zhang J, Krishnamoorthy M, Wang Y, Banerjee R, Samaha A, Munda R. Neointimal hyperplasia in early arteriovenous fistula failure. Am J Kidney Dis. 2007;50:782–790. [DOI] [PubMed] [Google Scholar]
- 8. Nassar GM, Nguyen B, Rhee E, Achkar K. Endovascular treatment of the “failing to mature” arteriovenous fistula. Clin J Am Soc Nephrol. 2006;1:275–280. [DOI] [PubMed] [Google Scholar]
- 9. Sivanesan S, How TV, Bakran A. Sites of stenosis in AV fistulae for haemodialysis access. Nephrol Dial Transplant. 1999;14:118–120. [DOI] [PubMed] [Google Scholar]
- 10. Daoui R, Asif A. Cephalic arch stenosis: mechanisms and management strategies. Semin Nephrol. 2012;32:538–544. [DOI] [PubMed] [Google Scholar]
- 11. Roy‐Chaudhury P, Wang Y, Krishnamoorthy M, Zhang J, Banerjee R, Munda R, Heffelfinger S, Arend L. Cellular phenotypes in human stenotic lesions from haemodialysis vascular access. Nephrol Dial Transplant. 2009;24:2786–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. TGF‐beta signaling mediates endothelial‐to‐mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6:227ra234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Castier Y, Lehoux S, Hu Y, Foteinos G, Tedgui A, Xu Q. Characterization of neointima lesions associated with arteriovenous fistulas in a mouse model. Kidney Int. 2006;70:315–320. [DOI] [PubMed] [Google Scholar]
- 14. Skartsis N, Manning E, Wei Y, Velazquez OC, Liu ZJ, Goldschmidt‐Clermont PJ, Salman LH, Asif A, Vazquez‐Padron RI. Origin of neointimal cells in arteriovenous fistulae: bone marrow, artery, or the vein itself? Semin Dial. 2011;24:242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang B, Janardhanan R, Vohra P, Greene EL, Bhattacharya S, Withers S, Roy B, Nieves Torres EC, Mandrekar J, Leof EB, Mukhopadhyay D, Misra S. Adventitial transduction of lentivirus‐shRNA‐VEGF‐A in arteriovenous fistula reduces venous stenosis formation. Kidney Int. 2014;85:289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang M, Wang Y, Liang A, Mitch WE, Roy‐Chaudhury P, Han G, Cheng J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires notch activation to form neointima. Kidney Int. 2015;88:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miano JM, Cserjesi P, Ligon KL, Periasamy M, Olson EN. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ Res. 1994;75:803–812. [DOI] [PubMed] [Google Scholar]
- 18. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser‐Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12‐G13‐LARG‐mediated signaling in vascular smooth muscle is required for salt‐induced hypertension. Nat Med. 2008;14:64–68. [DOI] [PubMed] [Google Scholar]
- 19. Wong CY, de Vries MR, Wang Y, van der Vorst JR, Vahrmeijer AL, van Zonneveld AJ, Roy‐Chaudhury P, Rabelink TJ, Quax PH, Rotmans JI. Vascular remodeling and intimal hyperplasia in a novel murine model of arteriovenous fistula failure. J Vasc Surg. 2014;59:192–201.e191. [DOI] [PubMed] [Google Scholar]
- 20. Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez‐Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M. Orai1‐mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res. 2011;109:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gomez D, Swiatlowska P, Owens GK. Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler Thromb Vasc Biol. 2015;35:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Long X, Slivano OJ, Cowan SL, Georger MA, Lee TH, Miano JM. Smooth muscle calponin: an unconventional CArG‐dependent gene that antagonizes neointimal formation. Arterioscler Thromb Vasc Biol. 2011;31:2172–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rothuizen TC, Wong C, Quax PH, van Zonneveld AJ, Rabelink TJ, Rotmans JI. Arteriovenous access failure: more than just intimal hyperplasia? Nephrol Dial Transplant. 2013;28:1085–1092. [DOI] [PubMed] [Google Scholar]
- 24. Herring BP, Hoggatt AM, Burlak C, Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell. 2014;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, Kaesler N, Chang‐Panesso M, Machado FG, Gratwohl S, Madhurima K, Hutcheson JD, Jain S, Aikawa E, Humphreys BD. Adventitial MSC‐like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. 2016;19:628–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S, Weiser‐Evans MC. SDF‐1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury‐induced neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee T, Wang Y, Arend L, Cornea V, Campos B, Munda R, Roy‐Chaudhury P. Comparative analysis of cellular phenotypes within the neointima from vein segments collected prior to vascular access surgery and stenotic arteriovenous dialysis accesses. Semin Dial. 2014;27:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bai H, Wang M, Foster TR, Hu H, He H, Hashimoto T, Hanisch JJ, Santana JM, Xing Y, Dardik A. Pericardial patch venoplasty heals via attraction of venous progenitor cells. Physiol Rep. 2016;4:e12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]