

Abstract
Background
Peripheral vascular disease is a major diabetes mellitus‐related complication. In this study, we noted that expressions of proapoptotic p53 gene and its downstream cascade gene such as p21 are upregulated in hyperglycemia. Therefore, we investigated whether p53‐ and p21‐silenced endothelial progenitor cells (EPCs) were able to survive in hyperglycemic milieu, and whether transplantation of either p53 knockout (KO) or p21KO or p53‐ and p21‐silenced EPCs could improve collateral vessel formation and blood flow in diabetic vaso‐occlusive peripheral vascular disease mouse models.
Methods and Results
We transplanted p53 and p21KO mouse EPCs (mEPCs) into streptozotocin–induced diabetic (type 1 diabetes mellitus model) C57BL/6J and db/db (B6.BKS(D)‐Leprdb/J) (type 2 model) post–femoral artery occlusion. Similarly, Ad‐p53–silenced and Ad‐p21–silenced human EPCs (CD34+) cells were transplanted into streptozotocin‐induced diabetic NOD.CB17‐Prkdcscid/J mice. We measured blood flow at 3, 7, and 10 days and hindlimb muscles were obtained postsacrifice for mRNA estimation and CD31 staining. Enhanced blood flow was noted with delivery of p53 and p21KO mEPCs in streptozotocin‐induced diabetic C57BL/6J mice. Similar results were obtained when human Ad‐p53shEPCs(CD34+) and Ad‐p21shEPCs(CD34+) were transplanted into streptozotocin‐induced nonobese diabetic severe combined immunodeficiency mice. Gene expression analysis of p53 and p21KO EPCs transplanted hindlimb muscles showed increased expression of endothelial markers such as endothelial nitric oxide synthase, vascular endothelial growth factor A, and platelet endothelial cell adhesion molecule 1. Similarly, quantitative reverse transcriptase polymerase chain reaction of human Ad‐p53shEPCs (CD34+)– and Ad‐p21shEPCs (CD34+)–transplanted hindlimb muscles also showed increased expression of endothelial markers such as vascular endothelial growth factor A, noted primarily in the p53‐silenced EPCs group. However, such beneficial effect was not noted in the db/db type 2 diabetic mouse models.
Conclusions
Transient silencing of p53 using adenoviral vector in EPCs may have a therapeutic role in diabetic peripheral vascular disease.
Keywords: adenovirus vector, apoptosis, diabetes mellitus, endothelial progenitor cells, gene therapy
Subject Categories: Peripheral Vascular Disease; Diabetes, Type 1; Diabetes, Type 2; Gene Therapy; Stem Cells
Introduction
Diabetes mellitus affects more than 11% of US adults and is projected to nearly double by 2025.1 The presence of moderate hyperglycemia in addition to mild and moderate obesity may confer significant cardiovascular risk.2 The American Diabetes Association has reported that coronary artery disease and stroke are 3 times more common in prediabetic compared with nondiabetic patients,3 and overt diabetes mellitus increases this risk 5‐fold. Many patients with prediabetes are either overweight or obese. Both diabetes mellitus and obesity are associated with cardiovascular complications such as endothelial dysfunction, oxidative stress, endothelial cell inflammation, and cardiovascular prothrombotic states.4 These complications are primary reasons of cardiovascular disease. Cardiovascular disease includes cerebral, cardiac, and peripheral vascular diseases (PVDs). In this study, our main focus will remain on PVD as a consequence of diabetes mellitus.
One of the major consequences of diabetes mellitus is PVD. PVD is the inadequate perfusion of blood in the peripheral arteries often triggered by atherosclerosis, one of the major consequences of diabetes mellitus. Every day, 230 patients undergo diabetes mellitus‐related amputation5 due to PVD. In these patients, PVD most often occurs secondarily to total occlusion of major vessels in the limb such as the femoral or tibial artery. Therefore, it is important to improve blood flow and neovascularization to reduce amputation and the use of cell therapy could be the possible solution in diabetes mellitus‐related PVD. Endothelial progenitor cells (EPCs), precursor of mature endothelium, are good candidates for cell therapy as they demonstrate regenerative properties. Therefore, use of EPCs is promising for the treatment of ischemia‐related complications in the diabetic population.
EPCs are stem/progenitor cell precursors from the hematopoietic stem cell lineage6 and are often referred to as circulating progenitor cells. Given the appropriate environmental conditions, EPCs can perform beneficial angiogenic and vascular repair functions, including incorporation into the endothelium and release of growth factors and cytokines that promote repair.7, 8 Cell sorting for CD34+ identifies stem‐like cells with endothelial/hematopoietic lineage markings.9, 10 Controversy regarding the precise definitions of endothelial progenitor and other angiogenic cells has slowed progress in the field.9 It is generally agreed upon that EPCs (CD34+) contribute to the maintenance of vascular endothelial integrity through actions of particular endothelial lineage–directed subsets or by acting as a pool of generic progenitors.10 In addition, EPCs (CD34+) have recently received a great deal of attention in the literature for their cell therapy applications.11
However, EPCs (CD34+) are susceptible to adverse in vivo environments. Cardiometabolic disease and diabetes mellitus reduces the mobilization and circulating number of EPCs (CD34+).12 Recently, EPCs (CD34+) and colony count were reported to be lower in prediabetes mellitus patients.13 In addition, EPCs (CD34+) isolated from diabetic patients were also unable to incorporate into damaged vessels and promote repair.14
Current studies have shown that the degree of collateral vessel formation is important for salvaging an ischemic leg15, 16 and having EPCs (CD34+) at or near the site of vascular occlusion has yielded a positive outcome in not only animal models8, 17, 18 but also human subjects in a setting of intractable angina.19, 20 However, for the EPCs (CD34+) to be effective posttransplantation, the progenitor cells need to survive in proapoptotic, hyperglycemic conditions.
In this study, we investigated whether EPCs (CD34+) can survive in hyperglycemia (20 mmol/L glucose) with genetic modification, and whether such modified EPCs (CD34+) can contribute effectively to revascularization in diabetic PVD.
Methods
In Vitro Cell Culture
Mouse EPCs (mEPCs) were obtained as described in our protocol previously.18 Briefly, mouse peripheral blood mononuclear cells were isolated from circulating whole blood (collected from 3 mice in each category) by the Ficoll separation method. These cells were then cultured in EGM‐2 media (Lonza Inc, Basel, Switzerland) for 5 to 7 days and trypsinized to collect and transplant to mice.
Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza and were cultured in growth factor–supplemented EGM‐2 media in a humidified 37°C incubator containing 5% CO2 using procedures previously described.21 Media was changed every 2 to 3 days. HUVECs were exposed to normal (5.5 mmol/L) and high glucose (20 mmol/L) for 28 days. Cells were collected for gene expression and mitochondrial respiration analysis at days 7, 14, and 28.
Human peripheral blood mononuclear cells were also isolated from circulating whole blood by the Ficoll separation method. Human EPCs (CD34+) were then magnetically sorted using CD34 microBeads (human) antibody (Miltenyi Biotec, Bergisch Glagbach, Germany) and cultured in StemSpan SFEM II media (STEMCELL Technologies, Vancouver, Canada) for 2 days. Cells were then exposed to normal (5.5 mmol/L) and high glucose (20 mmol/L) for 48 hours in EGM‐2 media before they were harvested for gene expression analysis. We followed institutional review board and Institutional Animal Care and Use Committee guidelines for this study and it was approved by appropriate institutional committees.
RNA Extraction, cDNA Synthesis, and Gene Expression in Human Umbilical Vein Endothelial Cells and Endothelial Progenitor Cells
Quantitative reverse transcriptase polymerase chain reaction was used for gene expression analysis. Total mRNA from both HUVECs and EPCs were isolated by RNeasy Mini Kit (Qiagen, Hilden, Germany). The T100 Thermal Cycler (Bio‐Rad, Hercules, CA) was used to convert mRNA to cDNA by using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The CFX96 Real‐Time qPCR System (Bio‐Rad) was used to analyze the genes of interest by using TaqMan Universal Master Mix II (Applied Biosystems, Foster City, CA). Individual gene expression was normalized to housekeeping genes 18S or GAPDH.
Oxygen Consumption Rate Analysis
Oxygen consumption rate (OCR) of HUVECs exposed to normal (5.5 mmol/L) and high (20 mmol/L) glucose was analyzed using Extracellular Flux Analyzers XFp (Seahorse Bioscience). Thirty‐thousand HUVECs per well were seeded in V7‐PS mini‐plates and cultured in a humidified 37°C incubator containing 5% CO2 for 24 hours. After washing with assay medium (nonbuffered XF Base Medium Minimum DMEM [Seahorse Bioscience] pH 7.4, containing 5.5 mmol/L glucose, 1 mmol/L pyruvate, and 2 mmol/L glutamine) cells were kept in a CO2‐free incubator at 37°C for 45 minutes before assay. During the mitochondrial stress test, 3 injections were performed: oligomycin (1.1 μmol/L), carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (2.08 μmol/L), and rotenone and antimycin (0.5 μmol/L). Basal respiration was subsequently measured.
Ex‐Vivo Lentiviral Gene Transduction
Replication‐deficient lentiviral construct was used to silence mouse p53 genes in mEPCs. The replication‐deficient lentiviral constructs containing green fluorescent protein that were used in this study have been previously developed and tested by Ventura et al.22 The plasmids used for the lentivirus construct were obtained from Dr Joseph Jerry's laboratory at Pioneer Valley Life Sciences Institute, Springfield, MA. The virus was packaged and propagated using standard NIH/3T3 fibroblasts and human embryonic kidney 293 cell lines. The vector titer used was 50 multiplicity of infection. Control for the Lenti‐GFP‐p53sh that is Lenti‐GFP‐Null constructs were obtained from Cell Biolabs Inc (San Diego, CA; catalog number LTV‐400).
Ex Vivo Adenoviral Gene Transduction
Ad‐human‐P53 (TP53)‐shRNA (Vector Biolabs, Malvern, PA) was used to silence p53 and AdU6‐Human‐CDKNIA‐shRNA (Vector Biolabs) was used to silence p21. Adenovirus was propagated and titered using human embryonic kidney 293 cells (Lonza). After isolation of EPCs (CD34+) from peripheral blood mononuclear cells, cells were cultured in StemSpan SFEM II media for 5 days. Cells were subsequently transduced by virus at 100 multiplicity of infection and kept in culture for another 4 to 5 days before transplantation in mouse.
Propidium Iodide, Annexin V Staining, and Fluorescence‐Activated Cell Sorting Analysis
Human EPCs (CD34+) following hyperglycemia (20 mmol/L glucose) exposure were stained with propidium iodide (Miltenyi Biotec, 130‐093‐233) and annexin V (Miltenyi Biotec, 130‐097‐928) along with suggested isotype control using Miltenyi Biotec standard protocols. Both human EPCs (CD34+) and HUVECs were stained with propidium iodide. The cells were analyzed poststaining using BD FACSCalibur (BD Biosciences) cell analyzer.
Animals Used
Adult male (aged 4–6 weeks), wild‐type (WT) mice (C57/BL6), p21 knockout (KO) B6.129S6(Cg)‐Cdkn1atm1Led/J), p53KO (B6.129S2‐Trp53tm1Tyj/J), db/db (B6.BKS(D)‐Leprdb/J), nonobese diabetic severe combined immunodeficiency (NOD‐SCID) mice (NOD.CB17‐Prkdcscid/J) were purchased from Jackson Laboratory. A normal diet (Harlan Teklab, Global 19% Protein Extruded Rodent Diet, catalog # 2019S) was provided throughout experimentation, and 12‐hour artificial light‐dark cycle was maintained while the room temperature was kept at 21°C. For C57/BL6 and NOD.CB17‐Prkdcscid/J as a PVD model, we used n=3 mice per group.
p53 global KO animals on C57Bl6 and Balb/c background were obtained from Dr Jerry's laboratory. The mice obtained from Dr Jerry's laboratory that were p53 silenced (either on C57Bl6 or BalB/c background) were genotyped for confirmation by tail‐snip genotyping. Only homozygous animals were used to obtain p53KO EPCs. Institutional guidelines were followed for all animal procedures (Baystate Medical Center, Pioneer Valley Life Sciences Institute, Springfield, MA, and The George Washington University, Washington, DC).
Streptozotocin‐Induced Hyperglycemia
Type 1 diabetes mellitus was induced in WT and NOD‐SCID mice by intraperitoneal injection of streptozotocin (STZ). Tail vein blood glucose level was tested (by contour next blood glucose meter) at day 0 after 6 hours of fasting before STZ injection. Doses of STZ 40 mg/kg body weight were administered for 3 to 5 consecutive days. By 1 to 2 weeks post‐STZ injection, mouse tail vein blood glucose was checked to verify hyperglycemia (>250 mg/dL). Before surgery and transplanting EPCs, mice blood glucose was rechecked to confirm hyperglycemia.
Animal Surgery and Postoperative Care
Mice were anesthetized by isoflurane (according to guidelines previously described18) before operation. The right hind leg was shaved and cleaned, followed by cleaning with 70% ethanol solution and exposure of the femoral artery. The femoral artery was ligated proximally and distally using absorbable sutures. The artery was cut in between the ligatures and the ends of the arteries were cauterized. Approximately 3×105 EPCs were injected around the ligature site for each genotype. For control animals, saline was injected instead of cells. The skin was closed and sutured back using nonabsorbable sutures in an interrupted pattern. Mice were carefully monitored postoperation for 3 days, and Buprenex analgesic at a dose of 0.1 mg/kg body weight was injected twice a day to relieve postoperative pain. Saline was also administered (subcutaneous) to maintain hydration.
Laser Doppler Imaging
Laser Doppler perfusion imaging was used to assay limb vasculature perfusion. Mice were anesthetized as described above (see Method: Animal Surgery and Postoperative Care) before imaging. Mice were placed on a ventral surface and exposed against black background, followed by shaving of both hindlimbs. Vascular perfusions in both right and left hindlimbs were measured and analyzed using a Moor LDI Laser Doppler Imager (version 5.3). Images were acquired at days 3, 7, and 10 postocclusion of femoral artery for most experiments. The blood perfusion value obtained from the analyzed image of the right hindlimb (surgery/ischemic) was normalized to value of left (no surgery/nonischemic) hindlimb to calculate the ratio of blood perfusion for each mouse.
RNA Extraction, cDNA Synthesis, and Gene Expression of Hindlimb
Mouse hindlimb quadriceps (right and left) were flash frozen in liquid nitrogen after harvesting at day 28 postsurgery and kept at −80°C. Frozen tissue was then homogenized and total RNA was isolated using an RNeasy Lipid Tissue Mini Kit (Qiagen). For cDNA conversion and quantitative reverse transcriptase polymerase chain reaction, we followed the same methodology as previously discussed (see Method: RNA Extraction, cDNA Synthesis, and Gene Expression in HUVECs and EPCs).
Immunohistochemistry
Immunohistochemistry was performed to quantify platelet endothelial cell adhesion molecule 1 (PECAM‐1; CD31) expression in right and left hindlimb muscles. CD31 antibody was diluted at a 1:100 ratio (Abcam) and horseradish peroxidase–conjugated anti‐Rat ImmPress (RTU) was used as a secondary antibody followed by 3,3′‐diaminobenzidin staining.
Statistical Analysis
Results were analyzed using multiple paired t test where *P<0.05, **P<0.01, and ***P<0.005 were considered significant. The Bonferroni‐Dunn method was used to correct multiple comparisons. Every statistical analysis was performed based on at least two biological or biochemical repetitions.
Results
In this study, one of our primary goals was to assess the effect of hyperglycemia on EPCs. One of the approaches was to measure apoptosis. To test the degree of apoptosis, human EPCs (CD34+) were exposed to normal glucose and high glucose concentrations (5.5 and 20 mmol/L, respectively) for 48 hours, followed by fluorescence‐activated cell sorting analysis. Human EPCs (CD34+) were also stained with annexin V, a marker for early stages of apoptosis, and propidium iodide, a marker for late or advanced stages of apoptosis. For both staining methods, the results showed features of apoptosis of EPCs (CD34+) upon hyperglycemic exposure conditions (Figure 1A through 1C). However, mature endothelial cells such as commercially obtained HUVECs did not exhibit any apoptotic effect, even after 10 days of hyperglycemic exposure (Figure 1D).
Figure 1.
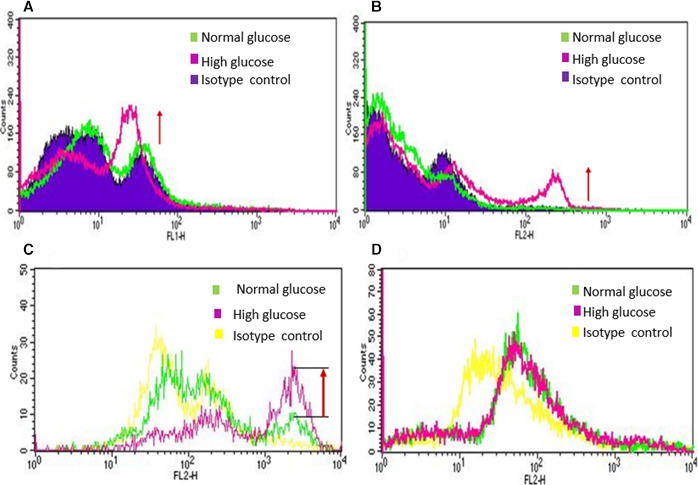
Fluorescence activated cell sorter (FACS) analysis of EPCs (CD34+) and HUVECs exposed to normal glucose (NG) and high glucose (HG): Human EPCs (CD34+) were exposed to NG (5.5 mmol/L) and HG (20 mmol/L) for 48 hours followed by FACS Analysis. A, Annexin V stained cells and B, Propidium iodide stained EPCs (CD34+) demonstrated significant degree of apoptosis takes place within 48 hours of high glucose exposure. To compare effect of NG and HG on endothelial progenitor and mature endothelial cells, C, human EPCs (CD34+) and D, HUVECs were exposed to NG and HG for 48 hours and 10 days, respectively, and cells were stained with propidium iodide. EPCs (CD34+) showed increased apoptosis by 48 hours of high glucose exposure. However, HUVECs did not show any apoptotic change even after 10 days of HG exposure compared to NG exposed cells. These data demonstrate that EPCs are more susceptible to HG mediated injury compared to mature endothelial cells (HUVECs).
Another approach was targeted gene expression analysis to examine the effect of high glucose on EPCs. For this purpose, quantitative reverse transcriptase polymerase chain reaction of EPCs (CD34+) was performed with a primary focus on apoptosis cascade and inflammation pathways. We observed that p53 gene was upregulated significantly when cells were exposed to high glucose. Moreover, p21 genes as well as certain vascular marker genes such as vascular endothelial growth factor receptor 2 (kinase insert domain receptor) and PECAM‐1 were also upregulated (Figure 2A). On the other hand, when HUVECs were exposed to hyperglycemic conditions, the upregulation of those genes (p53, p21, kinase insert domain receptor, PECAM‐1, and interleukin 6) was more evident at day 28 in comparison to days 7 and 14 (Figure 2B).
Figure 2.
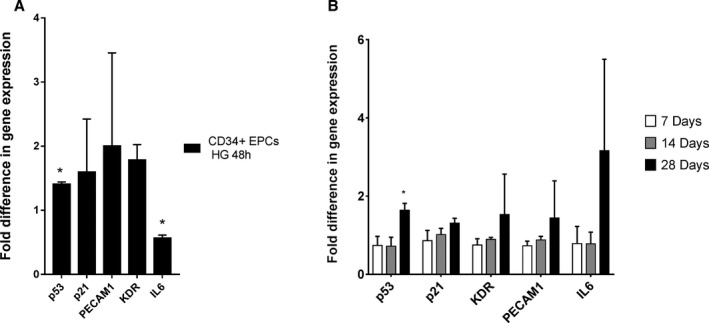
Gene expression analysis of EPCs (CD34+) and HUVECs exposed to high glucose (HG) and normal glucose (NG). Human EPCs (CD34+) were exposed to NG (5.5 mmol/L) and HG (20 mmol/L) for 48 hours. A, Upregulation of apoptotic genes (both p53 and p21) was observed due to HG exposure. B, HUVECs showed upregulation of apoptotic (p53 and p21), platelet endothelial cell adhesion molecule 1 (PECAM‐1) and inflammatory (IL6) genes after 28 days of HG exposure compared with 7 and 14 days, indicating that HG effects are not fully apparent in HUVECs until 28 days. *P<0.05.
Interestingly, mitochondrial function experiments using HUVECs demonstrated that OCR between cells exposed to normal and high glucose conditions becomes very distinct at day 28 when the OCR dropped significantly for high glucose–exposed cells compared with cells cultured in normal glucose (Figure 3).
Figure 3.
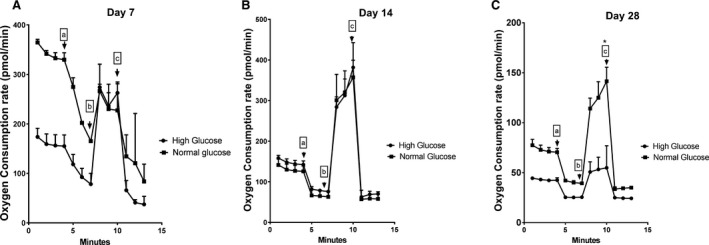
Oxygen consumption rate of HUVECs exposed to normal glucose (NG) and high glucose (HG). HUVECs were exposed to NG (5.5 mmol/L) and HG (20 mmol/L) for 7, 14, and 28 days followed by oxygen consumption rate measurement during a mitochondrial stress test. A and B, No significant change in between NG and HG exposed cells was observed at day 7 and 14. C, Lower respiration rate of HUVECs was noted only after 28 days of high glucose exposure. Additives injected are indicated with arrows: a. Oligomycin‐1.1 μmol/L, b. FCCP‐2.08 μmol/L and c. Rot/AA‐0.5 μmol/L. These data demonstrate reduced oxygen consumption rate of mature endothelial cells (HUVECs) exposed to hyperglycemic environment for a prolonged time (28 days). These results are representative of 3 independent experiments. *P<0.05.
Based on our results of fluorescence‐activated cell sorter and gene expression studies (Figures 1, 2 through 3), we noted that human EPCs (CD34+) are susceptible to hyperglycemic (20 mmol/L) injury (Figure 1A through 1C) at an earlier time point, compared with HUVEC. Therefore, for better survival of EPCs, in hyperglycemic milieu, we decided to silence p53 and p21 genes and verify whether KO or silencing of p53 and p21 (individually) would protect mEPCs and human EPCs (CD34+) from hyperglycemia‐mediated apoptosis.
p53KO mEPCs were identified microscopically as mature and robust (Figure 4A and 4B) and were able to differentiate further into mature endothelium‐like cells (cobblestone appearance) following 28 days of culture at normoglycemic conditions (5.5 mmol/L glucose) (Figure 4C). The improved survival of mouse p53KO EPCs were noted in mice with both C57Bl6 and BalbC background; however, only data with C57Bl6 mEPC are shown. The mouse p53KO EPCs progressed onto capillary‐like structures, indicating an endothelial cell–like property (Figure 4D). Lentivirus‐transduced p53sh‐mEPCs also showed improved survival compared with null‐transduced cells in hyperglycemic conditions, with cell survival differences more prominent as the number of days in culture increased from days 1 to 7 (Figure 5A).
Figure 4.
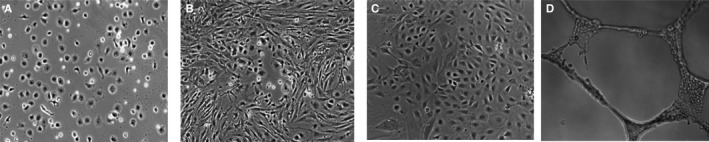
Comparison of survival between mouse peripheral blood‐derived wild type EPCs and p53KO EPCs. A, Rodent (wild type C57Bl/6 mice) peripheral blood‐derived EPCs exposed to normoglycemia showed poorly cell surviving in culture media at day 14. B, p53KO mice derived EPCs looked more mature and robust at day 14 of culture. C, At day 28, cells (shown at B) presented classical appearance of cobble stone endothelial cells. D, Cells shown in (C) have formed capillary‐like structures in matrigel, as a result of endothelial cell properties.
Figure 5.
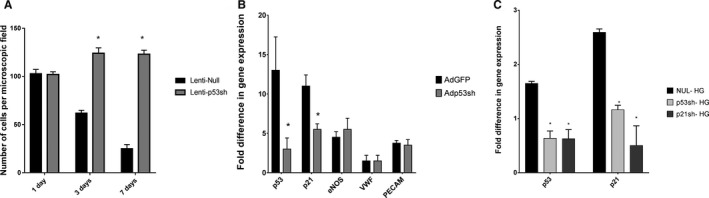
Effect of p53 and p21 silencing in EPCs. A, Silencing of p53 gene in mouse EPCs (mEPCs) by lentivirus as a vector demonstrates improved survival in hyperglycemic condition compared to null lenti (non‐silenced) transduced cells. Numbers of viable mEPCs are shown at days 1, 3, and 7 postexposures to high glucose (20 mmol/L). B, Gene expression analysis showed, p53 silencing in mouse EPCs by adenovirus down regulated p53 and p21 gene. Although, p53 silencing did not impair mRNA expression levels of critical endothelial marker gene such as endothelial nitric oxide synthase (eNOS), von Willebrand factor (vWF), and platelet endothelial cell adhesion molecule 1 (PECAM1). C, Human EPCs (CD34+) were transduced to silence p53 and p21 genes using adenovirus constructs and then exposed to high glucose for 48 hours. Silencing of both p53 and p21 in hEPCs were confirmed by the reduced gene expression in comparison to Ad‐null transduced cells, exposed to hyperglycemia. *P<0.05.
As the next step, we silenced p53 and p21 for a shorter period (transient silencing) using a DNA virus, such as adenovirus, rather than a long‐term silencer–like lentivirus, so that the long‐term silencing effects of p53 and p21 could be avoided. In fact, adenoviral p53 silencing allowed reduction of both p53 and p21 mRNA expression, while expression of key endothelial function genes such as endothelial nitric oxide synthase, von Willebrand factor, and PECAM‐1 were maintained even at 28 days of culture (Figure 5B). Moreover, Ad‐p53sh transduction prevented hyperglycemia‐mediated p53 and p21 mRNA overexpression. Similarly, Ad‐p21 silencing prevented p21 mRNA overexpression in hyperglycemia, and even reduced upstream p53 expression to a certain extent (Figure 5C).
To verify the beneficial effects of p53 and p21 silencing in vivo, STZ‐induced type 1 diabetic mice were subjected to a femoral artery occlusion (PVD model) and p53 and p21KO mEPCs (obtained from peripheral blood of global p53 and p21KO mice) were transplanted intramuscularly post–femoral artery occlusion. Laser Doppler imaging between days 3 and 10 showed improved vascular flow, most significantly in the p53KO mEPCs group (n=3) (Figure 6A and Figure S1). Post sacrifice, (four weeks following transplantation of EPCs), we observed upregulation of vascular marker genes such as vascular endothelial growth factor A, PECAM‐1, endothelial nitric oxide synthase, and kinase insert domain receptor by gene expression analysis of quadriceps muscles compared with saline and WT mEPCs transplanted mice (Figure 6B). However, CD31 staining did not show a significant difference, although a distinct trend of increased capillary density in hindlimb muscles were noted in mice that received p53‐silenced EPCs (Figure 6C). p21KO mEPCs were less efficacious than p53KO mEPCs.
Figure 6.
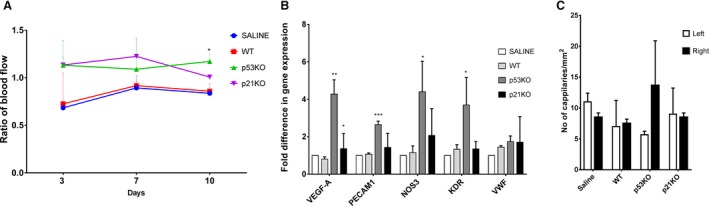
Enhanced vascularization in hind limb of Type 1 diabetes mellitus mouse model, posttransplantation of p53KO and p21KO EPCs. Type 1 diabetes mellitus was induced in C57BL/6J mice by STZ. p53KO and p21KO mouse EPCs were transplanted to mouse right hind limb quadriceps muscle, postocclusion of femoral artery. A, Blood flow was measured by Laser Doppler Perfusion Imager (LDPI) system after surgery (3, 7, 10 days). Increased blood flow was noted with delivery of p53KO (green line) and p21KO (purple line) EPCs, compared to WT (wild type; red line) and saline (blue line). B, Gene expression analysis was performed in quadriceps after mice were sacrificed at day 28. Upregulation of mature endothelial markers such as vascular endothelial growth factor A (VEGF‐A), platelet endothelial cell adhesion molecule 1 (PECAM1), endothelial nitric oxide synthase (eNOS), and kinase insert domain receptor (KDR) were observed in quadriceps from mice that received p53KO EPCs (statistical analysis was performed by using saline and WT EPC receiving muscle as control). C, Quadriceps were stained with CD31 antibody and an increased number of capillaries were observed in p53KO observed in quadriceps from mice that received p53KO EPCs. *P<0.05, **P<0.01 and ***P<0.005.
Evaluation of the efficacy of p53‐silenced mEPCs using a lentiviral vector in STZ mice showed similar findings as described for p53KO mEPCs. p53‐silenced mEPCs transplanted in STZ‐induced diabetic mice clearly improved vascular flow compared with both lenti‐null–transduced EPCs (≈1 million mEPCs) and the saline group (n=3) (Figure S2).
Next, we used ex vivo p53‐ and p21‐silenced (adenovirus mediated) human CD34+ cells for transplantation in STZ‐induced diabetic NOD‐SCID mice. The results for adenovirus‐transduced p53‐silenced peripheral blood–derived human EPCs (CD34+) transplantation to NOD‐SCID mice showed that secondary to p53 silencing, blood flow increased when p53‐silenced EPCs (CD34+) were transplanted into NOD‐SCID mice (Figure 7A and Figure S3), whereas p21‐silenced CD34+ cell transplantation did not show improved efficacy over adenovirus null EPCs (n=3). Vascular markers such as vascular endothelial growth factor A was upregulated significantly in the p53‐silenced group, more so than the p21‐silenced group (Figure 7B), in the quadriceps muscles compared with saline. Collectively, these results prove that p53‐silenced mEPCs or human EPCs (CD34+) are more efficacious for vascular regeneration compared with p21‐silenced cells. Interestingly, reverse transcriptase polymerase chain reaction of the quadriceps from this group for interleukin 6, a prominent inflammatory molecule, showed reduced expression in the animals that received p53‐silenced CD34+ cells compared with null‐transduced cells or saline, implying a reduction of inflammation post–p53‐silenced cell therapy in PVD (Figure S4).
Figure 7.
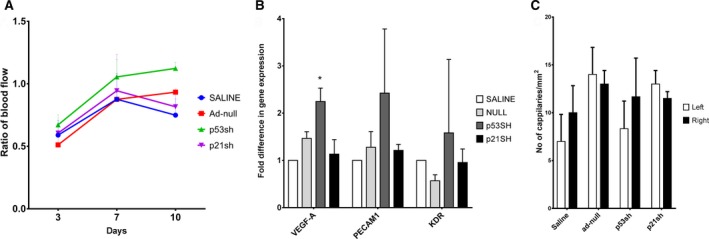
Enhanced vascularization in hind limb of diabetic mouse model posttransplantation of p53 and p21 silenced human EPCs. Type 1 diabetes mellitus was induced in NOD.CB17‐Prkdcscid/J mouse (NOD‐SCID) by STZ. Simultaneously, human EPCs were transduced with Adenovirus constructs to silence p53 and p21. Then, p53sh and p21sh human EPCs were transplanted to mouse right hind limb muscle postocclusion of femoral artery. A, Blood flow was measured by Laser Doppler Perfusion Imager (LDPI) system after surgery (3, 7, 10 days). Increased blood flow was noted with delivery of both p53 silenced EPCs (green line) and p21 silenced EPCs (purple line) compared to Adenovirus null transduced EPC (red line) delivery and saline (blue line). B, Gene expression analysis was performed in quadriceps after mice were sacrificed at day 28 postsurgery. Significant upregulation of mature endothelial marker such as vascular endothelial growth factor A (VEGF‐A) was observed in muscles that received p53sh (p53 silenced) observed in quadriceps from mice that received p53sh EPCs (statistical analysis was performed by using saline and wild type EPC as control). However, no difference was observed in muscles that received p21sh EPCs. C, No significant difference was noted in capillaries numbers when quadriceps were stained with CD31 antibody. *P<0.05.
However, in a type 2 diabetic obese mouse model (db/db), neither p53KO mEPCs nor p21KO mEPCs showed any advantage over WT mEPC delivery (Figure 8A through 8C) (n=2).
Figure 8.
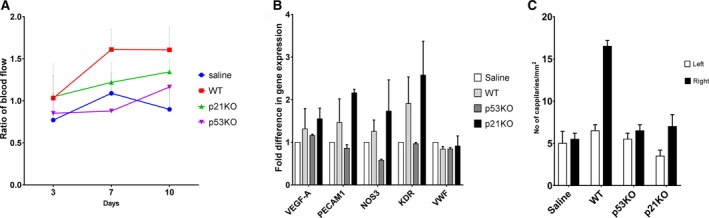
Enhanced vascularization in hind limb of obese diabetic Leptin resistant (db/db) mouse model posttransplantation of p53KO and p21KO mouse EPCs. p53KO and p21KO mouse EPCs were transplanted to mouse (B6.BKS(D)‐Leprdb/J) right hind limb muscle postocclusion of femoral artery. A, Blood flow was measured by Laser Doppler Perfusion Imager (LDPI) system after surgery (3, 7, 10 days). No statistical difference was noted with delivery of p53KO (purple line) and p21KO (green line) EPCs, compared to WT (wild type; red line) and saline (blue line). B, Similarly, gene expression analysis data unable to show any improvement due to gene silencing. C, No statistical difference in the number of capillaries were observed when quadriceps was stained with CD31 antibody between operated and non‐operated side.
Discussion
The hyperglycemic condition in diabetic patients is a major cause of endothelial dysfunction and cardiovascular disease.23 Previous studies have shown that EPC‐based cell therapy is useful for repairing cardiovascular injury.8, 17, 18, 19, 20 In our study, we reported increased cellular apoptosis in EPCs within 48 hours in response to hyperglycemia, whereas in HUVECs this effect was observed almost 4 weeks after cells were exposed to hyperglycemic condition. This outcome was confirmed by both gene expression (quantitative reverse transcriptase polymerase chain reaction) and OCR measurements. Collectively, these results indicate that EPCs (CD34+) are more susceptible to hyperglycemia compared with mature endothelial cells. Therefore, we can infer, diabetes mellitus and associated hyperglycemia is a proapoptotic condition that primarily affects EPCs more quickly (within 48 hours of hyperglycemia) than the mature endothelial cells, such as HUVECs. It takes 28 days to show a similar effect in HUVECs.
Additionally, our results suggest that by the time hyperglycemia affects endothelium such as HUVECs, endothelial progenitors have already undergone apoptosis and are possibly already depleted. Thus, it can be predicted that endothelial damage and vascular complications of hyperglycemia take time to develop, but, once the damage has occurred, the chances of vascular regeneration are poor as the progenitor pool is already depleted. This observation is similar among epidemiological studies, where vascular complications or endothelial dysfunction takes about 5 to 7 years to manifest, but, once the vascular complications are noted, the repair or vascular regeneration is difficult as the progenitor cell pool is depleted and the vascular complications are progressive from there on.1, 3, 14 This highlights the fact that interventions to treat endothelial dysfunction needs to precede EPC damage and needs to start early.
Interestingly, when we tried to culture mouse peripheral blood–derived EPCs in normoglycemic EBM‐2 media, we noted significant and progressive cell death. However, when p53KO mEPCs were cultured, we noted that the mouse endothelial cells appeared to retain all properties of mature endothelium, including tube formation in Matrigel. This finding indicates that suitable culture media for mEPCs have not yet been formulated, but protection from apoptosis does help cells grow to maturity within 28 days. Simultaneously, it also indicates that knocking down or suppression of p53 could be beneficial for cell survival in a proapoptotic condition. We decided to use this phenomenon to address diabetes mellitus‐associated PVD. We used an STZ‐induced diabetic PVD mouse model and db/db mice models to examine the effect of p53 and p21 suppression. We decided to test p21 silencing along with p53 silencing as we had hoped that silencing a downstream protein from p53 would reduce the off‐target effects of p53 silencing.
To evaluate the effect of transplanted p53KO and p21KO (downstream to p53 in apoptotic cascade) mEPCs in STZ‐induced diabetic mice, we used 3 outcome measures: blood flow, mRNA gene expression of the right hind muscle at the site of occlusion (that received EPC or saline deliver) compared with the untouched left leg, and CD31 capillary density measurement of the right leg compared with the right. We observed that p53KO rather than p21KO had more consistent improved results compared with WT cell therapy showing improved blood flow and vascularization. However, we were unable to observe statistically significant change in capillary density measurement, although our experiments showed improved trend with p53‐silenced cell therapy.
Taking all of our outcome measures in an STZ mouse model using genetically modified mEPCs, our results indicate that p53KO and p21KO EPC transplantation showed better viability than WT EPCs and proved beneficial to promote increased blood flow to ischemic hindlimb, although p53KO showed superiority over p21KO.
Since knocking down of apoptotic genes such as p53 and p21 long term is not a realistic clinical approach as a cell therapy, we investigated whether silencing of p53 would indicate the same outcome as knocking down. We noted that lentivirus‐mediated p53 silencing in mEPC did improve cell survival in high glucose, where cell depletion was rapid otherwise, over 7 days. Similarly, mEPCs with lentivirus‐mediated p53 silencing showed improved blood flow by laser Doppler (Figure S2). In order to obtain a translational benefit, we speculated that one can use similarly modified human cells for therapy in diabetic PVD. However, we decided not to use a lentiviral construct, as application of a mutagenic lentivirus in humans and suppressing p53 for long periods may not be safe in spite of efficacy.
To solve this problem, we used short‐term silencing of p53 where the viral construct would not be viable beyond 28 days. Effects of adenoviral silencing usually persist ≈28 days.24, 25 This time span is perfect because the time required by EPCs to transform to a mature endothelium is also ≈28 days. Simultaneously, we did not want to use a turn‐on, turn‐off gene promoter, with another DNA virus like adeno‐associated virus,18, 21 as the effect of silencing could be considered as “leaky.”24, 26 Thus, the use of an adenovirus as a gene modification vector was the best selection, with a transduction efficacy profile better than liposomes.24, 25
Adenovirus, unlike lentivirus, is a DNA virus and does not integrate with the host genome.24, 25, 27 It can also provide gene expression or a silencing effect for up to 4 weeks24, 27 which should be enough for EPCs transition to mature endothelium. In our experience, efficacy with adenovirus is about 30% to 40% compared with lentivirus, where the efficacy is about 70% to 80%. For the purposes of this study, adenovirus was chosen as the ideal vector for transient silencing of p53 and p21. Using adenoviral constructs, we were able to protect EPCs in hyperglycemia. Transplanting p53‐ and p21‐silenced human EPCs (CD34+) by adenoviral vector improved blood flow, gene expression, and capillary formation (Figure 7A and 7B) similar to our results using genetically modified mEPCS (Figure 6A and 6B). However, p53sh human CD34+ cells showed superiority over p21sh human CD34+ cells.
When we used p53KO and p21KO mEPCs to treat vaso‐occlusive disease in a db/db mouse model (type 2 diabetes mellitus mouse model), we did not note improvement in blood flow by laser Doppler or in capillary density estimation, indicating that p53 or p21 silencing may not be enough to protect mEPCs in a type 2 rather than type 1 diabetes mellitus mouse model. Type 2 diabetes mellitus has added adipocyte inflammation along with hyperglycemia as a proapoptotic agent. This could also mean that a higher number of modified cells for therapy may be required to obtain a discernable improvement compared with a type 1 model.
Our data also suggest that p53 silencing is not only superior to unmodified EPCs but also has better outcome efficacy than p21 silencing, particularly in a type 1 model. Moreover, p53 silencing offers protection predominantly in a type 1 diabetes mellitus environment where hyperglycemia is the primary apoptotic agent.
We expected that p21 silencing would afford protection and vascular regeneration effectiveness similar to p53 silencing so that off‐target effects of p53 silencing would be minimized. Although p21‐silenced EPCs did not work as expected, p53 silencing showed positive results and can emerge as a promising therapeutic intervention for ischemic type 1 diabetic complication. p53 silencing provided protection and improved survival of EPCs, which subsequently improved vascular regeneration postocclusion, which could be secondary to its paracrine properties.
Conclusions
Our data indicate that using genetically modified EPCs (CD34+) transplantation can usher in new hope for reducing vascular damage in type 1 diabetic patients where microvascular disease such as retinopathy, nephropathy, and neuropathy are more common.
Sources of Funding
This work was funded by an American Heart Association Scientific Development Grant awarded to S. Sen (grant No. 12050535).
Disclosures
None.
Supporting information
Figure S1. Images of enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced type 1 diabetic mouse model (C57BL/6J), posttransplantation of p53 knockout (KO) and p21KO endothelial progenitor cells (EPCs). Serial laser Doppler images showing improved circulation at day 10 in the right leg, particularly in the group that received p53KO EPCs.
Figure S2. Images of enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model, following transplantation of p53‐silenced (by lentivirus) mouse endothelial progenitor cells (EPCs). Serial laser Doppler images showing improved circulation at day 11 in the right leg, particularly in the group that received p53‐silenced EPCs. Lentivirus was used to silence mouse p53 and the results were compared with mouse that received lenti‐null–transduced EPCs or saline.
Figure S3. Enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model (nonobese diabetic severe combined immunodeficiency type 1 diabetes mice), posttransplantation of p53sh and p21sh human endothelial progenitor cells (EPCs; silenced by adenovirus). Serial laser Doppler images showing improved circulation at day 10 in the right leg, particularly in the group that received p53sh EPCs.
Figure S4. Reduced inflammation in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model (nonobese diabetic severe combined immunodeficiency [NOD‐SCID] type 1 diabetes mice), posttransplantation of p53sh human endothelial progenitor cells (EPCs; silenced by adenovirus). Type 1 diabetes was induced in NOD.CB17‐Prkdcscid/J mouse (NOD‐SCID) by STZ. Simultaneously, human EPCs were transduced with adenovirus constructs to silence p53 and p21. Then, p53sh and p21sh human EPCs were transplanted to mouse right hindlimb muscle postocclusion of femoral artery. Gene expression analysis was performed in the quadriceps after mice were sacrificed at day 28 postsurgery. Downregulation of inflammatory marker interleukin 6 was observed in p53sh EPCs transplanted muscles (statistical analysis was performed by using saline and null as control). These data suggest reduction of inflammation due to silencing of p53.
Acknowledgments
We would like to acknowledge the help and support of the animal facilities at Baystate Medical Center and The George Washington University. The project was approved by the Institutional Animal Care and Use Committee at both Baystate Medical Center and The George Washington University. We would also like to acknowledge the histopathology laboratory at Pioneer Valley Life Sciences Institute, Baystate Medical Center, and The George Washington University.
(J Am Heart Assoc. 2017;6:e005146 DOI: 10.1161/JAHA.116.005146.)28365567
References
- 1. Department of Health, and Human Services National Institutes of Health, and National Center for Chronic Disease Prevention and Health Promotion National Diabetes Statistics . Fact sheet. 2011.
- 2. Rask‐Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. [DOI] [PubMed] [Google Scholar]
- 3. American Diabetes Association standards of medical care in diabetes. Diabetes Care. 2012;35:11–63. [Google Scholar]
- 4. Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA. 2002;288:2579–2588. [DOI] [PubMed] [Google Scholar]
- 5. National Institute of Diabetes and Digestive and Kidney Diseases . National diabetes statistics: 2007 and 2011 fact sheet. Bethesda, MD: Department of Health and Human Services, NIH, 2008 and National Center for Chronic Disease Prevention and Health Promotion. 2011. [Google Scholar]
- 6. Wilson A, Trumpp A. Bone‐marrow haematopoietic‐stem‐cell niches. Nat Rev Immunol. 2006;6:93–106. [DOI] [PubMed] [Google Scholar]
- 7. Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012;110:624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. [DOI] [PubMed] [Google Scholar]
- 9. Yoder MC. Endothelial progenitor cell: a blood cell by many other names may serve similar functions. J Mol Med (Berl). 2013;91:285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sen S, McDonald SP, Coates PT, Bonder CS. Endothelial progenitor cells: novel biomarker and promising cell therapy for cardiovascular disease. Clin Sci (Lond). 2011;120:263–283. [DOI] [PubMed] [Google Scholar]
- 11. Fadini GP, Albiero M, Vigili de Kreutzenberg S, Boscaro E, Cappellari R, Marescotti Ml. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care. 2013;36:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caballero S, Sengupta N, Afzal A, Chang KH, Li Calzi S, Guberski DL. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sen S, Witkowski S, Lagoy A, Islam AM. A six‐week home exercise program improves endothelial function and CD34+ circulating progenitor cells in patients with pre‐diabetes. J Endocrinol Metab. 2015;5:163–171. [Google Scholar]
- 14. Povsic TJ, Sloane R, Green JB, Zhou J, Pieper CF, Pearson MP. Depletion of circulating progenitor cells precedes overt diabetes: a substudy from the VA enhanced fitness trial. J Diabetes Complications. 2013;27:633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalka C, Isner J, Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci USA. 2000;97:3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou X, Patel D, Sen S, Shanmugam V, Sidawy A, Mishra L, Nyugen BN. Poly‐ADP‐ribose polymerase inhibition enhances ischemic and diabetic wound healing by promoting angiogenesis. J Vasc Surg. 2017;65:1161–1169. [DOI] [PubMed] [Google Scholar]
- 17. Kawamoto A, Losordo DW, Isner JM, Asahara T. Intramyocardial transplantation of autologous endothelial progenitor cells for therapeutic neovascularization of myocardial ischemia. Circulation. 2003;107:461–468. [DOI] [PubMed] [Google Scholar]
- 18. Sen S, Merchan J, Dean J, Ii M, Gavin M, Silver M, Tkebuchava T, Yoon YS, Rasko JE, Aikawa R. Autologous transplantation of endothelial progenitor cells genetically modified by adeno‐associated viral vector delivering IGF‐1 gene following myocardial infarction. Hum Gene Ther. 2010;21:1327–1334. [DOI] [PubMed] [Google Scholar]
- 19. Losordo DW, Henry TD, Davidson C, Sup Lee J, Costa MA, Bass T, Mendelsohn F, Fortuin FD, Pepine CJ, Traverse JH, Amrani D, Ewenstein BM, Riedel N, Story K, Barker K, Povsic TJ, Harrington RA, Schatz RA. Intramyocardial autologous CD34+cell therapy for refractory angina: ACT34‐CMI Investigators. Circ Res. 2011;109:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Losordo DW, Schatz RA, White CJ, Udelson JE, Veereshwarayya V, Durgin M, Poh KK, Weinstein R, Kearney M, Chaudhry M, Burg A, Eaton L, Heyd L, Thorne T, Shturman L, Hoffmeister P, Story K, Zak V, Dowling D, Traverse JH, Olson RE, Flanagan J, Sodano D, Murayama T, Kawamoto A, Kusano KF, Wollins J, Welt F, Shah P, Soukas P, Asahara T, Henry TD. Intramyocardial transplantation of autologous CD34+ stem cells for intractable angina: a phase I/IIa double‐blind, randomized controlled trial. Circulation. 2007;115:3165–3172. [DOI] [PubMed] [Google Scholar]
- 21. Sen S, Conroy S, Hynes SO, McMahon J, O'Doherty A, Bartlett JS, Akhtar Y, Adegbola T, Connolly CE, Sultan S, Barry F, Katusic ZS, O'Brien T. Gene delivery to the vasculature mediated by low‐titre adeno‐associated virus serotypes 1 and 5. J Gene Med. 2008;10:143–151. [DOI] [PubMed] [Google Scholar]
- 22. Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre‐lox‐regulated conditional RNA interference from transgenes. Proc Natl Acad Sci USA. 2004;101:10380–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Standards of medical care in diabetes. Diabetes Care. 2011;34:11–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Domingues CC, Kundu N, Dore F, Sen S. Genetic modification of stem cells in diabetes and obesity In: Farrukh J, ed. Genetic Engineering—An Insight Into the Strategies and Applications. Rijeka: InTech; 2016:75–84. [Google Scholar]
- 25. Sen S, Strappe PM, O'Brien T. Gene transfer in endothelial dysfunction and hypertension. Methods Mol Med. 2005;108:299–314. [DOI] [PubMed] [Google Scholar]
- 26. Kappel S, Matthess Y, Strebhardt K. Silencing of mammalian genes by tetracycline‐inducible shRNA expression. Nat Protoc. 2007;2:3257–3269. [DOI] [PubMed] [Google Scholar]
- 27. Sen S, Domingues CC, Rouphael C, Chou C, Kim C, Yadava N. Genetic modification of human mesenchymal stem cells helps to reduce adiposity and improve glucose tolerance in an obese diabetic mouse model. Stem Cell Res Ther. 2015;6:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Images of enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced type 1 diabetic mouse model (C57BL/6J), posttransplantation of p53 knockout (KO) and p21KO endothelial progenitor cells (EPCs). Serial laser Doppler images showing improved circulation at day 10 in the right leg, particularly in the group that received p53KO EPCs.
Figure S2. Images of enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model, following transplantation of p53‐silenced (by lentivirus) mouse endothelial progenitor cells (EPCs). Serial laser Doppler images showing improved circulation at day 11 in the right leg, particularly in the group that received p53‐silenced EPCs. Lentivirus was used to silence mouse p53 and the results were compared with mouse that received lenti‐null–transduced EPCs or saline.
Figure S3. Enhanced vascularization in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model (nonobese diabetic severe combined immunodeficiency type 1 diabetes mice), posttransplantation of p53sh and p21sh human endothelial progenitor cells (EPCs; silenced by adenovirus). Serial laser Doppler images showing improved circulation at day 10 in the right leg, particularly in the group that received p53sh EPCs.
Figure S4. Reduced inflammation in the hindlimb of a streptozotocin (STZ)–induced diabetic mouse model (nonobese diabetic severe combined immunodeficiency [NOD‐SCID] type 1 diabetes mice), posttransplantation of p53sh human endothelial progenitor cells (EPCs; silenced by adenovirus). Type 1 diabetes was induced in NOD.CB17‐Prkdcscid/J mouse (NOD‐SCID) by STZ. Simultaneously, human EPCs were transduced with adenovirus constructs to silence p53 and p21. Then, p53sh and p21sh human EPCs were transplanted to mouse right hindlimb muscle postocclusion of femoral artery. Gene expression analysis was performed in the quadriceps after mice were sacrificed at day 28 postsurgery. Downregulation of inflammatory marker interleukin 6 was observed in p53sh EPCs transplanted muscles (statistical analysis was performed by using saline and null as control). These data suggest reduction of inflammation due to silencing of p53.