

Abstract
Background
The significance of brain angiotensin II in Alzheimer disease (AD) is unclear.
Methods and Results
To examine the role of brain angiotensin II in AD, intracerebroventricular angiotensin II infusion was performed on 5XFAD mice, a mouse model of AD, and wild‐type mice, and the detrimental effects of brain angiotensin II was compared between the 2 strains of mice. Intracerebroventricular angiotensin II infusion significantly impaired cognitive function in 5XFAD mice but not in wild‐type mice. This vulnerability of 5XFAD mice to brain angiotensin II was associated with enhancement of hippocampal inflammation and oxidative stress and with increased cerebrovascular amyloid β deposition. We also compared the effect of brain angiotensin II on the heart and skeletal muscle between the 2 strains because AD is associated with heart failure and sarcopenia. We found that cardiac compensatory response of 5XFAD mice to brain angiotensin II–induced hypertension was less than that of wild‐type mice. Brain angiotensin II caused skeletal muscle atrophy and injury in 5XFAD mice more than in wild‐type mice.
Conclusions
Brain angiotensin II seems to be involved in cognitive impairment and brain injury in AD, which is associated with oxidative stress, inflammation, and cerebral amyloid angiopathy. Further, brain angiotensin II may participate in cardiac disease and sarcopenia observed in AD.
Keywords: Alzheimer disease, angiotensin II, cerebrovascular disease, cognitive impairment, dementia, sarcopenia
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Basic Science Research, Cognitive Impairment
Introduction
Alzheimer disease (AD) is the most common form of dementia and is a severe neurodegenerative disease neuropathologically defined by amyloid plaques and neurofibrillary tangles.1 AD represents a major public health concern and has been identified as a research priority.1 Emerging evidence supports the notion that vascular risk factors, such as hypertension, diabetes mellitus, or dyslipidemia, are closely linked to the progression of AD.1, 2, 3 Furthermore, cerebrovascular injuries, such as cerebral amyloid angiopathy (CAA),2, 4, 5 are significantly associated with the severity and exacerbation of AD. However, the precise role of vascular factors in AD pathophysiology remains to be defined.
Accumulating evidence suggest that the brain renin‐angiotensin system plays an important role in the pathophysiology of AD or cognitive impairment.6, 7 Preclinical studies show that renin‐angiotensin system blockers such as angiotensin receptor blockers or angiotensin‐converting enzyme inhibitors ameliorate cognitive impairment in an animal model of AD.8, 9 Prospective cohort analysis10 and a population‐based cohort study11 indicate that the use of angiotensin receptor blockers is significantly associated with a significant reduction in the incidence and progression of AD. Treatment with angiotensin receptor blockers is associated with less AD‐related pathology on autopsy evaluations.12 Furthermore, small‐scale double‐blind randomized clinical trials suggest that antihypertensive therapy with angiotensin receptor blockers is associated with improvement of cognitive function in older hypertensive patients.13 Therefore, brain angiotensin II is proposed to be involved in the pathophysiology of AD. However, a definite role of brain angiotensin II in AD is unclear. The vulnerability of AD to central angiotensin II remains to be determined. To test our hypothesis that AD mice have more susceptibility to brain angiotensin II than do control mice, in the present study we examined the effect of centrally administered angiotensin II on cognitive function and brain pathology in a mouse model of AD. Furthermore, we also investigated the effect of brain angiotensin II on cardiac and skeletal muscle tissue of AD mice because AD is closely associated with heart failure14, 15 and sarcopenia.15, 16, 17
Methods
Animals
All experiments were approved by the Kumamoto University Committee for Laboratory Animal Care and Use. All procedures were in accordance with institutional guidelines for the care and use of laboratory animals. Cryopreserved embryos, which were purchased from Jackson Laboratory (Bar Harbor, ME), were implanted into pseudopregnant foster mice to make 5XFAD mice.18 5XFAD mice overexpress human amyloid precursor protein (APP695) with Swedish (K607N, M671L), Florida (I716V), and London (V717I) familial AD mutations, also with mutant human presenilin‐1 (M146L, L286V) controlled by murine Thy‐1 promotor.19 5XFAD mice generate amyloid β (Aβ)1‐42 almost exclusively, rapidly accumulate amyloid plaque formation and also recapitulate major pathologic and behavioral characteristics of AD.19, 20, 21 5XFAD mice were backcrossed into the C57BL/6J background for at least 10 generations to reduce genetic variation.18 Male wild‐type mice were purchased from SLC Japan (Shizuoka, Japan). All mice were housed under a 12‐hour light/darkness cycle in an animal facility and were given standard chow and water ad libitum.
Experimental Protocol
Twelve‐month‐old male 5XFAD (n=24) and control C57BL/6J wild‐type mice (n=24) were randomly divided into 2 groups and received continuous intracerebroventricular (ICV) infusion of (1) vehicle (saline) or (2) angiotensin II (20 μg/(kg·h) (Peptide Institute, Inc, Osaka, Japan) for 4 weeks (n=12, in each group). The dosage of angiotensin II was determined according to previous reports.22, 23, 24 The detailed experimental protocol is shown in Figure 1.
Figure 1.
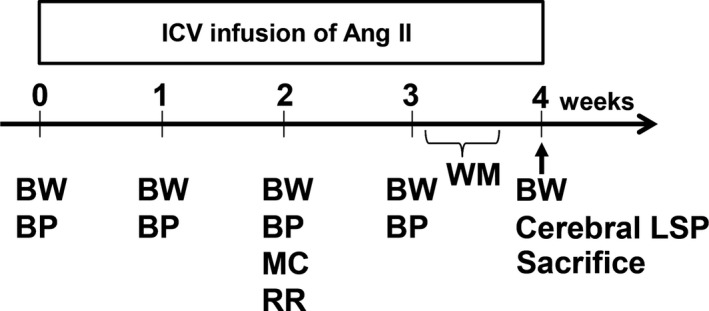
Schematic illustration of the experimental protocol. Intracerebroventricular (ICV) infusion of angiotensin II (Ang II) in wild‐type (C57/BL6J) and 5XFAD mice was performed for 4 weeks. Measurement of each parameter was carried out at specified time points during angiotensin II infusion, as shown. BP indicates blood pressure measurement; BW, body weight measurement; LSP, laser speckle perfusion; MC, housing in metabolic cage; RR, rotarod test; WM, water maze test.
Implantation of the ICV Infusion Cannula and Osmotic Pump
Implantation of an infusion cannula into the cerebral ventricles and an osmotic pump were performed on mice according to our previously reported method.18 Briefly, the mice were anesthetized with 1.5% to 2% isoflurane through a face mask, and stainless steel cannulas (ALZET Brain Infusion Kit3, Durect Co, Cupertino, CA) were implanted in the right lateral ventricle, placed 1.0 mm lateral and 0.5 mm posterior from the bregma. The cannulas were fixed on the skull, and catheters were attached to extension tubes and linked to the ALZET osmotic pumps (Model 1004, Durect Co, Cupertino, CA), which were installed in subcutaneous pockets on the lateral backs of the mice.
Measurement of Body Weight and Blood Pressure
The mice were weighed every week, and their systolic blood pressure was measured at 0, 1, 2, and 3 weeks after start of the ICV infusion, using the tail cuff method (BP‐98A; Softron Co, Tokyo, Japan), as previously described.25
Measurement of Food and Water Intake and Urinary Volume
Individual mice were housed in a metabolic cage at 2 weeks after start of the ICV infusion to evaluate food intake, water intake, and urine volume.
Rotarod Test
To assess motor function and sensorimotor coordination, rotarod tests were performed on the mice according to our previous protocol.26 Briefly, the mice were placed on the horizontal drum (Muromachi Kikai, Tokyo, Japan) and were allowed to walk forward for a maximum 60 seconds as a training session. Then, the mice were subjected to the trial on the accelerating spindle (4 to 40 rpm) for 5 minutes. The mean times for 3 trials of the tests were calculated for each animal.
Morris Water Maze Test
To assess spatial learning and memory function, from 21 to 25 days after the start of ICV infusion, the Morris water maze test was performed according to our previous protocol.27 Briefly, swimming paths were video‐tracked with a camera fixed on the ceiling of the room and analyzed by the software (Muromachi Kikai, Tokyo, Japan). On the hidden test, the mice had 4 sessions per day on the following 4 consecutive days (days 1 to 4). On the probe test (day 5), the hidden platform was moved away, and the mice swam freely for 100 seconds and then the number of times the mice crossed the original platform location was simultaneously recorded. On the visible platform test, the platform was elevated above the water surface and placed in a different position, which was done after the probe test on day 5. The mice were given 4 sessions of a visible trial with an intersession interval of 20 minutes.
Measurement of Cerebral Laser Speckle Perfusion
Cerebral laser speckle perfusion (LSP) was measured using a laser speckle blood flow imager (Omega Zone; Omegawave, Tokyo, Japan), as previously described.28 The skull of each mouse was exposed by a midline scalp incision under 1.5% to 2.0% isoflurane, and the ICV infusion cannula and osmotic pump were removed. Then, LSP on the surface of the region of left cerebral hemisphere was measured.
Histology
At the end of the ICV infusion, the brains were removed and divided at the point of bregma after perfusion with phosphate‐buffered saline, and the caudal side of each brain was immediately frozen in Tissue‐Tek OCT embedding medium (Sakura Finetek, Tokyo, Japan). An 8‐μm slice was made at 1.43 to 2.43 mm caudally from the bregma for the following histological evaluations. In addition, the cardiac left ventricle and gastrocnemius muscle from each mouse were removed, weighed, and frozen in Tissue‐Tek OCT embedding medium (Sakura Finetek, Tokyo, Japan) and in paraffin for the following histological evaluations.
Measurement of Tissue CD68‐Positive Cells
To determine CD68‐positive cells of brain, left ventricle, and gastrocnemius muscle, 8‐μm frozen sections from those organs were incubated overnight with the primary antibody (1:300, rat anti‐mouse CD68; AbD Serotec, Oxford, UK) followed by anti‐rat secondary antibody (Biosource, Camarillo, CA), as previously described.29 To evaluate brain macrophage/microglia, the number of CD68‐positive cells per square millimeter was counted in the 2 fields of the hippocampal CA1 region and somatosensory cortex on both sides for each animal at ×200 magnification. For estimation of left ventricular macrophage, the number of CD68‐positive cells per square millimeter was counted by examining >10 fields per section using a microscope with ×200 magnification. In gastrocnemius muscle, 3 fields per mouse were randomly selected, and the cells were counted under a microscope with ×200 magnification. The average CD68‐positive cell number was calculated in each mouse.
Measurement of Brain Superoxide
Dihydroethidium was used to evaluate brain superoxide levels in situ as previously described.30 Dihydroethidium fluorescence of each tissue section in the 2 fields of the hippocampal CA1 region and somatosensory cortex on both sides for each animal at ×200 magnification was quantified by WinROOF version 5.8 analysis software (Mitani Corporation, Fukui, Japan). Mean fluorescence was calculated and expressed relative to values obtained from a vehicle group of wild‐type mice.
Assessment of Blood‐Brain Barrier Disruption
Immunoglobulin G (IgG) extravasation in each brain section was evaluated according to our previous protocol.31, 32, 33 The brain sections were incubated with anti‐IgG antibody (1:500; Invitrogen, Carlsbad, CA) for 1 hour, and then the reaction product was visualized with diaminobenzidine. IgG stainability was quantified by the WinROOF version 5.8 analysis software (Mitani Corporation, Fukui, Japan).
Immunohistochemical Analysis for Deposition of Amyloid β Proteins
To determine the deposition of Aβ1‐40 and Aβ1‐42, brain sections were immunostained with anti‐Aβ1‐40 antibody (1:500, Code No. 18580; Immuno‐Biological Laboratories Co, Ltd, Gunma, Japan) and anti‐Aβ1‐42 antibody (1:500, Code No. 18582; Immuno‐Biological Laboratories Co, Ltd, Gunma) overnight at 4°C, reacted with horseradish peroxidase‐conjugated anti‐rabbit IgG secondary antibody for 1 hour at room temperature, and visualized with 3,3′‐diaminobenzidine (Dako Cytomation, Glostrup, Denmark), according to our previous method.18 The deposition of Aβ1‐40 and Aβ1‐42 in the 2 fields of the hippocampal CA1 region and somatosensory cortex on both sides for each animal at ×200 magnification were calculated by WinRoof version 5.8 analysis software (Mitani Corporation, Fukui, Japan) and were expressed as a percentage of the positive area per region of interest.
For assessment of CAA, brain sections were incubated with an antibody against the basement membrane marker collagen IV (Col IV; 1:200, rabbit; Abcam, Cambridge, MA) followed by Alexa Fluor 568 goat anti‐rabbit secondary IgG (1:200; Invitrogen, Carlsbad, CA), immersed in a solution of thioflavin S (0.05% in 50% ethanol) (Sigma‐Aldrich, St. Louis, MO).34 To quantify the thioflavin S–positive cortical arteries, photos were taken in 2 cortical branches of the middle cerebral artery on the cortical surface of each hemisphere (total of 4 cortical arteries) at ×400 magnification using WinRoof Version 5.8 (Mitani Corporation, Fukui, Japan), and they were expressed as the mean of percentage area of thioflavin S deposition in each mouse.
Measurement of Cardiac Fibrosis
Five‐micrometer slices from left ventricles were stained with Sirius Red F3BA (0.5% wt/vol in saturated aqueous picric acid; Aldrich Chemical Company, St. Louis, MO) for the measurement of cardiac interstitial fibrosis. The positive area of fibrosis per field area was assessed by examining at least 10 fields per mouse using WinRoof Version 5.8 (Mitani Corporation, Fukui, Japan), as previously described.29
Measurement of Gastrocnemius Muscle Fiber Size
Five‐micrometer slices from the gastrocnemius muscle were stained with hematoxylin‐eosin. For assessment of muscle fiber size, we used the minimal Feret diameter concept.35 To evaluate size of gastrocnemius, at least 100 gastrocnemius muscle fibers were measured by Image J software (National Institutes of Health, Bethesda, MD) using the minimal Feret diameter.35
Statistical Analysis
All measurements described above were made by an examiner without knowledge of the group to which the mice belonged. All data were expressed as mean±SEM, and statistical analysis was performed using GraphPad Prism (version 6.01) for Windows (GraphPad Software Inc, La Jolla, CA) and Statcel (OMS Publication, Saitama, Japan). Data on time course experiments were analyzed by 2‐way ANOVA with repeated measures followed by Tukey post hoc test for multiple comparisons. Statistical significance was tested with 1‐way ANOVA followed by Tukey post hoc test between groups. When a normal distribution was not confirmed among comparison groups, or similar variances were not obtained among comparison groups, data were analyzed with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test. Unpaired Student t test was used for comparison between 2 groups. Differences were considered statistically significant at a value of P<0.05 in all the tests. The method of statistical analysis in each measurement is described in the figure legends.
Results
Body Weight, Blood Pressure, and Metabolic Cage Data
Because 1 mouse in the 5XFAD group with angiotensin II ICV infusion showed brain injury attributed to surgical procedure for ICV cannula implantation, that mouse was excluded from this analysis. Body weight of wild‐type mice was significantly decreased by angiotensin II ICV infusion for 4 weeks compared to vehicle (P<0.01), but there was no significant decrease in body weight of 5XFAD mice by angiotensin II ICV infusion compared with vehicle (Figure 2A). Systolic blood pressure was significantly increased by angiotensin II ICV infusion compared to vehicle in either 5XFAD (P<0.01) or wild‐type mice (P<0.01), and blood pressure elevation by angiotensin II was comparable between the 2 strains (Figure 2B). There was no significant difference in food intake between 5XFAD and wild‐type mice regardless of angiotensin II infusion (Figure 2C). Compared to vehicle, water intake was significantly increased by angiotensin II ICV infusion in wild‐type (P<0.01) or 5XFAD (P<0.05) mice, and the increase in water intake by angiotensin II was comparable between the strains (Figure 2D). The increase in urine volume by angiotensin II ICV infusion was similar between 5XFAD and wild‐type mice (Figure 2E).
Figure 2.
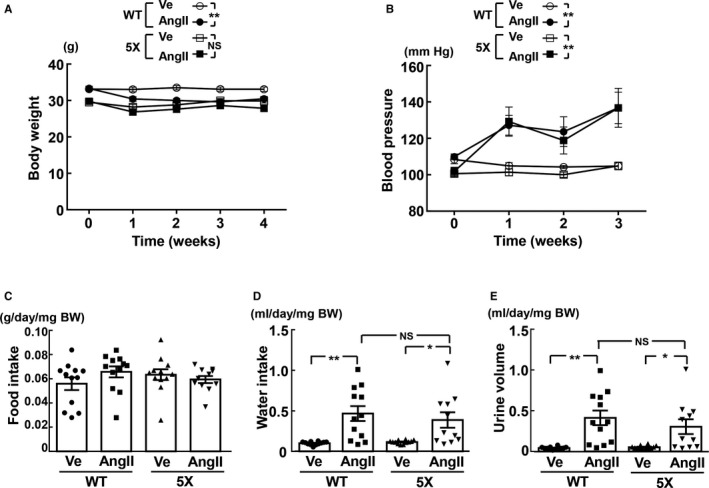
Time course of body weight (A), time course of blood pressure (B), 24‐hour food intake (C), 24‐hour water intake (D), and 24‐hour urine volume (E). 5X indicates 5XFAD mice; AngII, angiotensin II infusion; BW, body weight; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice. *P<0.05; **P<0.01. Values are means±SEM. n=12 in WT‐Ve, n=12 in WT‐AngII, n=12 in 5X‐Ve, n=11 or 12 in 5X‐AngII. A and B, Statistical analysis was performed by 2‐factor ANOVA with repeated measures followed by Tukey post hoc test between groups. C through E, Statistical significance was tested with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test.
Cognitive Function
As shown by the hidden platform test on the Morris water maze test in Figure 3A and 3B, in 5XFAD mice, escape latency (P<0.05) and cumulative escape latency (P<0.05) of the angiotensin II group were significantly greater than those of the vehicle group. On the other hand, there was no significant difference in escape latency and cumulative escape latency between the angiotensin II and vehicle groups of wild‐type mice. As shown by the probe test of the Morris water maze test in Figure 3C, in 5XFAD mice, the angiotensin II group exhibited fewer times across the platform than the vehicle group (P<0.05), although the number was not significantly different between angiotensin II and vehicle groups of wild‐type mice. As shown in Figure 3D, there was no significant difference in escape latency of the visible test between angiotensin II and vehicle groups in both strains.
Figure 3.
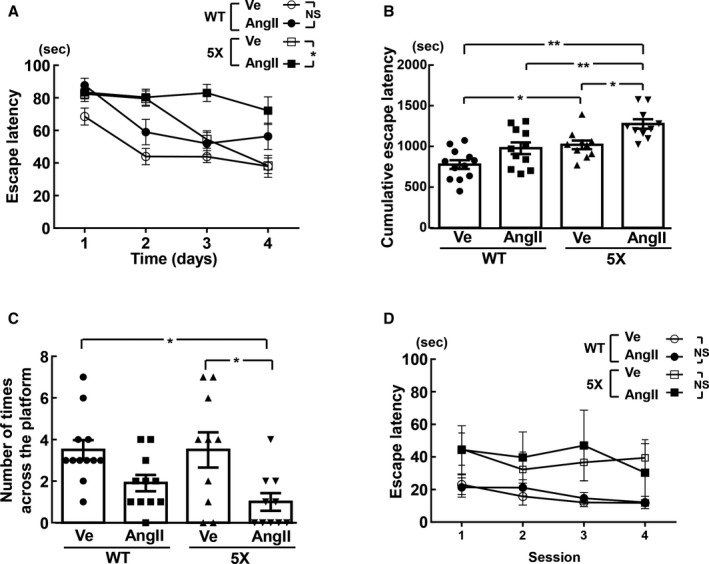
Escape latency and cumulative escape latency of the hidden platform test (A and B, respectively), number of times across the platform of the probe test (C), and visible test (D) estimated by Morris water maze test. A and B, In hidden test, escape latency at each day and cumulative escape latency, respectively, are shown for each group of mice. C, Probe trial indicates number of times across the platform for each group of mice. *P<0.05; **P<0.01. Values are means±SEM; n=12 in WT‐Ve, n=11 in WT‐AngII, n=10 in 5X‐Ve, n=10 in 5X‐AngII. A and D, Statistical analysis was performed by 2‐factor ANOVA with repeated measures followed by Tukey post hoc test between each group. B and C, Statistical significance was tested with 1‐way ANOVA followed by the Tukey multiple‐comparison post hoc test between groups. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; BW, body weight; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
Cerebral Macrophage/Microglia and Oxidative Stress
As shown in Figure 4A and 4B, in 5XFAD mice, angiotensin II ICV infusion significantly enhanced hippocampal CD68‐positive cell (macrophage/microglia) numbers (P<0.05) and increased superoxide levels (P<0.05) compared with vehicle, but angiotensin II failed to increase them in wild‐type mice. In addition, as shown in Figure 5A and 5B, there was a trend toward an increase in cortical macrophage/microglia and superoxide levels in the angiotensin II group compared with the vehicle group in 5XFAD mice, although the difference did not reach statistical significance.
Figure 4.
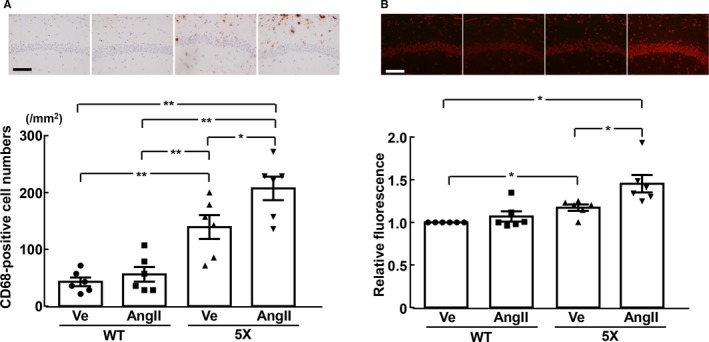
Hippocampal inflammatory cell (CD68‐positive cell) number (A) and relative fluorescence of dihydroethidium‐stained hippocampus (B). Upper panels (A and B) indicate representative microphotographs of hippocampal sections stained with anti‐CD68 antibody and dihydroethidium, respectively. *P<0.05; **P<0.01. Values are means±SEM; n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. A, Statistical significance was tested with 1‐way ANOVA followed by the Tukey multiple‐comparison post hoc test between groups. B, Statistical significance was tested with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test. Scale bar=100 μm. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
Figure 5.
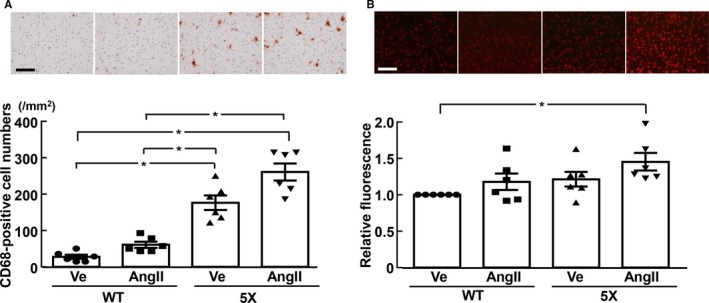
Cortical inflammatory cell (CD68‐positive cell) number (A) and relative fluorescence of dihydroethidium‐stained cortical sections (B). A and B, Upper panels indicate representative microphotographs of cortical sections stained with anti‐CD68 antibody and dihydroethidium, respectively. *P<0.05. Values are means±SEM; n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. Statistical significance was tested with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test. Scale bar=100 μm. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
Cerebrovascular and Parenchymal Amyloid Deposition and Cerebral LSP
As shown by quantification of the collagen IV and thioflavin S double‐stained area in Figure 6A, the percentage area of Aβ deposition in the cortical branch of the middle cerebral artery was significantly greater in 5XFAD mice infused with angiotensin II than in those infused with vehicle (P<0.01). On the other hand, cerebrovascular Aβ deposition was not detectable in wild‐type mice, irrespective of angiotensin II infusion (data not shown). As shown in Figure 6B, cerebral LSP was significantly less in 5XFAD mice infused with angiotensin II than that in wild‐type mice infused with vehicle (P<0.05).
Figure 6.
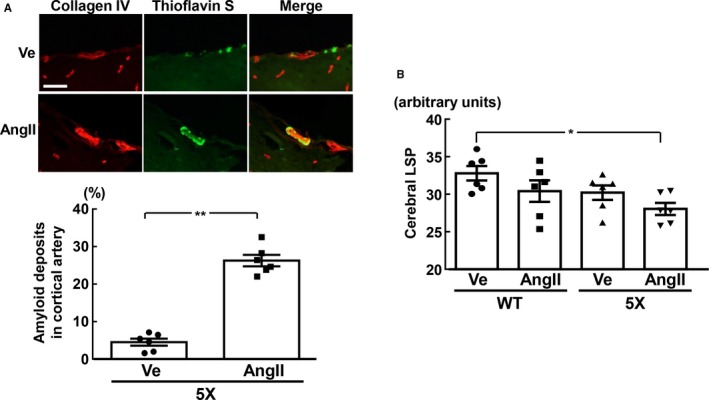
Deposition of amyloid β in cerebral cortical arteries (A) and cerebral laser speckle perfusion (LSP; B). A, Upper panels indicate representative photomicrographs of collagen IV (red) and thioflavin S (green) double staining in cortical branch of middle cerebral artery. B, Cerebral LSP was measured in the left hemisphere because ICV infusion in this study was performed in the right hemisphere. *P<0.05; **P<0.01. Values are means±SEM; n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. A, Statistical analysis was performed by unpaired Student t test. B, Statistical significance was tested with 1‐way ANOVA followed by the Tukey multiple comparison post hoc test between groups. Scale bar=50 μm. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
As shown in Figure 7, there was no significant difference between vehicle and angiotensin II groups of 5XFAD mice regarding percentage area of Aβ1‐42 or Aβ1‐40 deposits in hippocampus or cortex. In both groups of wild‐type mice, hippocampal or cortical Aβ1‐42 or Aβ1‐40 deposits were undetectable (data not shown).
Figure 7.
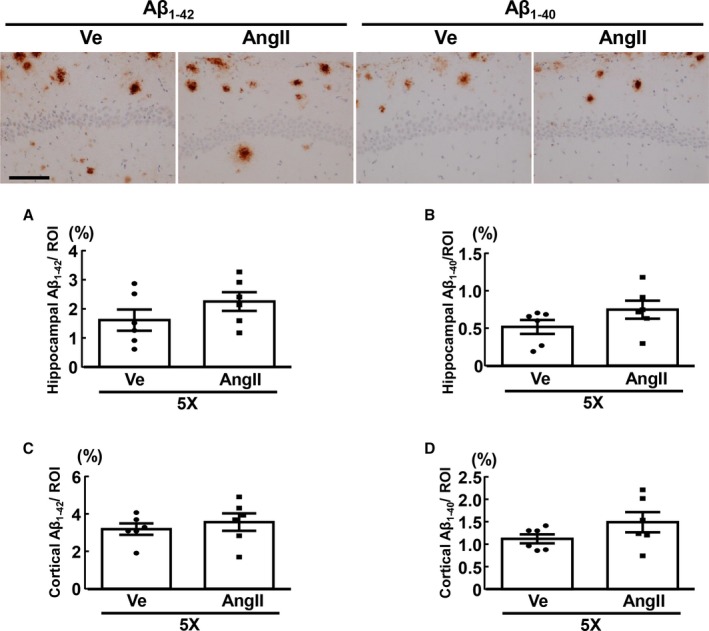
Deposition of hippocampal Aβ1‐42 (A) and Aβ1‐40 (B), and cortical Aβ1‐42 (C) and Aβ1‐40 (D) of 5XFAD mice. Upper panels indicate representative photomicrographs of hippocampal sections stained with anti‐Aβ1‐42 (left 2 panels) and anti‐Aβ1‐40 (right 2 panels) antibodies from 2 groups of 5XFAD. Values are means±SEM; n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. Statistical analysis was performed by unpaired Student t test. Scale bar=100 μm. 5X indicates 5XFAD mice; Aβ, amyloid β; AngII, angiotensin II infusion; ROI, region of interest; Ve, vehicle infusion.
Cerebral IgG Extravasation
As shown in Figure 8, cerebral IgG extravasation in 5XFAD mice was not significantly increased by angiotensin II, but cerebral IgG extravasation of angiotensin II‐infused 5XFAD mice was greater than that of vehicle‐infused wild‐type mice (P<0.05).
Figure 8.
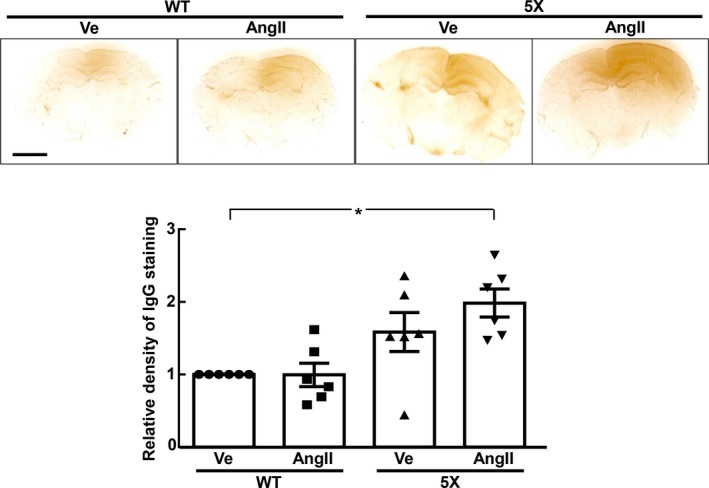
Cerebral IgG extravasation of each group of mice. Upper panels indicate representative brain sections immunostained with IgG antibody. *P<0.05. Values are means±SEM; n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. Statistical significance was tested with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; IgG, immunoglobulin G; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
Tibia Length, Cardiac Weight, Cardiac Fibrosis, and Cardiac Macrophage
As shown in Figure 9A, in wild‐type mice, body weight at the end of the infusion for 4 weeks was smaller in the angiotensin II group than in the vehicle group (P<0.05), but body weight did not significantly differ between vehicle and angiotensin II groups of 5XFAD mice. Tibia length was similar among all 4 groups of mice (Figure 9B). As shown in Figure 9C, left ventricular weight of wild‐type mice was greater in the angiotensin II group than in the vehicle group (P<0.05), but left ventricular weight did not significantly differ between vehicle and angiotensin II groups of 5XFAD mice. As shown in Figure 9D and 9E, cardiac fibrosis (P<0.01) and macrophage numbers (P<0.05) of wild‐type mice were greater in the angiotensin II group than in the vehicle group. On the other hand, cardiac fibrosis and macrophage numbers did not differ between vehicle and angiotensin II groups of 5XFAD mice.
Figure 9.
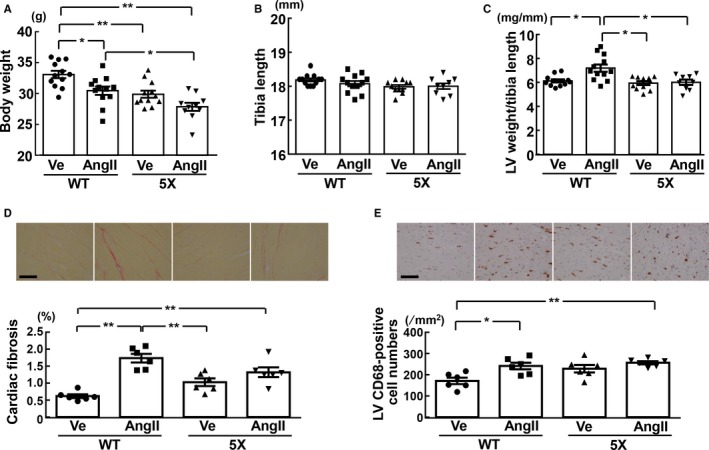
Body weight (A), tibia length (B), left ventricular (LV) weight per tibia length (C), LV fibrosis (D), and LV CD68‐positive cell numbers (E) of each group of mice. D, Upper panels show representative photomicrographs of Sirius red–stained cardiac sections. E, Upper panels show representative photomicrographs of CD68‐stained cardiac sections. *P<0.05; **P<0.01. Values are means±SEM. A through C, n=12 in WT‐Ve, n=12 in WT‐AngII, n=11 or 12 in 5X‐Ve, n=9 or 11 in 5X‐AngII. D and E, n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. A, B, D, and E, Statistical analysis was performed by 1‐way ANOVA followed by the Tukey multiple‐comparison post hoc test between groups. C, Statistical significance was tested with Kruskal‐Wallis test followed by Steel‐Dwass post hoc test. Scale bar=100 μm. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; Ve, vehicle infusion; WT, control wild‐type (C57/BL6J) mice.
Motor Activity, and Weight, Minimum Feret Diameter, and Macrophage of Gastrocnemius Muscle
As shown by the Rotarod test in Figure 10A, 5XFAD mice infused with angiotensin II tended to have less latency to fall compared with those infused with vehicle and had a shorter latency to fall than wild‐type mice infused with either vehicle (P<0.01) or angiotensin II (P<0.01).
Figure 10.
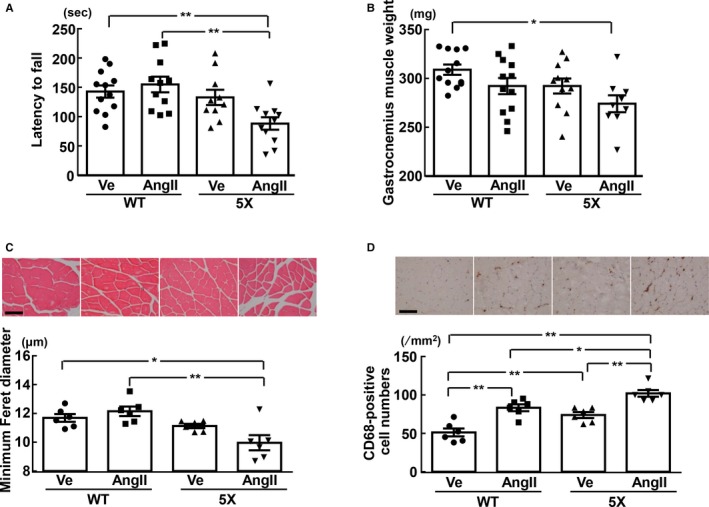
Latency to fall in rotarod test (A), and the weight (B), minimum Feret diameter (C), and CD68‐positive cell numbers (D) of gastrocnemius muscle. C and D, Upper panels show representative photomicrographs of gastrocnemius muscle sections stained with hematoxylin‐eosin and CD68 antibody, respectively. *P<0.05; **P<0.01. Values are means±SEM. A and B, n=12 in WT‐Ve, n=11 or 12 in WT‐AngII, n=10 or 11 in 5X‐Ve, n=9 or 11 in 5X‐AngII. C and D, n=6 in WT‐Ve, n=6 in WT‐AngII, n=6 in 5X‐Ve, n=6 in 5X‐AngII. Statistical significance was tested with 1‐way ANOVA followed by the Tukey multiple‐comparison post hoc test between groups. Scale bar=100 μm. 5X indicates 5XFAD mice; AngII, angiotensin II infusion; Ve, vehicle infusion; WT, control wild (C57/BL6J) mice.
Gastrocnemius muscle weight in 5XFAD mice infused angiotensin II was smaller than that in wild‐type mice infused with vehicle (P<0.05) (Figure 10B). 5XFAD mice infused with angiotensin II had smaller minimum Feret diameter of gastrocnemius muscle fiber than wild‐type mice infused with vehicle (P<0.05) or angiotensin II (P<0.01) (Figure 10C). As shown in Figure 10D, gastrocnemius muscle macrophage numbers in 5XFAD mice subjected to angiotensin II infusion were significantly larger than those subjected to vehicle infusion (P<0.01), and were also greater than those of the angiotensin II group (P<0.05) and vehicle group (P<0.01) of wild‐type mice.
Discussion
The major findings of our present work were that AD mice exhibited more susceptibility to brain angiotensin II–induced cognitive impairment than control mice, and this vulnerability of AD mice to brain angiotensin II was associated with the enhancement of hippocampal inflammation and oxidative stress and with the increase in cerebrovascular Aβ deposition. Therefore, our present work provided the evidence suggesting the critical role of brain angiotensin II in cognitive impairment and brain injury in AD. Other intriguing novel findings of this work were that AD mice had less cardiac compensatory response to brain angiotensin II–induced hypertension than control mice and that AD mice were more susceptible to brain angiotensin II–induced skeletal muscle atrophy and injury than control mice, thereby suggesting the potential contribution of brain angiotensin II to cardiac and skeletal muscle injuries in AD.
In the present study, we used 5XFAD mice because 5XFAD mice recapitulate major features of AD pathology and are regarded as 1 of the popular models of AD.18, 19, 20, 27 To examine spatial learning and memory function of 5XFAD mice, we used the Morris water maze test because it is the most popular behavioral assay for detecting AD‐relevant cognitive impairments across species.36, 37 We found that 5XFAD mice were more prone to angiotensin II–induced cognitive impairment than control mice, as evidenced by the findings of the water maze test. Cognitive function estimated by the Morris water maze test is closely related to hippocampal function.37 Of note, centrally administered angiotensin II significantly increased hippocampal oxidative stress and inflammation in 5XFAD mice but not in control mice, as evidenced by the findings of hippocampal dihydroethidium staining and CD68 immunohistochemistry. Taken together with the fact that inflammation or oxidative stress plays a key role in the development of cognitive impairment in AD,3, 38, 39, 40 our present observations suggested that the vulnerability of 5XFAD mice to brain angiotensin II–induced cognitive impairment may be attributed to the enhancement of hippocampal oxidative stress and inflammation by brain angiotensin II, although further study investigating the impact of antioxidant in 5XFAD mice is required to define our proposal.
Cerebrovascular Aβ accumulation, ie, CAA, plays a pivotal role in the pathogenesis of AD through the disturbance of cerebral blood flow and disruption of the blood‐brain barrier.3, 4, 5 Therefore, we examined the effect of centrally administered angiotensin II on cerebrovascular Aβ deposition. Of note are the observations that the deposition of Aβ in cerebral cortical artery was significantly increased by centrally administered angiotensin II in 5XFAD mice, whereas cerebrovascular Aβ deposition was not detectable in wild‐type mice regardless of central angiotensin II infusion. Cerebral LSP was significantly less in 5XFAD mice subjected to central angiotensin II infusion than control mice with vehicle infusion, although the difference between angiotensin II and vehicle groups of 5XFAD did not reach statistical significance. Furthermore, we also examined the effect of cerebral angiotensin II on blood‐brain barrier of AD mice because the blood‐brain barrier is involved in cognitive impairment of AD,41, 42 and the inhibition of endogenous angiotensin II with an angiotensin receptor blocker lessens the disruption of the blood‐brain barrier in 5XFAD mice with chronic kidney disease.43 In the present study, the disruption of blood‐brain barrier in AD mice with central angiotensin II administration significantly occurred compared with wild‐type mice, as evidenced by cerebral IgG extravasation. Taken together with the fact that CAA plays a causal role in the impairment of cerebral blood flow and disruption of the blood‐brain barrier, the enhancement of CAA by central angiotensin II might be partially involved in angiotensin II–induced cognitive impairment in 5XFAD mice. On the other hand, brain angiotensin II infusion did not significantly enhance cerebral parenchymal Aβ deposition in 5XFAD mice, thereby suggesting the preferential involvement of brain angiotensin II in CAA rather than parenchymal Aβ deposition.
Solid evidence indicates that angiotensin II ICV infusion causes blood pressure elevation and thirst in rodents.23, 24, 44 In the present study, the extent of blood pressure elevation and the increase in water intake by central angiotensin II infusion did not significantly differ between 5XFAD mice and wild‐type mice. Thus, in contrast to more susceptibility of 5XFAD mice regarding cognitive impairment, 5XFAD mice showed normal pressor response or dipsogenic response to brain angiotensin II. Heart failure is one of the most frequent comorbidities prevailing in AD patients.14 Furthermore, the renin‐angiotensin system is well known to play a key role in the pathogenesis of cardiovascular diseases and heart failure.45 Therefore, in the present study, we also examined the effect of centrally administered angiotensin II on cardiac tissue of 5XFAD mice compared to control mice. Notably, centrally administered angiotensin II significantly increased cardiac left ventricular weight in control mice but failed to increase left ventricular weight in 5XFAD mice despite comparable blood pressure elevation between the strains. It is possible that cardiac hypertrophy in control mice subjected to central angiotensin II infusion might be attributed to angiotensin II leaked into the peripheral circulation from cerebrospinal fluid. However, previous studies confirm that the dose (20 μg/[kg·h]) of angiotensin II infused ICV in the present study does not significantly increase the circulating angiotensin II levels.23, 24 Thus, in the present study, it is unlikely that significant amounts of angiotensin II might leak into the peripheral circulation from cerebrospinal fluid. These findings indicate that cardiac hypertrophy observed in control mice subjected to angiotensin II ICV infusion is mainly secondary to angiotensin II–induced blood pressure elevation rather than a direct cardiac action of leaked angiotensin II. Further, centrally administered angiotensin II significantly enhanced cardiac fibrosis and inflammatory cell numbers in control mice but did not significantly enhance them in 5XFAD mice. Therefore, our present study suggests that 5XFAD mice may exhibit less compensatory cardiac response to pressure overload (angiotensin II–induced hypertension) than control mice. Collectively, these findings shed new light on the potential link of angiotensin II to cardiac disease in AD, although further study is needed to define our proposal.
Epidemiological and observational studies15, 16, 17 suggest that AD is significantly associated with not only heart failure but also sarcopenia or reduced muscle mass and function. It is assumed that AD and sarcopenia appear to share common underlying mechanisms. Interestingly, preclinical and clinical studies suggest that the renin‐angiotensin system may participate in the pathophysiology of sarcopenia.46, 47, 48 Systemic infusion of angiotensin II is well known to cause skeletal muscle atrophy and wasting in rodents, and this skeletal muscle injury is attributed to the enhancement of oxidative stress47 and inflammation48 by angiotensin II. These findings encouraged us to examine the effect of angiotensin II ICV infusion on skeletal muscle of 5XFAD mice. We obtained the evidence that brain angiotensin II caused the disturbance of motor activity in 5XFAD mice compared to control mice as evidenced by the findings of Rotarod test, and this motor disturbance in 5XFAD mice by brain angiotensin II was associated with skeletal muscle atrophy and greater skeletal muscle inflammation in 5XFAD mice. Thus, our present work suggests that AD mice are susceptible to brain angiotensin II–induced skeletal muscle wasting and that brain angiotensin II may be one of the contributing factors to shared mechanisms common to both AD and sarcopenia. However, further study is needed to define the significance of brain angiotensin II in comorbidity of sarcopenia and AD.
Detailed investigations regarding the effect of centrally administered angiotensin II in an animal model of AD are very scarce. To date, there is 1 detailed recent report investigating the long‐term effect of systemic angiotensin II infusion (but not ICV infusion) on brain functional and structural alterations in the AD mouse model (APPPS1).49 The previous report49 showed that systemic infusion of angiotensin II (3‐fold greater dose than our present work) for 2.5 months (versus 4 weeks in our study) in AD mice caused ≈60 mm Hg rise in blood pressure (versus 20 mm Hg rise in our study) and chronic angiotensin II–induced severe hypertension impaired cognitive function in AD mice. Differing from the purpose of our present work addressing to the role of brain angiotensin II, the previous report49 was focused on the long‐term effect of severe hypertension induced by peripheral angiotensin II rather than angiotensin II itself in AD. Furthermore, the effect on oxidative stress, inflammation, or CAA and on cardiac and skeletal muscle tissues of AD mice was not examined in the previous report.49 Therefore, there is the novelty in our present work, which investigated the comprehensive effect of brain angiotensin II in an AD animal model.
Study Limitations
There are several study limitations in this study. First, the present study examined the effect of exogenous angiotensin II in AD mice because the main purpose of this study was to examine the susceptibility of AD mice to central angiotensin II. Therefore, further study is needed to confirm whether our present findings can apply to the role of endogenous angiotensin II. Second, the prevalence of AD is greater in women than in men. However, the sexual cycle of female mice differs among individual mice and potentially affects mouse behavior and the action of angiotensin II. Therefore, in the present study, we used only male mice. It is a key issue to confirm that our present findings can apply to female mice. Third, C57BL/6J mice instead of littermates were used as the control mice in the present study because 5XFAD mice were backcrossed with their C57BL/6J background at least 10 generations to reduce genetic variation. Fourth, the present study did not allow us to determine the concentrations of angiotensin II in cerebrospinal fluid of mice because of technical difficulty. However, in humans, cerebrospinal fluid angiotensin II levels are not so different from plasma angiotensin II levels, and cerebrospinal angiotensin II levels are independent of plasma angiotensin II levels.50, 51 Therefore, the measurement of cerebrospinal fluid angiotensin II in mice is valuable to define the clinical implication of our experimental work. Fifth, the measurement of oxygenation, end‐tidal CO2, etc was important for interpretation of cerebral LSP data because cerebral blood flow is highly dependent on O2 and CO2 status. However, the measurement of such parameters was not performed in our present study. Finally, the present study addressed only the association of cognitive impairment with oxidative stress or inflammation and provided no causal relationships between them.
In conclusion, we obtained evidence supporting the concept that brain angiotensin II is involved in the progression of cognitive impairment in AD, which is associated with the enhancement of oxidative stress and inflammation and the progression of CAA. Furthermore, our work suggests that angiotensin II may be involved in the pathophysiology of cardiac disease or skeletal muscle injury in AD. Thus, our present work provides a novel insight into the potential role of angiotensin II in AD‐related functional and structural abnormalities not only in the brain but also in peripheral organs.
Sources of Funding
This work was partially supported by research grants from Mitsui Sumitomo Insurance Welfare Foundation.
Disclosures
Kim‐Mitsuyama received lecture fees and research grant from Astellas, AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, Novartis, Sionogi, Takeda, Kyowa Hakko Kirin. All other co‐authors declare that they have no financial competing interests. No nonfinancial conflicts of interest exist for any of the authors.
(J Am Heart Assoc. 2017;6:e004897 DOI: 10.1161/JAHA.116.004897.)28428194
References
- 1. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011;377:1019–1031. [DOI] [PubMed] [Google Scholar]
- 2. Iadecola C. Hypertension and dementia. Hypertension. 2014;64:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. [DOI] [PubMed] [Google Scholar]
- 4. Nicoll JA, Yamada M, Frackowiak J, Mazur‐Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro‐CAA position statement. Neurobiol Aging. 2004;25:589–597; discussion 603‐584. [DOI] [PubMed] [Google Scholar]
- 5. Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kehoe PG, Passmore PA. The renin‐angiotensin system and antihypertensive drugs in Alzheimer's disease: current standing of the angiotensin hypothesis? J Alzheimers Dis. 2012;30(suppl 2):S251–S268. [DOI] [PubMed] [Google Scholar]
- 7. Wright JW, Kawas LH, Harding JW. A role for the brain RAS in Alzheimer's and Parkinson's diseases. Front Endocrinol (Lausanne). 2013;4:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ongali B, Nicolakakis N, Tong XK, Aboulkassim T, Papadopoulos P, Rosa‐Neto P, Lecrux C, Imboden H, Hamel E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer's disease model. Neurobiol Dis. 2014;68:126–136. [DOI] [PubMed] [Google Scholar]
- 9. Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K, Kano M, Ogihara T, Rakugi H, Morishita R. Angiotensin receptor blocker prevented β‐amyloid‐induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension. 2009;54:1345–1352. [DOI] [PubMed] [Google Scholar]
- 10. Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiu WC, Ho WC, Lin MH, Lee HH, Yeh YC, Wang JD, Chen PC; Health Data Analysis in Taiwan (hDATa) Research Group . Angiotension receptor blockers reduce the risk of dementia. J Hypertens. 2014;32:938–947. [DOI] [PubMed] [Google Scholar]
- 12. Hajjar I, Brown L, Mack WJ, Chui H. Impact of angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol. 2012;69:1632–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hajjar I, Hart M, Chen YL, Mack W, Milberg W, Chui H, Lipsitz L. Effect of antihypertensive therapy on cognitive function in early executive cognitive impairment: a double‐blind randomized clinical trial. Arch Intern Med. 2012;172:442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cermakova P, Eriksdotter M, Lund LH, Winblad B, Religa P, Religa D. Heart failure and Alzheimer's disease. J Intern Med. 2015;277:406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beydoun MA, Beydoun HA, Gamaldo AA, Rostant OS, Dore GA, Zonderman AB, Eid SM. Nationwide inpatient prevalence, predictors, and outcomes of Alzheimer's disease among older adults in the United States, 2002–2012. J Alzheimers Dis. 2015;48:361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burns JM, Johnson DK, Watts A, Swerdlow RH, Brooks WM. Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol. 2010;67:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tolea MI, Galvin JE. Sarcopenia and impairment in cognitive and physical performance. Clin Interv Aging. 2015;10:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uekawa K, Hasegawa Y, Senju S, Nakagata N, Ma M, Nakagawa T, Koibuchi N, Kim‐Mitsuyama S. Intracerebroventricular infusion of angiotensin‐(1‐7) ameliorates cognitive impairment and memory dysfunction in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2016;53:127–133. [DOI] [PubMed] [Google Scholar]
- 19. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet‐Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maarouf CL, Kokjohn TA, Whiteside CM, Macias MP, Kalback WM, Sabbagh MN, Beach TG, Vassar R, Roher AE. Molecular differences and similarities between Alzheimer's disease and the 5XFAD transgenic mouse model of amyloidosis. Biochem Insights. 2013;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bahner U, Geiger H, Palkovits M, Luft FC, Heidland A. Angiotensin II‐induced hypertension: effects on central and peripheral atrial natriuretic peptide. Hypertens Res. 1995;18:279–284. [DOI] [PubMed] [Google Scholar]
- 23. Bruner CA, Weaver JM, Fink GD. Sodium‐dependent hypertension produced by chronic central angiotensin II infusion. Am J Physiol. 1985;249:H321–H327. [DOI] [PubMed] [Google Scholar]
- 24. Di Nicolantonio R, Mendelsohn FA, Hutchinson JS, Takata Y, Doyle AE. Dissociation of dipsogenic and pressor responses to chronic central angiotensin II in rats. Am J Physiol. 1982;242:R498–R504. [DOI] [PubMed] [Google Scholar]
- 25. Toyama K, Koibuchi N, Hasegawa Y, Uekawa K, Yasuda O, Sueta D, Nakagawa T, Ma M, Kusaka H, Lin B, Ogawa H, Ichijo H, Kim‐Mitsuyama S. ASK1 is involved in cognitive impairment caused by long‐term high‐fat diet feeding in mice. Sci Rep. 2015;5:10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasegawa Y, Nakagawa T, Uekawa K, Ma M, Lin B, Kusaka H, Katayama T, Sueta D, Toyama K, Koibuchi N, Kim‐Mitsuyama S. Therapy with the combination of amlodipine and irbesartan has persistent preventative effects on stroke onset associated with BDNF preservation on cerebral vessels in hypertensive rats. Transl Stroke Res. 2016;7:79–87. [DOI] [PubMed] [Google Scholar]
- 27. Lin B, Hasegawa Y, Takane K, Koibuchi N, Cao C, Kim‐Mitsuyama S. High‐fat‐diet intake enhances cerebral amyloid angiopathy and cognitive impairment in a mouse model of Alzheimer's disease, independently of metabolic disorders. J Am Heart Assoc. 2016;5:e003154 DOI: 10.1161/JAHA.115.003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toyama K, Koibuchi N, Uekawa K, Hasegawa Y, Kataoka K, Katayama T, Sueta D, Ma MJ, Nakagawa T, Yasuda O, Tomimoto H, Ichijo H, Ogawa H, Kim‐Mitsuyama S. Apoptosis signal‐regulating kinase 1 is a novel target molecule for cognitive impairment induced by chronic cerebral hypoperfusion. Arterioscler Thromb Vasc Biol. 2014;34:616–625. [DOI] [PubMed] [Google Scholar]
- 29. Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, Ogawa H, Kim‐Mitsuyama S. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens. 2010;28:340–352. [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto E, Lai ZF, Yamashita T, Tanaka T, Kataoka K, Tokutomi Y, Ito T, Ogawa H, Kim‐Mitsuyama S. Enhancement of cardiac oxidative stress by tachycardia and its critical role in cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:2057–2069. [DOI] [PubMed] [Google Scholar]
- 31. Nakagawa T, Hasegawa Y, Uekawa K, Ma M, Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Maeda M, Kuratsu J, Kim‐Mitsuyama S. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J Am Heart Assoc. 2013;2:e000375 DOI: 10.1161/JAHA.113.000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richmon JD, Fukuda K, Maida N, Sato M, Bergeron M, Sharp FR, Panter SS, Noble LJ. Induction of heme oxygenase‐1 after hyperosmotic opening of the blood‐brain barrier. Brain Res. 1998;780:108–118. [DOI] [PubMed] [Google Scholar]
- 33. Tsubokawa T, Solaroglu I, Yatsushige H, Cahill J, Yata K, Zhang JH. Cathepsin and calpain inhibitor E64d attenuates matrix metalloproteinase‐9 activity after focal cerebral ischemia in rats. Stroke. 2006;37:1888–1894. [DOI] [PubMed] [Google Scholar]
- 34. Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post‐mortem Alzheimer's disease brain. Neurobiol Dis. 2015;74:281–294. [DOI] [PubMed] [Google Scholar]
- 35. Briguet A, Courdier‐Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx‐mouse. Neuromuscul Disord. 2004;14:675–682. [DOI] [PubMed] [Google Scholar]
- 36. Possin KL, Sanchez PE, Anderson‐Bergman C, Fernandez R, Kerchner GA, Johnson ET, Davis A, Lo I, Bott NT, Kiely T, Fenesy MC, Miller BL, Kramer JH, Finkbeiner S. Cross‐species translation of the Morris maze for Alzheimer's disease. J Clin Invest. 2016;126:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han BH, Zhou ML, Johnson AW, Singh I, Liao F, Vellimana AK, Nelson JW, Milner E, Cirrito JR, Basak J, Yoo M, Dietrich HH, Holtzman DM, Zipfel GJ. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci USA. 2015;112:E881–E890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luque‐Contreras D, Carvajal K, Toral‐Rios D, Franco‐Bocanegra D, Campos‐Pena V. Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer's disease? Oxid Med Cell Longev. 2014;2014:497802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu X, Smith MA, Honda K, Aliev G, Moreira PI, Nunomura A, Casadesus G, Harris PL, Siedlak SL, Perry G. Vascular oxidative stress in Alzheimer disease. J Neurol Sci. 2007;257:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Popescu BO, Toescu EC, Popescu LM, Bajenaru O, Muresanu DF, Schultzberg M, Bogdanovic N. Blood‐brain barrier alterations in ageing and dementia. J Neurol Sci. 2009;283:99–106. [DOI] [PubMed] [Google Scholar]
- 42. Wardlaw JM, Sandercock PA, Dennis MS, Starr J. Is breakdown of the blood‐brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke. 2003;34:806–812. [DOI] [PubMed] [Google Scholar]
- 43. Nakagawa T, Hasegawa Y, Uekawa K, Kim‐Mitsuyama S. Chronic kidney disease accelerates cognitive impairment in a mouse model of Alzheimer's disease, through angiotensin II. Exp Gerontol. 2017;87:108–112. [DOI] [PubMed] [Google Scholar]
- 44. McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FA, Chai SY. The brain renin‐angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35:901–918. [DOI] [PubMed] [Google Scholar]
- 45. Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II‐mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 46. Sartiani L, Spinelli V, Laurino A, Blescia S, Raimondi L, Cerbai E, Mugelli A. Pharmacological perspectives in sarcopenia: a potential role for renin‐angiotensin system blockers? Clin Cases Miner Bone Metab. 2015;12:135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Semprun‐Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez‐Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun. 2011;409:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE. IL‐6 and serum amyloid a synergy mediates angiotensin II‐induced muscle wasting. J Am Soc Nephrol. 2009;20:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cifuentes D, Poittevin M, Dere E, Broqueres‐You D, Bonnin P, Benessiano J, Pocard M, Mariani J, Kubis N, Merkulova‐Rainon T, Levy BI. Hypertension accelerates the progression of Alzheimer‐like pathology in a mouse model of the disease. Hypertension. 2015;65:218–224. [DOI] [PubMed] [Google Scholar]
- 50. Kawamura M, Kawano Y, Yoshida K, Imanishi M, Akabane S, Matsushima Y, Kuramochi M, Shimamoto K, Ito K, Omae T. Alterations in cerebrospinal fluid angiotensin II by sodium intake in patients with essential hypertension. Clin Sci (Lond). 1989;77:389–394. [DOI] [PubMed] [Google Scholar]
- 51. Kawamura M, Kawano Y, Yoshida K, Imanishi M, Akabane S, Matsushima Y, Kuramochi M, Shimamoto K, Yamamoto K, Ito K. Cerebrospinal fluid angiotensin II in patients with essential hypertension. J Hypertens Suppl. 1988;6:S508–S510. [DOI] [PubMed] [Google Scholar]