

Abstract
Background
We have identified the cardiomyopathy‐susceptibility gene vinculin (VCL) mutation M94I may account for a sudden unexplained nocturnal death syndrome (SUNDS) case. We addressed whether VCL common variant D841H is associated with SUNDS.
Methods and Results
In 8 of 120 SUNDS cases, we detected an East Asian common VCL variant p.Asp841His (D841H). Comparing the H841 allele frequency of the general population in the local database (15 of 1818) with SUNDS victims (10 of 240) gives an odds ratio for SUNDS of 5.226 (95% CI, 2.321, 11.769). The VCL‐D841H variant was engineered and either coexpressed with cardiac sodium channel (SCN5A) in HEK293 cells or overexpressed in human induced pluripotent stem‐cell–derived cardiomyocytes to examine its effects on sodium channel function using the whole‐cell patch‐clamp method. In HEK293 cells, under physiological pH conditions (pH 7.4), D841H caused a 29% decrease in peak IN a amplitude compared to wild type (WT), whereas under acidotic conditions (pH 7.0), D841H decreased further to 43% along with significant negative shift in inactivation compared to WT at pH 7.4. In induced pluripotent stem‐cell‐derived cardiomyocytes, similar effects of D841H on IN a were observed. VCL colocalized with SCN5A at the intercalated disk in human cardiomyocytes. VCL was also confirmed to directly interact with SCN5A, and VCL‐D841H did not disrupt the association of VCL and SCN5A.
Conclusions
A VCL common variant was genetically and biophysically associated with Chinese SUNDS. The aggravation of loss of function of SCN5A caused by VCL‐D841H under acidosis supports that nocturnal sleep respiratory disorders with acidosis may play a key role in the pathogenesis of SUNDS.
Keywords: cardiac sodium channel, sudden cardiac death, sudden unexplained nocturnal death syndrome, vinculin
Subject Categories: Sudden Cardiac Death
Introduction
Sudden unexplained nocturnal death syndrome (SUNDS), first reported in 1917 in the Philippines, predominantly prevails in Southeast Asia.1 The most impressive and unique clinic phenotype for SUNDS is sudden unexpected death during night sleep in apparently healthy young adult males with structurally normal heart.1, 2, 3 Usually, it is diagnosed by pathologists when it remains unexplained after a comprehensive analysis of cause of death, including autopsy examination and toxicological screening.1, 2, 3 Although considerable hypotheses (such as primary arrhythmia,2, 3 bacterial infection,4 potassium deficiency,5 coronary arteries defect,6 and sleep respiratory disorders7) have been proposed as potential pathogenic mechanisms for SUNDS, this syndrome, so far, is a puzzling entity with unknown etiology.8
The clinical features of SUNDS are very similar to those of Brugada syndrome (BrS). Previous molecular studies of 10 families with SUNDS have identified 3 cases with loss‐of‐function mutations in cardiac sodium channel (SCN5A).3 Furthermore, in a large cohort of over 120 SUNDS victims,8, 9, 10 we successively identified mutations in the BrS susceptible genes (SCN5A, SCN1B, SCN3B, PKP2, and SCN10A) as plausible genetic causes of ≈20% of SUNDS cases. These results highlight the importance of uncovering a possible pathogenic gene for SUNDS in BrS‐related genes and indicate that the search of novel susceptibility genes for the majority of SUNDS cases is needed.
Primary arrhythmias, such as BrS,11, 12 are known to share some pathogenic genes (PKP2, SCN5A, and RYR2) with cardiomyopathy. Vinculin (VCL) and its muscle splice variant isoform, metavinculin, which encodes a cytoskeletal protein that connects actin microfilaments to the intercalated disk and membrane costameres in the heart, have been confirmed as a susceptible gene for dilated and hypertrophic cardiomyopathy.13, 14, 15 The high incidence of sudden death (attributed to defective myocardial conduction and ventricular tachycardia), before the appearance of cardiomyopathy, was found in cardiomyocyte‐specific inactivation of the VCL gene in mouse.16 Most recently, we characterized the VCL rare variant, M94I, identified in a SUNDS victim with a structurally normal heart, which implicated VCL as a new susceptibility gene for SUNDS.17 Based on all these findings above, we hypothesized that with VCL, some certain common variants may increase the risk of susceptible individuals for SUNDS.
Material and Methods
Chinese SUNDS Cohort
One‐hundred twenty unrelated SUNDS cases were collected from March 1, 2006 to November 30, 2014 at the National Forensic Autopsy Center at Sun Yat‐sen University (Guangzhou, China). The SUNDS inclusion criteria were as previously reported8, 9, 10: (1) an apparently healthy Han Chinese with age older than 15 years who suffered a sudden unexpected death during nocturnal sleep; (2) without history of significant disease; (3) and a negative forensic autopsy (gross and microscopic examination), toxicology, and death‐scene investigation, resulting in an unexplained death. Victims with significant disease or pathological alterations to elucidate the death were excluded. Approval for human research protocols were obtained from the ethics committee of Sun Yat‐sen University. The principles outlined in the Declaration of Helsinki were followed. Informed consent was obtained from the legal representatives of the victims.
Genetic Analysis
Genomic DNA was extracted from blood samples using the DNA IQ Casework Pro Kit for Maxwell 16 (Promega Corporation, Madison, WI). Genetic screening of the VCL gene (Genbank accession no. NM_014000.2) was performed on SUNDS cases using direct Sanger sequencing. A total of 80 genes associated with primary arrhythmia/cardiomyopathy were genetically screened in VCL missense variant carriers based on target captured next‐generation sequencing technology, and then the identified variants were confirmed by direct Sanger sequencing as described previously.18 Sequences were compared with the corresponding reference cDNA sequence of the VCL gene using SeqManII expert sequence analysis software (DNASTAR, Inc, Madison, WI). All suspicious variants were sequenced in both sense and antisense directions. Rare variants (minor allele frequency [MAF] <1%) and polymorphisms (MAF >1%) resulting in nonsynonymous amino acid changes (missense, nonsense, frame‐shift insertion/deletions, in‐frame insertion/deletions, or splice errors) observed in any ethnic group among population databases, including a local database (n=2087, n=989 Han Chinese without cardiac arrhythmia), the National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project (n=6503), the 1000 Genome Project (n=2504), and Exome Aggregation Consortium (ExAC; n=60 706 all ethnicities, n=4327 East Asian) were considered for genetic analysis. Rare nonsynonymous variants and polymorphisms were characterized according to the strict variant interpretation guidelines outlined by the American College of Medical Genetics (ACMG).19
Plasmid Constructions of Expressing Vectors
The cDNA of the wild‐type (WT) human VCL gene was subcloned into mEmerald vector (Plasmid #54304; from Addgene, Cambridge, MA). The VCL‐p.Asp841His (D841H) polymorphism was introduced into VCL‐WT using a site‐directed mutagenesis kit (Stratagene, La Jolla, CA). All clones were sequenced to ensure the existence of the target mutation and the absence of other substitutions caused by polymerase chain reaction.
HEK293 Cells Transfection
The VCL‐WT or ‐D841H in mEmerald vector was transiently cotransfected with expressing vectors containing SCN5A (hNav1.5; Genbank accession no. AB158469) at a ratio of 1:4 into HEK293 cells using FuGENE6 reagent (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions.
Human Induced Pluripotent Stem‐Cell–Derived Cardiomyocytes Transfection
The previously and extensively characterized human induced pluripotent stem‐cell–derived cardiomyocytes (iPS‐CMs) were obtained from Cellular Dynamics International (Madison, WI) and handled according to manufacturer specifications.20, 21 The iPS‐CMs were split 24 hours before cellular electrophysiology experiments and plated on 12‐mm precoated coverslips (BD Biosciences, San Jose, CA). The VCL‐WT or ‐D841H vectors were transiently transfected into iPS‐CMs using TransIT‐LT1 transfection reagent (Mirus Bio LLC, Madison, WI), according to manufacturer's instructions.
Histological Study of the Heart and Immunoprecipitation
Paraffin‐embedded left ventricle sections (6 μm) at the midventricular level, from the SUNDS victims with VCL‐D841H and their age‐ and sex‐matched controls without structural heart disease, were stained with hematoxylin and eosin, Masson's trichrome, and immunofluorescent staining as described previously.22 Sections were stained with antibodies: mouse anti‐VCL (monoclonal, 1:200; Sigma‐Aldrich, St. Louis, MO) and Guinea Pig anti‐Nav1.5 (polyclonal, 1:200; Alomone Labs Ltd, Jerusalem, Israel). Fluorescent images were acquired using a Leica SP5 laser confocal microscope system (Leica Microsystems, Wetzlar, Germany). VCL and SCN5A antibodies were excited at 561 and 488 mm, respectively, and emission light was filtered by 605±10‐ and 525±20‐mm band‐pass filter for VCL and SCN5A signals, respectively. Signal intensity profile of both VCL and SCN5A were calculated along the transverse direction (t‐tubule direction), aligned, and plotted. Immunoprecipitations were performed in both adult mouse cardiac homogenates and cell lysates from HEK293 cells overexpressing VCL and SCN5A using a Pierce direct IP kit (#26148; Thermo Fisher Scientific Inc, Waltham, MA), which directly immobilized 25 μg of VCL (#MA5‐11690; Invitrogen, Carlsbad, CA) or 25 μg of SCN5A (#PA5‐34190; Invitrogen) antibodies on agarose‐resin support to improve specificity. The immunoprecipitated samples were analyzed by western blotting by probing with anti‐SCN5A (#SAB2107930; Sigma‐Aldrich) or anti‐VCL (#V4139; Sigma‐Aldrich).
Electrophysiological Measurements
Macroscopic voltage‐gated sodium current (I Na) was measured 24 hours after transfection with the standard whole‐cell patch clamp method at 22°C in both HEK293 cells and iPS‐CMs under both normal (pH 7.4) and moderate acidosis (extracellular and intracellular pH 7.0) conditions. For HEK293 cells, the pipette solution contained (in mmol/L) EGTA 2, CsF 120, CsCl2 20, HEPES 5, and NaCl 5 and was adjusted to pH 7.4 or 7.0 with CsOH; the bath solution contained (in mmol/L) CaCl2 1.8, NaCl 140, HEPES 5, MgCl2 0.75, and KCl 4 and was adjusted to pH 7.4 or 7.0 with NaOH.20, 21 For iPS‐CMs, the pipette solution contained (in mmol/L) HEPES 10, NaCl 5, CaCl2 2, CsCl2 135, MgATP 5, and EGTA 10 and was adjusted to pH 7.4 or 7.0 with CsOH; the bath solution contained (in mmol/L) CsCl2 105, NaCl 60, glucose 10, CaCl2 1.8, MgCl2 1, HEPES 5, and Nifedipine 0.001 and was adjusted to pH 7.4 or 7.0 with CsOH (modified from reference).22 The resistances of microelectrodes ranged from 1.0 to 2.3 MΩ. Voltage clamp data were generated with pClamp software 10.5 and an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) with series‐resistance compensation. Membrane current data were digitalized at 100 kHz, low‐pass filtered at 5 kHz, and then normalized to membrane capacitance.
Activation was measured by clamp steps of −120, −110, −100, −90, −80, −70, −60, −50, −40, −30, −20, −10, 0, 10, 30, and 60 mV from a holding potential of −140 mV. The midpoint of activation was obtained using a Boltzmann function of GNa=[1+exp (V1/2−V)/k]−1, where V1/2 and k are the midpoint and slope factor, respectively. G/GNa=I Na (norm)/(V−Vrev) where Vrev is the reversal potential and V is the membrane potential. Steady‐state inactivation was measured in response to a test depolarization to 0 mV for 24 ms from a holding potential of −140 mV, following a 1‐second conditioning pulse from −150 to 0 mV in 10‐mV increments. Voltage‐dependent availability from inactivation relationship was determined by fitting the data to the Boltzmann function: I Na=I Na‐max [1+exp (Vc−V1/2)/k]−1, where V1/2 and k are the midpoint and the slope factor, respectively, and Vc is the membrane potential. Late I Na was measured as the mean between 600 and 700 ms after the initiation of the depolarization from −140 to −20 mV for 750 ms after passive leak subtraction, as previously described.22 Time course of recovery from inactivation was elicited using the protocol: holding potential of −140 mV, conditioning pulse to 0 mV for 1 second, followed by different recovery intervals (from 0.1 to 2000 ms), and then a test pulse to 0 mV for 24 ms. Data were analyzed by fitting data with a 2‐exponential (exp) function: normalized I Na (t)=A f [1−exp(−t/τf)]+A S [1−exp(−t/τS)], where t is time, A f and A S are fractional amplitudes of fast and slow components, respectively, and τf and τS are fast and slow time constants, respectively. The cycle length of the used voltage clamp protocols is 2 ms.
Statistical Analysis
Data points are reported as the mean value and the SEM. Determinations of statistical significance were performed using a Student t test for comparisons of 2 means or using ANOVA for comparisons of multiple groups. Differences in genotype frequency and allele frequency between SUNDS cases and controls were tested by chi‐square test or Fisher's exact probability test. Statistical significance was determined by a value of P<0.05.
Results
Demographics of SUNDS Cohort
All 120 unrelated SUNDS cases (1 female) were from the Chinese Han population. The average death age was 31.1±7.6 years (range, 17–52). No family history of sudden death and no clinic records existed for any of these apparently previously healthy SUNDS cases. Comprehensive autopsy examination showed no significant pathological changes or diseases to explain the sudden death. The hearts of the 120 SUNDS victims were structurally normal. The average heart weight and left (LV) and right ventricle (RV) thickness were 365±52 g, 1.20±0.16 cm, and 0.31±0.05 cm, respectively. DNA samples and detailed clinic information of family members of the SUNDS cases were not available, because data were collected as a retrospective study on forensic autopsy cases and follow‐up study was not possible.
Vinculin Mutational Analysis
Overall, 9 of 120 SUNDS cases carried VCL missense variants. One case had a rare variant M94I on 1 VCL allele. Six cases were heterozygous for a common variant where a C replaces G (NM_014000.2:c.2521G>C, rs150385900) in the first position of codon 841 on 1 copy of VCL to yield a change from aspartic acid (D) to histidine (H) (p.Asp841His; Figure 1), indicating the genotype D841/H841 (DH). Two cases were homozygous for the variant genotype, H841/H841 (HH). The MAF of H841 in 120 SUNDS is 0.04167 (10 of 240). The p.Asp841His (D841H) substitution involves an amino acid that is highly conserved among species (Figure 1).
Figure 1.
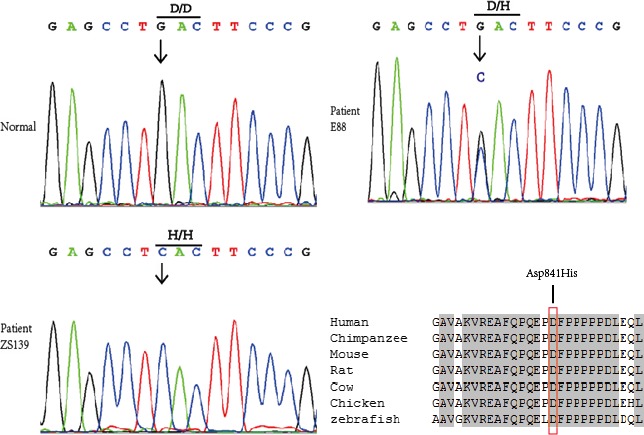
Genotyping of sudden unexplained nocturnal death syndrome (SUNDS) victim. The common variant, Asp841His, of the vinculin (VCL) gene is confirmed by Sanger sequencing in 6 SUNDS cases with heterozygous G/C and in 2 SUNDS cases with homozygosis C/C at nucleotide 841. An arrow shows the position of the mutation. The Asp841His substitution involves an amino acid that is highly conserved among species. VCL accession numbers: human, NP_054706.1; chimpanzee, XP_507854; mouse, NP_033528.3; rat, NP_001100718.1; cow, NP_001178299.1; chicken, NP_990772.1; zebrafish, NP_001122153.1.
Only 909 of 989 Han Chinese in the local database underwent VCL screening. We set the MAF of H841 in these 909 Han Chinese (15 of 1818=0.00825) as a control. In the ExAc database, H841 was absent in the European population and the MAF presented a prevalence of 0.01322, 0.0002424, and 0.0001928 in the East Asian, South Asian, and African populations, respectively (Table 1). No individual was homozygous (HH) in either controls or the ExAC database.
Table 1.
Frequencies of the D841H in VCL in Different Populations From the ExAC
| Population | Allele Count | Allele No. | Allele Frequency |
|---|---|---|---|
| East Asian | 114 | 8626 | 0.01322 |
| African | 2 | 10376 | 0.0001928 |
| South Asian | 4 | 16500 | 0.0002424 |
| European (non‐Finnish) | 0 | 66634 | 0 |
| European (Finnish) | 0 | 6606 | 0 |
| Latino | 0 | 11560 | 0 |
| Other | 0 | 908 | 0 |
| Total | 120 | 121210 | 0.00099 |
ExAC indicates Exome Aggregation Consortium; VCL, vinculin.
No pathogenic rare variants were identified in 79 known primary arrhythmia/cardiomyopathy–causing genes18 screened in these 8 SUNDS cases with VCL‐D841H, except for cases E9 (hosted KCNE1‐F54V) and E88 (carried DMPK‐P623L; Table 2). D841H was predicted to be malignant by in silico tools SIFT, Polyphen2, and CONDEL. However, using the strict ACMG guideline‐based definition for pathogenicity, D841H was considered as a variant of uncertain significance. D841H localized to the proline‐rich zone (amino acids 837–878), which links the head domain (residues 1–836, harbors binding sites of talin, α‐actin, or α‐catenin) to tail domain (residues 879–1066, harbors paxillin‐binding site).23, 24, 25
Table 2.
Gross Autopsy Findings of Heart From D841H Carriers
| Case | D841H Zygosity | Age (yr) | Height (cm) | Heart Weight (g) | LV Thickness (cm) | RV Thickness (cm) | Tricuspid Valve (cm) | Pulmonary Valve (cm) | Mitral Valve (cm) | Aortic Valve (cm) |
|---|---|---|---|---|---|---|---|---|---|---|
| E88 | Heterozygous | 38 | 160 | 380 | 1.1 | 0.3 | 12 | 8 | 9 | 7.5 |
| ZS139 | Homozygous | 21 | 174 | 395 | 1.2 | 0.3 | 13 | 8 | 10 | 7.5 |
| E131 | Heterozygous | 33 | 173 | 380 | 1.2 | 0.3 | 12 | 7.2 | 10.5 | 6.5 |
| E137 | Heterozygous | 36 | 170 | 360 | 1.2 | 0.3 | 12 | 7 | 9.5 | 6.5 |
| E147 | Heterozygous | 31 | 166 | 390 | 1.3 | 0.4 | 14 | 9 | 10 | 8 |
| 41 | Heterozygous | 23 | 160 | 350 | 1.3 | 0.3 | 12.5 | 8 | 10.5 | 7 |
| E9 | Heterozygous | 52 | ··· | 385 | 1.3 | 0.3 | ··· | ··· | ··· | ··· |
| 15 | Homozygous | 38 | ··· | 380 | 1.2 | 0.3 | ··· | ··· | ··· | ··· |
Ellipses (···) indicates lacking the raw data because of incomplete records; LV, left ventricle; RV, right ventricle.
Hardy–Weinberg Equilibrium
Observed and expected genotypic frequencies, assuming Hardy–Weinberg equilibrium, for each genotype in the Han Chinese population, were as follows: In SUNDS victims (n=120): DD 112, 117.3 expected; DH 6, 2.4 expected; HH 2, 0.2 expected. In controls (n=909): DD 894, 888.7 expected; DH 15, 18.6 expected; HH 0, 1.8 expected. SUNDS case genotype frequencies were not in Hardy–Weinberg equilibrium (χ2=16.78; P<0.001), whereas control frequencies did not significantly deviate from Hardy–Weinberg equilibrium (χ2=0.063; P=0.802). In SUNDS cases, there was a 10‐fold excess of HH genotypes over the expected amount. In the controls, there were no HH individuals, whereas 1 or 2 were expected. Comparing HH frequency in SUNDS cases to controls, 2 of 120 versus 0 of 909, indicated that the overpresentation of HH in SUNDS was statistically significant (P=0.013). While comparing allele frequency of H in SUNDS cases to controls, 10 of 240 versus 15 of 1818, gives an odds ratio for SUNDS of 5.226 (95% CI, 2.321, 11.769), indicating an ≈5‐fold increase in risk for SUNDS with the H allele type (P<0.001). All above evidences demonstrate that there is congregation of H in the SUNDS population when compared to the general population.
The Morphological Examination and Immunoprecipitations
All of the 8 SUNDS victims with D841H were identified to have sudden tachypnea preceding their sudden death during nocturnal sleep at 1 to 5 am. The heart weight and ventricle thickness for these 8 SUNDS victims (Table 2) were comparable to the SUNDS cases without D841H (data not shown). Hematoxylin and eosin and Masson's trichrome staining for myocardial sections of SUNDS cases showed no signs of dilated or hypertrophic cardiomyopathy and no significant infiltration of inflammatory cells and fibrous/fatty tissue in myocardium (data not shown). These cases were compared to sex‐ and age‐matched controls (death caused by traffic accident, no pathogenic rare variants identified in VCL and 79 known primary arrhythmia/cardiomyopathy–causing genes). Colocalization of SCN5A and VCL from human LV tissue slices was detected at the intercalated disk (ID) area in an immunofluorescent staining experiment (Figure 2 shows samples from a 38‐year‐old homozygous D841H carrier and control). The yellow color pixels in the Figure 2A and 2B overlay panel suggest that SCN5A and VCL were colocalized at the ID. In the signal intensity profile presented in Figure 2C and 2D, SCN5A was observed to be expressed at both the ID and t‐tubules (blue line) in both control and case groups, whereas VCL (red line) only expressed at the ID. No significant difference of expression and distribution of both SCN5A and VCL were observed between the controls and SUNDS cases. As previously reported,25 we identified that VCL directly interacts with SCN5A in vivo and in vitro using coimmunoprecipitations (Figure 3A and 3B). Furthermore, VCL‐D841H does not affect this physical association between VCL and SCN5A (Figure 3C and 3D).
Figure 2.
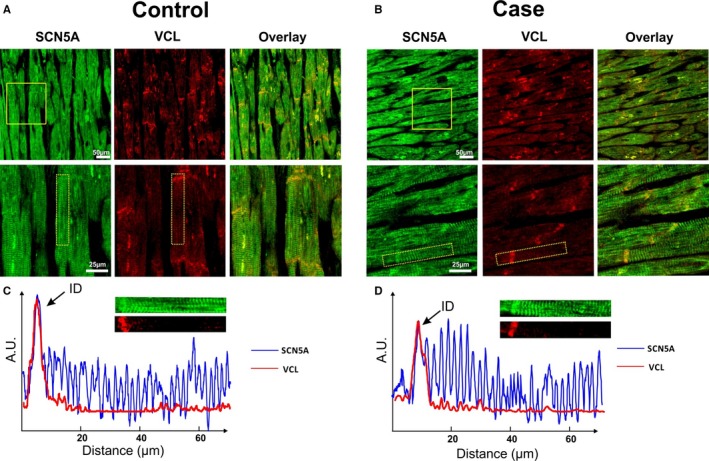
Colocalization of vinculin (VCL) and cardiac sodium channel (SCN5A) in human ventricular tissue slice. A and B, Immunofluorescent staining of SCN5A (green), VCL (red), and overlay channel from control and case (a homozygous D841H carrier) human ventricular tissue sections, presented from a normal and magnification field of view, respectively. Magnification field of view were labeled by yellow solid boxes in the panel. Yellow color from overlay panel indicates the colocalization of VCL and SCN5A. C and D, Signal intensity profile of signals from selected areas (shown in the panel) were calculated and plotted from control and case slices, respectively. The selected areas were labeled by yellow dashed boxes in the magnification in (A) and (B). Integrity of the fluorescence signals of both SCN5A (blue line) and VCL (red line) along transverse direction was aligned and plotted. A.U. indicates arbitrary units; ID, intercalated disk.
Figure 3.
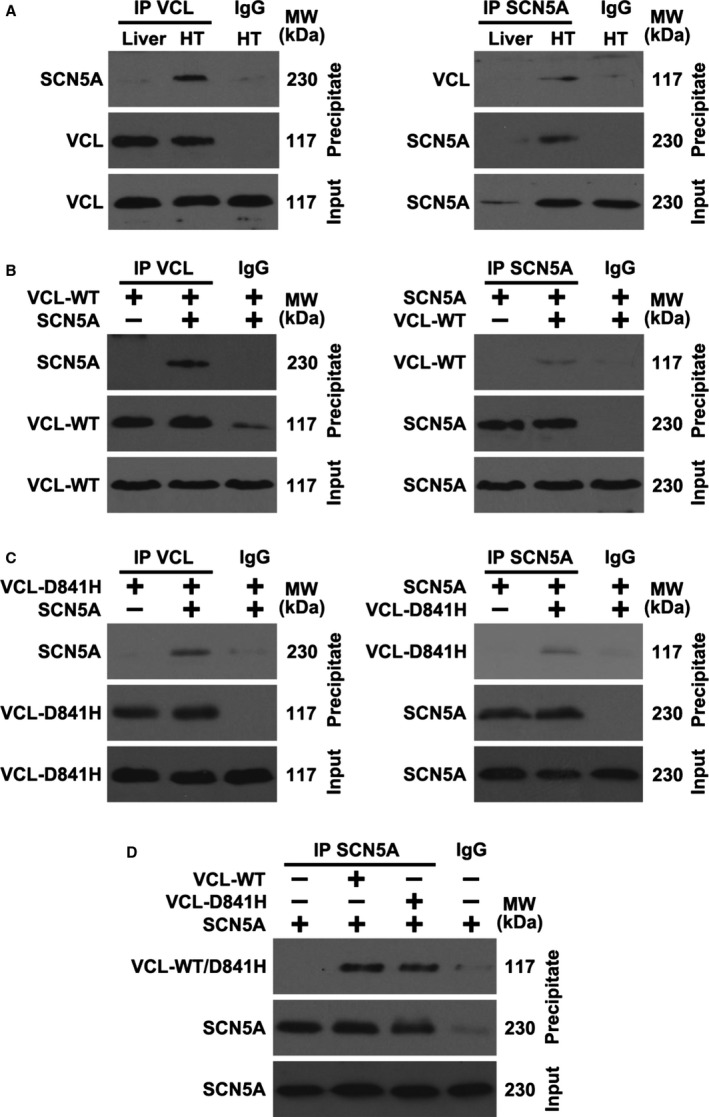
Vinculin (VCL) interacts with cardiac sodium channel (SCN5A) in vivo and in vitro. A, Heart tissue (HT) lysates from mouse were immunoprecipitated (IP) using SCN5A or VCL antibody and analyzed by western blotting using the indicated antibodies. Because of the negative expression of SCN5A, mouse liver tissue was used as a control. (B) SCN5A and wild‐type VCL (VCL‐WT) were transfected into HEK293 cells for 1 day, and cell lysates were IP and analyzed by western blotting. (C) VCL‐D841H and SCN5A were transfected into HEK293 cells for 1 day, and cell lysates were IP and analyzed by western blotting. (D) VCL‐WT, VCL‐D841H, and SCN5A were transfected into HEK293 cells for 1 day, and cell lysates were IP and analyzed by western blotting. IgG indicates immunoblobulin G; MW, molecular weight.
Vinculin Common Variant D841H Caused the Sodium Channel Loss of Function in Both HEK293 Cells and iPSC‐CMs
First, we investigated the baseline effect of coexpressed VCL‐WT on sodium channel function in HEK293 cells. As compared to the expression of SCN5A alone, coexpression of both SCN5A and VCL can increase sodium current density by ≈30% but did not change the sodium channel's activation and inactivation properties (Table 3). Then, we biophysically characterized the effects of coexpressed VCL‐D841H on I Na in HEK293 cells. Under physiological pH conditions (pH 7.4), D841H caused a 29% decrease in peak I Na amplitude compared to WT (Figure 4A and 4B; Table 4). The level of late I Na was measured as a percentage of peak I Na. The late I Na for D841H was comparable to WT (Table 4). The analysis on the kinetic parameters exhibited that there was no significant difference between WT and D841H in both activation and inactivation (Table 4; Figure 5A and 5B). However, D841H showed slower recovery from inactivation and had significantly larger slow time constant (τS) values compared to WT (Table 5).
Table 3.
Biophysical Properties of Sodium Channels in HEK293 Cells Coexpressing SCN5A and Either VCL‐WT or Empty Vector
| Samples | Peak I Na | Activation | Inactivation | |||||
|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2 (mV) | k | n | V1/2 (mV) | k | n | |
| SCN5A+VCL | −145±8 | 13 | −41.5±0.9 | 5.0 | 13 | −84.4±1.5 | 5.1 | 13 |
| SCN5A+empty vector | −113±10a | 13 | −41.2±0.8 | 5.0 | 13 | −85.1±1.2 | 5.1 | 13 |
Values are mean±SE for n experiments. I Na indicates sodium current; k, slope factor; pA/pF, current density; V1/2, voltage of half‐maximal activation/inactivation; VCL, vinculin; WT, wild type.
P=0.02 vs SCN5A+VCL.
Figure 4.
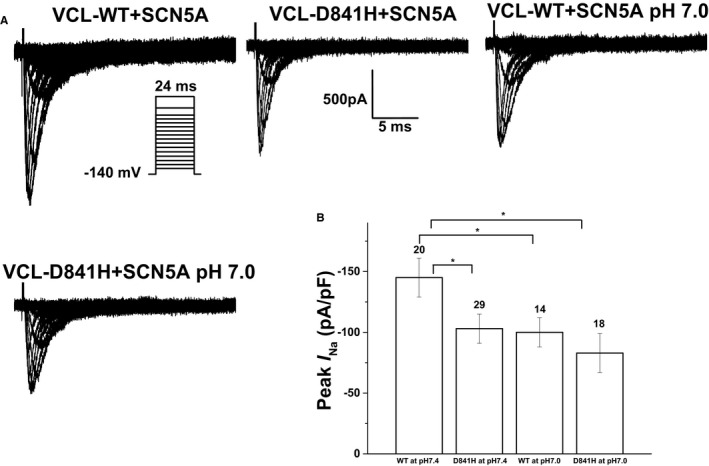
Electrophysiological properties of cardiac sodium channel (SCN5A) in HEK293 cells coexpressing SCN5A and either wild‐type vinculin (VCL‐WT) or VCL‐D841H. A, Representative whole‐cell current traces showing peak sodium current (IN a) under both normal (pH 7.4) and acidosis (pH 7.0) conditions in HEK293 cells expressing SCN5A and either WT or variant VCL‐D841H. B, Summary data of peak IN a densities from every group. The number of tested cells is indicated above the bar. *P<0.05.
Table 4.
Biophysical Properties of Sodium Channels in HEK293 Cells Coexpressing SCN5A and Either VCL‐WT or ‐D841H
| Samples | Peak I Na | Activation | Inactivation | Late I Na | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2 (mV) | k | n | V1/2 (mV) | k | n | % | n | |
| WT at pH 7.4 | −145±16 | 20 | −41.9±0.8 | 5.0 | 20 | −84.9±1.4 | 5.1 | 26 | 0.31±0.06 | 16 |
| D841H at pH 7.4 | −103±12a | 29 | −41.0±0.7 | 5.0 | 26 | −85.5±1.1 | 5.0 | 29 | 0.32±0.05 | 16 |
| WT at pH 7.0 | −100±12a | 24 | −43.8±0.8 | 5.5 | 18 | −87.1±0.9 | 5.0 | 20 | 0.45±0.23 | 7 |
| D841H at pH 7.0 | −83±16a | 18 | −44.9±1.5 | 5.4 | 18 | −87.9±0.6a | 5.0 | 15 | 0.39±0.19 | 5 |
Values are mean±SE for n experiments. The late I Na level was described as a percentage of peak I Na. All parameters were analyzed using 1‐way ANOVA followed by a Bonferroni test. I Na indicates sodium current; k, slope factor; pA/pF, current density; V1/2, voltage of half‐maximal activation/inactivation; VCL, vinculin; WT, wild type.
P<0.05 vs WT at pH 7.4.
Figure 5.
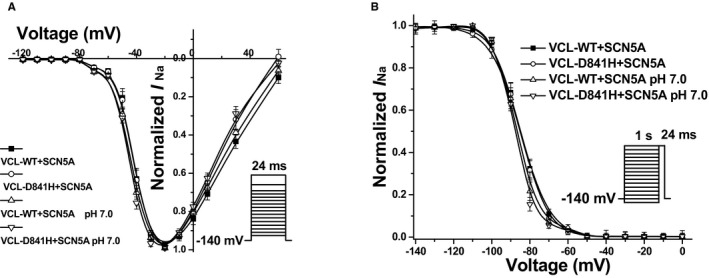
Voltage‐dependent gating for cardiac sodium channel (SCN5A) coexpressed with vinculin (VCL) in HEK293 cells. A, Between each group, no significant difference in activation of SCN5A was observed. B, Under pH 7.0, D841H showed a significant repolarizing shift by 3.0 mV in inactivation of SCN5A compared to WT at pH 7.4.
Table 5.
Recovery of Sodium Channels in HEK293 Cells Coexpressing SCN5A and Either WT or Mutant VCL
| Samples | Recovery | |||
|---|---|---|---|---|
| τf (ms) | τS (ms) | A S, % | n | |
| WT at pH 7.4 | 1.82±0.13 | 29.5±3.3 | 0.16±0.01 | 20 |
| D841H at pH 7.4 | 2.10±0.16 | 45.7±5.3a | 0.18±0.01 | 24 |
| WT at pH 7.0 | 2.72±0.23a | 62.5±7.3b | 0.21±0.01 | 11 |
| D841H at pH 7.0 | 2.89±0.12a | 70.5±8.2b | 0.21±0.03 | 7 |
A S is fractional amplitudes of slow components. τf and τS are fast and slow time constants. All parameters were analyzed using 1‐way ANOVA followed by a Bonferroni test. SCN5A indicates cardiac sodium channel; VCL, vinculin; WT, wild type.
P<0.05 vs WT at pH 7.4.
P<0.01 vs WT at pH 7.4.
Compared with WT at pH 7.4, WT and D841H at pH 7.0 decreased peak I Na by 31% and 43%, respectively (Figure 4A and 4B; Table 4). At low pH, D841H caused a significant negative shift by 3 mV in inactivation compared to WT at pH 7.4 (Table 4; Figure 5B) and had significantly larger fast and slow time constant values in recovery from inactivation compared with WT at pH 7.4 (Table 5), which may account for the statistically significant reduction in peak I Na for D841H. The impaired inactivation and recovery would be predicted to further decrease I Na at more‐physiological heart rates and resting membrane potentials.
To address whether VCL‐D841H exerts effects on cardiomyocytes, we then tested induced pluripotent stem‐cell‐derived cardiomyocytes (iPSC‐CMs) overexpressing VCL. Under physiological pH conditions, D841H tended to decrease peak I Na compared to WT (Figure 6A and 6B; Table 6), without reaching statistical significance (P=0.22). With acidosis at pH 7.0, D841H showed a significant 41% reduction in peak I Na amplitude compared with WT at pH 7.4 (Figure 6A and 6B; Table 6). Consistent with the alteration in HEK293 cells, there was no significant difference in activation (Table 6; Figure 7A) and recovery from inactivation (data not shown) between each group. Notably, compared to WT at pH 7.4, D841H at pH 7.0 showed a significant negative shift in inactivation (Table 6; Figure 7B), which may be responsible for remarkable decrease in peak I Na for D841H under acidosis.
Figure 6.

Electrophysiological properties of cardiac sodium channels in induced pluripotent stem‐cell–derived cardiomyocytes (iPSC‐CMs) expressing either wild‐type vinculin (VCL‐WT) or VCL‐D841H. A, Representative whole‐cell current traces showing peak sodium current (IN a) under both normal (pH 7.4) and acidosis (pH 7.0) conditions in iPSC‐CMs expressing either WT or variant VCL‐D841H. B, Summary data of peak IN a densities from every group. The number of tested cells is indicated above the bar. *P<0.05.
Table 6.
Biophysical Properties of Sodium Channels in iPSC‐CMs Overexpressing Either WT or Mutant VCL
| Samples | Peak I Na | Activation | Inactivation | |||||
|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2 (mV) | k | n | V1/2 (mV) | k | n | |
| WT at pH 7.4 | −71±13 | 17 | −43.5±3.0 | 6.2 | 13 | −80.9±5.5 | 6.3 | 5 |
| D841H at pH 7.4 | −53±14 | 16 | −43.7±2.1 | 5.6 | 10 | −84.4±6.2 | 5.7 | 5 |
| WT at pH 7.0 | −55±8 | 8 | −48.4±1.2 | 5.0 | 7 | −84.8±5.5 | 6.5 | 5 |
| D841H at pH 7.0 | −42±6a | 8 | −46.7±1.0 | 6.5 | 5 | −106.5±4.3a | 7.3 | 5 |
Values are mean±SE for n experiments. All parameters were analyzed using 1‐way ANOVA followed by a Bonferroni test. I Na indicates sodium current; iPSC‐CMs, induced pluripotent stem‐cell–derived cardiomyocytes; k, slope factor; pA/pF, current density; V1/2, voltage of half‐maximal activation/inactivation; VCL, vinculin; WT, wild type.
P<0.05 vs WT at pH 7.4.
Figure 7.
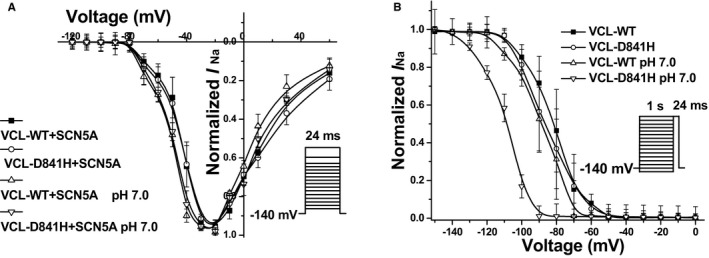
Voltage‐dependent gating for cardiac sodium channel in induced pluripotent stem‐cell–derived cardiomyocytes expressing with vinculin (VCL). A, There was no statistically significant difference in activation of cardiac sodium channel between each group. B, Under pH 7.0 conditions, D841H caused a significant repolarizing shift by 25.6 mV in inactivation of cardiac sodium channel compared to wild type (WT) at pH 7.4.
Vinculin‐D841H Combined With SCN5A Common Variant H558R Caused Increased Late I Na in HEK293 Cells
Attributed to the biophysical effect of SCN5A common variant His558Arg (H558R) on SCN5A function and detection of H558R in 1 of the VCL‐D841H carriers, we tested whether SCN5A‐H558R affects the biophysical phenotype of VCL‐D841H under physiological pH conditions.
Surprisingly, the variant, SCN5A‐H558R, reversed the decrease of peak I Na amplitude caused by VCL‐D841H in HEK293 cells. However, SCN5A‐H558R significantly increased the late I Na for VCL‐D841H in HEK293 cells (Figure 8). For channel kinetics, SCN5A‐H558R did not disrupt both activation and inactivation for VCL‐D841H in HEK293 cells (Table 7).
Figure 8.
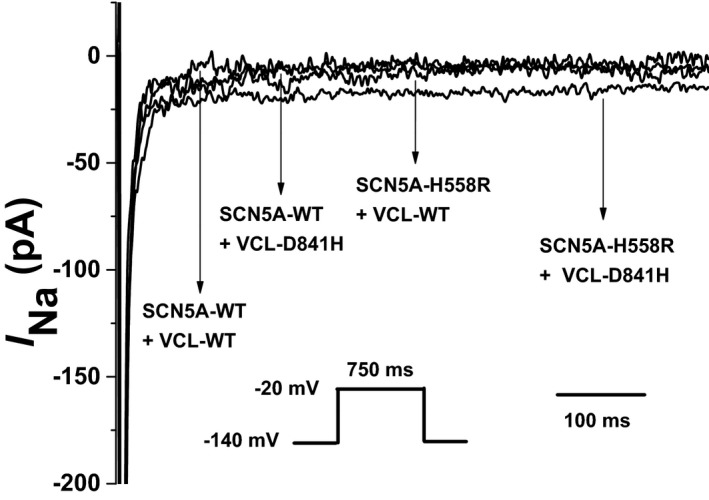
Late sodium current characterized in HEK293 cells coexpressing cardiac sodium channel (SCN5A) and vinculin (VCL). Representative traces showing increased late sodium current (IN a) associated with SCN5A‐H558R+VCL‐D841H compared with SCN5A‐WT+VCL‐WT, or SCN5A‐WT+VCL‐D841H, or SCN5A‐H558R+VCL‐WT.
Table 7.
Effect of Polymorphisms of SCN5A and VCL on Late Sodium Current in HEK293 Cells
| Samples | Peak I Na | Activation | Inactivation | Late I Na | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2 (mV) | k | n | V1/2 (mV) | k | n | % | n | |
| SCN5A‐WT+VCL‐WT | −101±11 | 17 | −39.4±0.7 | 5.0 | 24 | −84.4±0.9 | 5.0 | 22 | 0.32±0.04 | 29 |
| SCN5A‐WT+VCL‐D841H | −70±9a | 24 | −38.2±0.6 | 5.0 | 26 | −84.8±0.8 | 5.0 | 26 | 0.34±0.05 | 15 |
| SCN5A‐H558R+VCL‐WT | −105±9 | 25 | −38.5±1.1 | 5.0 | 32 | −85.4±1.2 | 5.0 | 30 | 0.43±0.05 | 19 |
| SCN5A‐H558R+VCL‐D841H | −102±7 | 29 | −39.4±0.5 | 5.0 | 38 | −86.0±0.8 | 5.0 | 31 | 0.60±0.06a | 23 |
Values are mean±SE for n experiments. The late I Na level was described as a percentage of peak I Na. All parameters were analyzed using 1‐way ANOVA followed by a Bonferroni test. I Na indicates sodium current; k, slope factor; pA/pF, current density; SCN5A indicates cardiac sodium channel; V1/2, voltage of half‐maximal activation/inactivation; VCL, vinculin; WT, wild type.
P<0.05 vs SCN5A‐WT+VCL‐WT.
Discussion
Vinculin‐D841H was Associated With SUNDS in Chinese Han Population
VCL, as well as its muscle isoform, metavinculin, has been identified as a susceptibility gene for cardiomyopathy13, 14, 15 and has also been linked to sudden arrhythmia death in VCL knockout mice preceding the formation of cardiomyopathy.16 Most recently, VCL mutation M94I was identified to be associated with the genetic cause of 1 SUNDS victim.17 In the present study, we genetically associated a common VCL variant D841H with SUNDS in the Chinese Han population, in which those individuals carrying the H841 allele showed a 5‐fold increased risk for SUNDS. Biophysical studies of VCL‐D841H in both HEK293 cells and human iPSC‐CMs showed that this polymorphism tended to decrease peak sodium current under physiological conditions, and this trend becomes a significant sodium channel loss of function under slight acidosis conditions. Moreover, the SCN5A common variant, H558R, interacted with VCL‐D841H to cause significant increased late I Na. All these results suggest that VCL‐D841H may be an independent risk factor for Chinese SUNDS through the interaction between VCL and SCN5A by which VCL‐D841H causes SCN5A either loss or gain of function in the setting of environmental and/or genetic factors. Notably, whether the high prevalence of H841 in the East Asian population is associated with the high occurrence of SUNDS in Asians requires further investigation.
Vinculin Physically and Functionally Interacts With SCN5A
In cardiomyocytes, VCL and its muscle isoform, metavinculin, are predominantly expressed at costameres and IDs and serve to maintain normal cell‐cell and cell‐matrix junctions as well as cardiac rhythm.16, 26, 27 So far, whereas several VCL mutations have been identified to account for cardiomyopathy,13, 14, 15, 28, 29 the functional effect of VCL on cardiac arrhythmia remains unclear. In both hemizygous null VCL mice26 and cardiomyocyte‐specific excision of VCL mice16 models, the gap junctional protein, connexin 43 (Cx43), within the ID was found to be abnormally distributed.16, 26 This regulatory effect of VCL on Cx43 was observed to be associated with the direct interaction between VCL and zonula occludens‐1 at the ID.27 In this investigation, we biophysically identified a functional effect of VCL on SCN5A by showing that the loss of function of SCN5As resulted from VCL variant D841H. According to our findings that VCL directly interacts with SCN5A in vivo and in vitro and that VCL and SCN5A were colocalized at the ID in the human heart, this functional effect may result from the direct association between VCL and SCN5A.17 However, the fact that VCL‐D841H did not affect the interaction between VCL and SCN5A suggested that the molecular mechanism for this functional interaction remains to be addressed.
SUNDS and Abrupt Breathing Abnormalities During Nocturnal Sleep
An early sleep‐monitoring experiment indicated that nocturnal hypoxia might be the primary abnormality in individuals with SUNDS family history.30 The high prevalence of sleep apnea and paralysis were regarded to be related to the high occurrence of SUNDS in Hmong immigrants in the United States.7 All this clinical evidence as well as forensic epidemiological studies on SUNDS1, 2, 3, 4, 5, 6, 7, 31 show that respiratory disorders during nocturnal sleep (such as sleep apnea, gasping, and unusual snoring) may play an important role in the pathogenesis of SUNDS. Plant et al32 previously reported that sleep respiratory abnormality‐associated intracellular acidosis significantly increased the late I Na caused by the sudden infant death syndrome susceptibility SCN5A common variant, S1103Y, in blacks. Recently, the SCN5A mutation, R1512W, found in a Chinese SUNDS victim with sudden nocturnal tachypnea before death was identified to show obvious SCN5A loss of function only under acidosis conditions.33 In this study, we identified, once again, that breathing‐disorder–related acidosis plays a crucial role for VCL‐D841H in aggravating SCN5A loss of function. All these findings strongly implicate the possible important contribution of sleep respiratory disorders in the occurrence of SUNDS.7, 30, 31, 33
Cardiomyopathy and SUNDS
Studies with VCL knockout mice revealed a chronic pathological progression from an apparently normal heart to cardiomyopathy, whereas sudden arrhythmia death could happen at any life stage.16, 26 Consistently, these 8 SUNDS cases identified in this study with VCL‐D841H did not show any pathological characteristics for cardiomyopathy, but suffered sudden unexplained death. The genetic and functional data implicated VCL‐D841H as a susceptible polymorphism for Chinese SUNDS.
Increasing lines of evidence show that primary arrhythmia disorders (such as BrS and idiopathic atrial fibrillation) with a structurally normal heart share some common clinical phenotypes and susceptible or plausible pathogenic genes (such as SCN5A, PKP2, ABCC9, RYR2, DSG2, CASQ2, JUP, and DSP) with cardiomyopathy.12, 13, 18, 34 These findings suggest that there must be an intrinsic genetic association between cardiomyopathy and primary arrhythmias. Most recently, there was new evidence showing a morphological association of cardiomyopathy and primary arrhythmia syndromes. Structural alterations, such as fibrosis and loss of gap junctions, were found in BrS patients elucidating the primary arrhythmia.35 Our group also observed the previously unrecognized significant cardiac structural changes (slightly increased heart weight, enlarged circumference of cardiac valves) in SUNDS victims.36
All these findings support the idea that some of the previously recognized primary arrhythmias with apparently intact heart, such as BrS and SUNDS, might be a subtype or early stage of cardiomyopathy.35, 36, 37, 38
Study Limitations
This work has several limitations. First, the absence of clinic (such as ECG) records is always a study limitation that is inherent to the forensic medical investigation on SUNDS victims, where, by definition, victims are apparently healthy individuals who suffer sudden unexpected death. This limits a deeper analysis of association between clinical phenotype, genetic findings, and functional data. Second, the functional study was conducted by in vitro experiments using both HEK293 cells and iPSC‐CMs, which is different from the genuine in vivo environment. It would be helpful to assess the impacts of VCL‐D841H in vivo by characterizing mouse models established by CRISPR technology. Last, we would not be able to quickly investigate another replication cohort of SUNDS in the near future because of the difficulty of collecting new samples. Moreover, it is hard for us to estimate the effect of this VCL‐D841H variant on SUNDS accurately because of the rarity of the variant in both SUNDS victims and general population. Although this is the largest SUNDS cohort reported, it is necessary to confirm the prevalence of VCL‐D841H in a replication SUNDS cohort or primary arrhythmia disorder in the future attributed to the possible genetic heterogeneity for SUNDS.
Conclusions
In summary, a VCL common variant, D841H, was associated with Chinese SUNDS. Molecular, biochemical, and biophysical charcterizations revealed VCL‐D841H as likely causing loss of function of SCN5A. The significant aggravation of loss of function of SCN5A caused by VCL‐D841H under acidosis further supports the idea that nocturnal sleep respiratory disorders with acidosis may play a crucial role in triggering the deadly arrhythmia underlying SUNDS victims.
Sources of Funding
This work was supported by the Key Program (81430046) from the National Natural Science Foundation of China (Cheng) and the grants R56 HL71092 and R01HL128076‐01 from the National Institutes of Health (Makielski).
Disclosures
None.
(J Am Heart Assoc. 2017;6:e005330 DOI: 10.1161/JAHA.116.005330.)28373245
References
- 1. Gaw AC, Lee B, Gervacio‐Domingo G, Antzelevitch C, Divinagracia R, Jocano F Jr. Unraveling the enigma of Bangungut: is sudden unexplained nocturnal death syndrome (SUNDS) in the Philippines a disease allelic to the brugada syndrome? Philipp J Intern Med. 2011;49:165–176. [PMC free article] [PubMed] [Google Scholar]
- 2. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595–2600. [DOI] [PubMed] [Google Scholar]
- 3. Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, Nademanee K, Brugada R, Brugada J, Veerakul G, Li H, Bowles NE, Brugada P, Antzelevitch C, Towbin JA. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–345. [DOI] [PubMed] [Google Scholar]
- 4. Yap EH, Chan YC, Goh KT, Chao TC, Heng BH, Thong TW, Singh M, Jacob E. Pseudomonas pseudomallei and sudden unexplained death in Thai construction workers. Lancet. 1990;336:376–377. [DOI] [PubMed] [Google Scholar]
- 5. Feest TG, Wrong O. Potassium deficiency and sudden unexplained nocturnal death. Lancet. 1991;338:1406. [DOI] [PubMed] [Google Scholar]
- 6. Nakajima K, Takeichi S, Nakajima Y, Fujita MQ. Pokkuri death syndrome; sudden cardiac death cases without coronary atherosclerosis in South Asian young males. Forensic Sci Int. 2011;207:6–13. [DOI] [PubMed] [Google Scholar]
- 7. Young E, Xiong S, Finn L, Young T. Unique sleep disorders profile of a population‐based 367 sample of 747 Hmong immigrants in Wisconsin. Soc Sci Med. 2013;79:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu C, Tester DJ, Hou Y, Wang W, Lv G, Ackerman MJ, Makielski JC, Cheng J. Is sudden unexplained nocturnal death syndrome in Southern China a cardiac sodium channel dysfunction disorder? Forensic Sci Int. 2014;236:38–45. [DOI] [PubMed] [Google Scholar]
- 9. Huang L, Tang S, Peng L, Chen Y, Cheng J. Molecular autopsy of desmosomal protein plakophilin‐2 in sudden unexplained nocturnal death syndrome. J Forensic Sci. 2016;61:687–691. [DOI] [PubMed] [Google Scholar]
- 10. Zhang L, Zhou F, Huang L, Wu Q, Zheng J, Wu Y, Yin K, Cheng J. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int J Legal Med. 2017;131:53–60. [DOI] [PubMed] [Google Scholar]
- 11. Liu M, Yang KC, Dudley SC Jr. Cardiac sodium channel mutations: why so many phenotypes? Nat Rev Cardiol. 2014;11:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cerrone M, Lin X, Zhang M, Agullo‐Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin‐2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maeda M, Holder E, Lowes B, Valent S, Bies RD. Dilated cardiomyopathy associated with deficiency of the cytoskeletal protein metavinculin. Circulation. 1997;95:17–20. [DOI] [PubMed] [Google Scholar]
- 14. Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105:431–437. [DOI] [PubMed] [Google Scholar]
- 15. Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006;87:169–174. [DOI] [PubMed] [Google Scholar]
- 16. Zemljic‐Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac‐myocyte‐specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng J, Kyle JW, Wiedmeyer B, Lang D, Vaidyanathan R, Makielski JC. Vinculin variant M94I identified in sudden unexplained nocturnal death syndrome decreases cardiac sodium current. Sci Rep. 2017;7:42953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Q, Chen Y, Peng L, Gao R, Liu N, Jiang P, Liu C, Tang S, Quan L, Makielski JC, Cheng J. Identification of rare variants of DSP gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Int J Legal Med. 2016;130:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT. High purity human‐induced pluripotent stem cell‐derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301:H2006–H2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, Janaury CT, Eckhardt LL. IK1‐enhanced human induced pluripotent stem cell‐derived cardiomyocytes: an improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol. 2016;310:H1611–H1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC. Caveolin‐3 suppresses late sodium current by inhibiting nNOS‐dependent S‐nitrosylation of SCN5A. J Mol Cell Cardiol. 2013;61:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakolitsa C, Cohen DM, Bankston LA, Bobkov AA, Cadwell GW, Jennings L, Critchley DR, Craig SW, Liddington RC. Structural basis for vinculin activation at sites of cell adhesion. Nature. 2004;430:583–586. [DOI] [PubMed] [Google Scholar]
- 24. Kim LY, Thompson PM, Lee HT, Pershad M, Campbell SL, Alushin GM. The structural basis of actin organization by vinculin and metavinculin. J Mol Biol. 2016;428:10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izard T, Brown DT. Mechanisms and functions of vinculin interactions with phospholipids at cell adhesion sites. J Biol Chem. 2016;291:2548–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zemljic‐Harpf AE, Ponrartana S, Avalos RT, Jordan MC, Roos KP, Dalton ND, Phan VQ, Adamson ED, Ross RS. Heterozygous inactivation of the vinculin gene predisposes to stress‐induced cardiomyopathy. Am J Pathol. 2004;165:1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zemljic‐Harpf AE, Godoy JC, Platoshyn O, Asfaw EK, Busija AR, Domenighetti AA, Ross RS. Vinculin directly binds zonula occludens‐1 and is essential for stabilizing connexin‐43‐containing gap junctions in cardiac myocytes. J Cell Sci. 2014;127:1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vasile VC, Ommen SR, Edwards WD, Ackerman MJ. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2006;345:998–1003. [DOI] [PubMed] [Google Scholar]
- 29. Wells QS, Ausborn NL, Funke BH, Pfotenhauer JP, Fredi JL, Baxter S, Disalvo TD, Hong CC. Familial dilated cardiomyopathy associated with congenital defects in the setting of a novel VCL mutation (Lys815Arg) in conjunction with a known MYPBC3 variant. Cardiogenetics. 2011;1:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Charoenpan P, Muntarbhorn K, Boongird P, Puavilai G, Ratanaprakarn R, Indraprasit S, Tanphaichitr V, Likittanasombat K, Varavithya W, Tatsanavivat P. Nocturnal physiological and biochemical changes in sudden unexplained death syndrome: a preliminary report of a case control study. Southeast Asian J Trop Med Public Health. 1994;25:335–340. [PubMed] [Google Scholar]
- 31. Tanchaiswad W. Is sudden unexplained nocturnal death a breathing disorder? Psychiatry Clin Neurosci. 1995;49:111–114. [DOI] [PubMed] [Google Scholar]
- 32. Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, Chen W, Kittles RA, Goldstein SA. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest. 2006;116:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng J, Zhou F, Su T, Huang L, Wu Y, Yin K, Wu Q, Tang S, Makielski JC, Cheng J. The biophysical characterization of the first SCN5A mutation R1512W identified in Chinese sudden unexplained nocturnal death syndrome. Medicine (Baltimore). 2016;95:e3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allegue C, Coll M, Mates J, Campuzano O, Iglesias A, Sobrino B, Brion M, Amigo J, Carracedo A, Brugada P, Brugada J, Brugada R. Genetic analysis of arrhythmogenic diseases in the era of NGS: the complexity of clinical decision‐making in Brugada syndrome. PLoS One. 2015;10:e0133037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, Makita N, Kowase S, Boonmee N, Vitayakritsirikul V, Ratanarapee S, Sharma S, van der Wal AC, Christiansen M, Tan HL, Wilde AA, Nogami A, Sheppard MN, Veerakul G, Behr ER. Fibrosis, connexin‐43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol. 2015;66:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang L, Tester DJ, Lang D, Chen Y, Zheng J, Gao R, Corliss RF, Tang S, Kyle JW, Liu C, Ackerman MJ, Makielski JC, Cheng J. Does sudden unexplained nocturnal death syndrome remain the autopsy negative disorder: a gross, microscopic, and molecular autopsy investigation in Southern China. Mayo Clin Proc. 2016;91:1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peters S. Ion channel diseases as a part in the definition and classification of cardiomyopathies recently confirmed in Brugada syndrome. Int J Cardiol. 2016;207:103. [DOI] [PubMed] [Google Scholar]
- 38. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ , Seidman CE, Young JB; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the Council on Clinical Cardiology; Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]