

Abstract
Background
The transition of aortic valve interstitial cells (AVICs) to myofibroblastic and osteoblast‐like phenotypes plays a critical role in calcific aortic valve disease progression. Several microRNAs (miRs) are implicated in stem cell differentiation into osteoblast. We hypothesized that an epigenetic mechanism regulates valvular pro‐osteogenic activity. This study examined miR profile in AVICs of calcified valves and identified miRs responsible for AVIC phenotypic transition.
Methods and Results
AVICs were isolated from normal and diseased valves. The miR microarray analysis revealed 14 upregulated and 12 downregulated miRs in diseased AVICs. Increased miR‐486 and decreased miR‐204 levels were associated with higher levels of myofibroblastic biomarker α‐smooth muscle actin and osteoblastic biomarkers runt‐related transcription factor 2 (Runx2) and osterix (Osx). Cotransfection of miR‐486 antagomir and miR‐204 mimic in diseased AVICs reduced their ability to express Runx2 and Osx. The miR‐486 mimic upregulated α‐smooth muscle actin expression in normal AVICs through the protein kinase B pathway and moderately elevated Runx2 and Osx levels. Knockdown of α‐smooth muscle actin attenuated Runx2 and Osx expression induced by miR‐486. The miR‐486 mimic and miR‐204 antagomir synergistically promoted Runx2 and Osx expression and calcium deposition in normal AVICs and normal aortic valve tissue.
Conclusions
In AVICs of calcified valves, increased levels of miR‐486 induce myofibroblastic transition to upregulate Runx2 and Osx expression and synergize with miR‐204 deficiency to elevate cellular and valvular pro‐osteogenic activity. These novel findings indicate that modulation of the epigenetic mechanism underlying valvular pro‐osteogenic activity has therapeutic potential for prevention of calcific aortic valve disease progression.
Keywords: aortic valve, calcification, fibroblasts, microRNA, myofibroblast, osteogenesis, phenotypic transition
Subject Categories: Valvular Heart Disease, Gene Expression & Regulation, Cell Signalling/Signal Transduction, Cell Biology/Structural Biology, Vascular Biology
Introduction
Calcific aortic valve disease (CAVD) affects a large number of elder people (65 or older) and is one of the leading cardiovascular diseases in older adults in industrial countries.1, 2 This disease is an active pathobiological process involving fibrosis and calcification of aortic valve leaflets.3 Severe CAVD causes morbidity and results in the second most common cardiovascular surgery performed.4 The growing prevalence of this disease, associated with prolongation of the human life span, and the unavailability of pharmacological intervention for limitation of disease progression emphasize the importance of investigations of pathobiological mechanisms. In particular, it is critical to elucidate the cellular and molecular mechanisms by which aortic valve leaflets become fibrotic and calcified.
Aortic valve interstitial cells (AVICs) play an important role in maintaining valvular structure and function. In normal human aortic valves, AVICs primarily assume a fibroblastic phenotype.5 They produce extracellular matrix proteins to maintain the valvular structure. In addition, AVICs synthesize and release growth factors and cytokines.6, 7 In calcified aortic valves, however, the majority of AVICs exhibit a myofibroblastic phenotype.8 Myofibroblasts express high levels of α‐smooth muscle actin (α‐SMA), possess elevated proliferative activity, and produce higher levels of extracellular matrix protein.9, 10 Elevated proliferative activity of myofibroblasts and overproduction of extracellular matrix protein by these cells lead to aortic valve fibrosis and valvular thickening. Currently, the molecular mechanism underlying human AVIC phenotypic transition remains unclear, although several growth factors, such as transforming growth factor‐β1 (TGF‐β1) and bone morphogenetic protein 2 (BMP‐2), have been shown to induce myofibroblastic transition in porcine AVICs.11, 12
AVICs are also capable of undergoing osteoblast‐like phenotypic transition in response to pro‐osteogenic stimulation.13, 14 Interestingly, AVICs of calcified valves display a pro‐osteogenic phenotype with enhanced expression of osteoblast biomarkers, such as runt‐related transcription factor 2 (Runx2), osterix (Osx), alkaline phosphatase, and osteopontin.15 Such AVICs are termed osteoblastic‐like cells, and they exhibit pro‐osteogenic activities at baseline and assume greater activities when exposed to a variety of stimuli, including TGF‐β1 and BMP‐2.16, 17 The osteoblast‐like activity of AVICs is believed to be responsible for valve mineralization associated with CAVD.13, 14, 15, 18 However, the mechanism underlying the transition of AVICs to a pro‐osteogenic phenotype is not well understood. In addition, the relationship between AVIC myofibroblastic transition and pro‐osteogenic activities is unclear. Understanding the molecular mechanism underlying AVIC phenotypic transition, particularly the mechanism responsible for the pro‐osteogenic phenotype of AVICs in calcified valves, is important for the development of therapeutic strategies for prevention of CAVD progression.
MicroRNAs (miRs) are a group of small noncoding 21‐ to 23‐nucleotide‐long RNA molecules.19 Several studies have reported that the expression of miRs can be modulated by Toll‐like receptor signaling, such as nuclear factor κB and MAPK pathways, and by pro‐osteogenic factors TGF‐β and BMP‐2.11, 12, 20 For example, miR‐9 expression is upregulated by lipopolysaccharide via the TLR4–MyD88–nuclear factor κB–dependent pathway in human monocytes and neutrophils.21 Our previous study found that Toll‐like receptor 4 or 2 mediates AVIC expression of TGF‐β1 or BMP‐2 in response to pro‐inflammatory stimulation and promotes AVIC pro‐osteogenic reprogramming.22 It is likely that pro‐inflammatory mechanisms modulate miR expression through regulation of the production of growth factors. The miRs regulate the expression of protein‐coding genes and thus play important roles in the epigenetic regulation of gene expression. Emerging evidence has demonstrated that miRs modulate a wide range of developmental and physiological processes, including cell proliferation and differentiation.23 Recently, miRs were implicated in the pathophysiology of several cardiovascular diseases and have become intriguing targets for therapeutic intervention.24 The search for miRs that target Runx2 led to the identification of roles of multiple miRs, including miR204, in the regulation of Runx2 expression in mesenchymal progenitor cells.25 These miRs downregulate Runx2 to inhibit osteoblast and chondrocyte differentiation.26 In addition, several studies found that altered expression of miR‐30b, miR‐26a, and miR‐195 is associated with CAVD.27, 28 Systematic analysis of miR profile in calcified aortic valves and investigation of the role of miRs in AVIC phenotypic transition will provide insights into the molecular mechanism underlying CAVD pathogenesis and may identify therapeutic targets for suppression of CAVD progression.
The purposes of this study were (1) to examine the altered miR expression profile in AVICs of calcified human aortic valves, (2) to identify miRs responsible for the myofibroblastic and osteoblast‐like transitions in human AVICs, and (3) to determine the mechanism by which miRs modulate AVIC and valvular pro‐osteogenic activity.
Materials and Methods
Materials
Antibody against human Osx was purchased from Santa Cruz Biotechnology, Inc. Antibodies against human α‐SMA were purchased from Abcam, Inc. Antibodies against human Runx2, vimentin, phosphatase and tensin homolog (PTEN), phosphorylated protein kinase B (AKT), and total AKT were purchased from Cell Signaling, Inc. The miR mimics and antagomirs, control miR, HiPerFect transfection reagent, other transfection‐related reagents, and the EndoFree Plasmid Maxi Kit were purchased from Qiagen. Lentivirus vector expressing miR mimic or antagomir and BLOCK‐iT Lentiviral Pol II miR RNAi expression plasmids were purchased from Invitrogen. Lenti α‐SMA short hairpin RNA (shRNA), lenti control shRNA, lenti α‐SMA open reading frame vector, lenti PTEN open reading frame vector, and lenti control vector were purchased from the Functional Genomics Facility of University of Colorado. Lipofectamine 2000 was purchased from Life Technologies, Inc. Runx2 3′ untranslated region (UTR) Lenti reporter–Luc vector, the Luciferase Reporter Assay Kit, and TransDux transduction reagent were purchased from System Biosciences. MK‐2206 was purchased from Selleck Chemicals. Medium 199 was purchased from Lonza. Osx 3′ UTR lenti reporter–Luc vector and all other chemicals and reagents were from Sigma‐Aldrich.
Isolation, Culture, and Treatment of Human AVICs
Calcified aortic valve leaflets were collected from 8 patients (7 male and 1 female, age 58.4±11.6 years) undergoing aortic valve replacement surgery due to CAVD (Table S1). Normal aortic valve leaflets were collected from explanted hearts of 8 patients (8 male, aged 50.3±16.1 years) undergoing heart transplantation due to late stage of cardiomyopathy (Table S1). The valve leaflets from transplant‐recipient hearts were thin and did not have histological abnormality. This study was approved by the University of Colorado Denver institutional review board. All patients gave informed consent for the use of their valves for this study.
AVICs were isolated and cultured using a previously described method.17, 29, 30 Briefly, valve leaflets were subjected to sequential digestions with collagenase. A high concentration of collagenase (2.5 mg/mL) for 0.5 hour was used to remove endothelial cells. The remaining tissue was then treated with a low concentration of collagenase (0.8 mg/mL) for 3 hours to free the interstitial cells. AVICs were collected by centrifugation. AVIC isolates, which are obtained using this modified protocol are lack of endothelial cells as being verified by von Willebrand factor staining.29 Cells were maintained in Medium 199 growth medium supplemented with 10% fetal bovine serum, penicillin G (100 U/mL), streptomycin (100 mg/mL), and amphotericin B (0.25 μg/mL). Cells of passages 3 to 6 were used when they reached 80% to 90% confluence.
To determine the role of miRs in regulating AVIC expression of α‐SMA, Runx2, and Osx, cells were transfected with miR mimic (5 nmol/L), miR antagomir (50 nmol/L), or control miR (50 nmol/L).
PTEN acts as a negative regulator of AKT,31 and AKT is known to play a role in myofibroblastic transition.32, 33 Prediction using mirSVR and TargetScanS confirmed that PTEN is a target of miR‐486. PTEN levels and AKT phosphorylation were determined after treatment of cells with miR‐486 mimic for 4 to 48 hours. To determine the role of AKT in miR‐486–induced α‐SMA expression, AKT inhibitor (MK2206, 2.5–5.0 μmol/L) was added to culture medium 60 minutes before the transfection with miR‐486 mimic.
Lentiviral expression of miRs was applied to determine their effect on calcium deposition. AVICs were incubated in a conditioning medium (growth medium supplemented with 10 mmol/L β‐glycerophosphate, 10 nmol/L vitamin D3, 10 nmol/L dexamethasone, and 8 mmol/L CaCl2) for 14 days to induce calcium deposition. Alizarin red S staining was applied to evaluate calcium deposition.
MiR Microarray
MiR expression profile was analyzed using Affymetrix GeneChip miRNA 3.0 array according to the manufacturer's instruction (Affymetrix). Biotin‐labeled samples were hybridized to miR arrays that were then washed and scanned using Affymetrix GeneChip Scanner 3000. The resulting data were processed with Partek Genomics Suite 6.6 (Partek Inc). Pairwise comparisons of average group values and 1‐way ANOVA were performed. Only probes that resulted in a fold change of ≥1.5 and a P value <0.05, with or without Benjamini and Hochberg multiple hypothesis correction, were considered significant.
Real‐Time Reverse Transcription–Polymerase Chain Reaction Analysis
Total RNA was isolated using Trizol reagent and a Qiagen miRNeasy Mini Kit. Reverse transcription and polymerase chain reaction were performed using the iScript cDNA Synthesis Kit (Bio‐Rad), Qiagen miScript II RT Kit, iQ SYBR Green Supermix, and Qiagen miScript SYB Green PCR Kit, according to the manufacturers’ instructions. The following primers were used to amplify specific cDNA fragments: miR‐204 (forward: 5′‐CCCCTTCCCTTTGTCATCCTATGCCT‐3′; reverse: miScript Universal Primer); miR‐486 (forward: 5′‐CCCCTCCTGTACTGAGCTGCCCCGAG‐3′; reverse: miScript Universal Primer); U6 (forward: 5′‐CTCGCTTCGGCAGCACA‐3′; reverse: miScript Universal Primer). The miR‐204 and miR‐486 levels were quantified by real‐time polymerase chain reaction using the iQ5 Multicolor Real‐Time PCR Detection System (Bio‐Rad). The miR‐204 and miR‐486 levels normalized to U6 were calculated using the 2−ΔΔT method.34
Luciferase Reporter Assay
Runx2 or Osx 3′ UTR lenti reporter–Luc vector was cotransfected in human AVICs with miR‐204 mimic, miR‐204 antagomir, or nontargeting miR. Luciferase assays were performed after 72 hours. Luciferase reporter gene assay solution (Roche) was added onto cell lysates and incubated for 15 minutes at room temperature, and luminescence was read for 2 seconds using a Synergy H1 microplate reader (BioTek). The activities of Runx2 3′ UTR and Osx 3′ UTR were determined by calculating the luciferase signal ratio of treatment with specific miR against that with nontargeting control miR.
Lentiviral Transduction for miR Expression, α‐SMA Knockdown, and Overexpression
BLOCK‐iT Lentiviral Pol II miR RNAi expression plasmids were amplified using standard bacterial transformation and purified using the EndoFree Plasmid Maxi Kit. Lentivirus expressing miR mimic or antagomir was generated by Lipofectamine 2000 cotransfection of 293 T cells. After 48 hours, lentiviral supernatants were collected and concentrated. AVICs were infected with lentivirus expressing miR mimic or antagomir and cotransfected with TransDux transduction reagent.35
Lentiviral expression of α‐SMA shRNA or α‐SMA open reading frame was applied to knock down or overexpress α‐SMA for evaluation of the role of myofibroblastic transition in the expression of osteogenic transcription factors Runx2 and Osx. Cells were infected with lenti α‐SMA shRNA, lenti control shRNA, lenti α‐SMA open reading frame, or lenti control vector. At 3 or 5 days later, the effects of knockdown and overexpression on cellular levels of α‐SMA were validated by immunoblotting.
Immunoblotting
Immunoblotting was performed, as described previously,36 to analyze α‐SMA, Runx2, and Osx, PTEN, and phosphorylated and total AKT. β‐actin levels were examined for normalization of loading. In brief, cell lysates were prepared with a sample buffer (100 mmol/L Tris‐HCl, pH 6.8, 2% SDS, 0.02% bromophenol blue, and 10% glycerol). Proteins in cell lysates were fractioned by 4% to 20% SDS‐PAGE and subsequently transferred to polyvinylidene difluoride membranes. After being blocked with 5% skim milk solution, membranes were incubated with primary antibodies followed by peroxidase‐linked secondary antibodies specific to the primary antibodies. Protein bands were visualized with the enhanced chemiluminescence system. Band density was analyzed using ImageJ software (National Institutes of Health).
Staining for Calcium Deposits
Alizarin red S staining was performed as described previously.8, 17 Briefly, cell monolayers and tissue section were washed twice with PBS and fixed for 15 minutes in 4% paraformaldehyde, followed by incubation with 0.2% Alizarin red S solution (pH 4.2) at room temperature. Cells and tissue section were washed with distilled water. Alizarin red S stains were examined and photographed with a Nikon Eclipse TS100 microscope. To quantitatively analyze Alizarin red S staining, Alizarin red S stains were bleached with 10% acetic acid at 85°C. Supernatant was spectrophotometrically analyzed at 450 nm.37
Immunofluorescence Staining
Immunofluorescence staining was performed, as described previously,8 to detect α‐SMA in human AVICs. After permeabilization with an ethanol/acetone mixture, cells on chamber slides were fixed in 4% paraformaldehyde and incubated with a rabbit polyclonal antibody against human α‐SMA overnight at 4°C. After washing with PBS, cells were incubated with Cy3‐tagged secondary antibody against the primary antibody (imaged on the red channel). Nuclei were stained with bis‐benzimide (4′,6‐diamidino‐2‐phenylindole, imaged on the blue channel) and glycoproteins on cell surfaces with Alexa 488–tagged wheat germ agglutinin (imaged on the green channel). Microscopy was performed with a Leica DM 5500 digital microscope (Leica Microscopy und System GmbH).
Human Aortic Valve Culture
Ex vivo culture of human aortic valve leaflet was performed following the method described by Peacock and colleagues.38 In brief, aortic valve leaflets from explanted hearts of 8 heart transplant recipients were used. The valvular tissue used was comparable in size. Each piece of tissue was placed on a filter membrane (0.2‐μm pore size) and immersed in Medium 199 growth medium. After overnight incubation, conditioning medium (growth medium with 10 mmol/L β‐glycerophosphate, 10 nmol/L vitamin D3, 10 nmol/L dexamethasone, and 8 mmol/L CaCl2) was applied. Lentivirus expressing miR‐486 mimic, miR‐204 antagomir, or irrelevant oligonucleotide was added to determine the effect of expression of miR‐486 and inhibition of miR‐204 function on aortic valve tissue osteogenic activity.
Statistical Analysis
The Kolmogorov–Smirnov test was used to test for normality of distribution. Results are expressed as mean±SE or as median and 95% CI when the Kolmogorov–Smirnov test indicated a nonnormal distribution. Comparisons between groups were performed using StatView software (Abacus Concepts) with 1‐way ANOVA with the post hoc Bonferroni/Dunn test and independent‐samples t test, and differences were confirmed with the Mann–Whitney U and Kruskal–Wallis H tests. A difference was considered significant at P≤0.05.
Results
The Pro‐Osteogenic Phenotype of AVICs of Calcified Valves Is Associated With Upregulated miR‐486 Expression and Downregulated miR‐204 Expression
AVICs from noncalcified tissue of calcified aortic valves had significantly higher levels of myofibroblastic biomarker α‐SMA (Figure 1), whereas the levels of fibroblastic biomarker vimentin were moderately but insignificantly elevated in these cells (Figure S1). AVICs of calcified valves also had elevated levels of osteogenic transcription factors Runx2 and Osx (Figure 1). Furthermore, such cells formed more calcium deposits when cultured in the conditioning medium (Figure 1).
Figure 1.
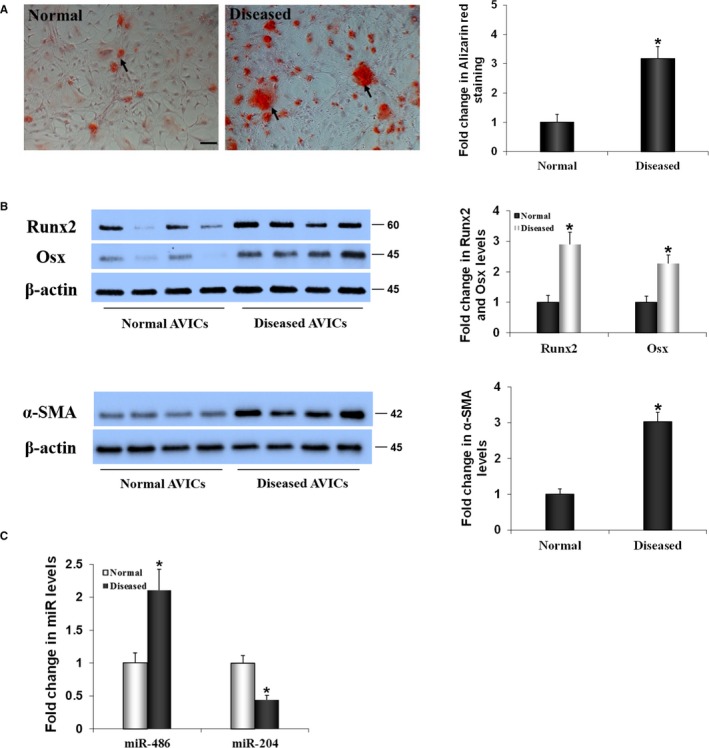
The pro‐osteogenic phenotype of aortic valve interstitial cells (AVICs) of calcified valves is associated with altered expression of microRNAs miR‐486 and miR‐204. A, Representative images and spectrophotometric data show that AVICs from calcified human aortic valves exhibit greater calcium deposition when incubated in a conditioning medium (growth medium supplemented with 10 mmol/L β‐glycerophosphate, 10 nmol/L vitamin D3, 10 nmol/L dexamethasone, and 8 mmol/L CaCl2) for 14 days. Scale bar=200 μm. B, Representative images and spectrophotometric data show that AVIC isolates from calcified valves have higher levels of runt‐related transcription factor 2 (Runx2) and osterix (Osx), and α–smooth muscle actin (α‐SMA). C, Real‐time quantitative reverse transcriptase–polymerase chain reaction data in the bar graph show that miR‐486 levels are increased and miR‐204 levels are decreased in AVICs of calcified valves. Data are expressed as mean±SE. n=8 cell isolates from different valves in each group; independent samples t test and nonparametric Mann–Whitney U test; *P<0.05 vs normal.
To understand whether the elevated pro‐osteogenic activity in AVICs of calcified valves is associated with altered miR expression, we applied microarray analysis to compare miR profiles in AVICs isolated from normal and calcified valves. Interestingly, microarray analysis identified only 26 miRs that are expressed differently in AVICs of calcified valves in comparison to AVICs from normal valves. Among them, 14 miRs were upregulated and 12 miRs were downregulated. The upregulation of miR‐486 and downregulation of miR‐204 were prominent (Tables 1 and 2). Real‐time quantitative reverse transcription–polymerase chain reaction data in Figure 1C confirmed that AVICs from noncalcified tissue of calcified valves have markedly higher levels of miR‐486 (2.1‐fold of those in AVICs of normal valves; P<0.05; independent‐samples t test and nonparametric Mann–Whitney U test) and significantly lower levels of miR‐204 (40% of those in AVICs of normal valves; P<0.05; independent‐samples t test and nonparametric Mann–Whitney U test).
Table 1.
Upregulated MicroRNAs in AVICs of Calcified Human Aortic Valves
| Probeset ID | Sequence | Fold Change (Calcified vs Normal) | P Value | |
|---|---|---|---|---|
| 1 | hsa‐miR‐486‐5p_st | UCCUGUACUGAGCUGCCCCGAG | 2.86792 | 0.011 |
| 2 | hsa‐miR‐602_st | GACACGGGCGACAGCUGCGGCCC | 2.09316 | 0.004 |
| 3 | hsa‐miR‐2277‐3p_st | UGACAGCGCCCUGCCUGGCUC | 2.06677 | 0.099 |
| 4 | hsa‐miR‐1972_st | UCAGGCCAGGCACAGUGGCUCA | 1.94952 | 0.011 |
| 5 | hp_hsa‐mir‐551b_st | AGAUGUGCUCUCCUGGCCCAUGAAAUCAAGCGUGGGUGAGACCUGGUGCAGAACGGGAAGGCGACCCAUACUUGGUUUCAGAGGCUGUGAGAAUAA | 1.88772 | 0.019 |
| 6 | hsa‐miR‐4695‐3p_st | UGAUCUCACCGCUGCCUCCUUC | 1.75907 | 0.017 |
| 7 | 14qI‐4_st | TGGACCAATGATGAGTACCATGGGGTATCTGAAACAGGATTTTTGATTAAACCCATATGCAATTCTGAGGTCCA | 1.74293 | 0.036 |
| 8 | U46_st | GTAGGGTGATGAAAAAGAATCCTTAGGCGTGGTTGTGGCCGTCTTGGTCACCTGTGTGCCACTTGCCAATGCAAGGACTTGTCATAGTTACACTGACT | 1.72971 | 0.033 |
| 9 | hsa‐miR‐4519_st | CAGCAGUGCGCAGGGCUG | 1.66571 | 0.045 |
| 10 | 14qII‐1_st | TGGACCTATGATGATGACTGGTGGCGTATGAGTCATTGACGGTGAATACAGGTCTGGAAGTCTGAGGTCCA | 1.58002 | 0.049 |
| 11 | Z17B_st | GGGTGCAAATGATGCATATGTTAGCGACCAAAGCCTGATCTTTGCTGATTAGTCATAATTAACTGACTGCACCC | 1.57415 | 0.033 |
| 12 | ENSG00000202252_st | TCGCTGTGATGAGTGATTGTTAAACATTCGTAGTTTCCACCAAAAGCTTGGCTAATGATGGCAACACCTTCCTTGGATGTCTGAGCGA | 1.53936 | 0.040 |
| 13 | hsa‐miR‐3620_st | UCACCCUGCAUCCCGCACCCAG | 1.52208 | 0.013 |
| 14 | hsa‐miR‐4710_st | GGGUGAGGGCAGGUGGUU | 1.51300 | 0.025 |
Table 2.
Downregulated MicroRNAs in AVICs of Calcified Human Aortic Valves
| Probeset ID | Sequence | Fold Change (Calcified vs Normal) | P Value | |
|---|---|---|---|---|
| 1 | hsa‐miR‐204_st | UUCCCUUUGUCAUCCUAUGCCU | −4.90955 | 0.001 |
| 2 | hsa‐miR‐411‐star_st | UAUGUAACACGGUCCACUAACC | −1.93579 | 0.025 |
| 3 | ACA47_st | ACGGTCTGGGGAAAGGCTCCTGTGTTGTTGAGCCTGCCAAGTTGTGGGTGCAGCATGGTACCAGGCAGCCCAACCCTGACCTAAAGTAAATTCCCGGAAGAAGTCTTTCCTGGGTTTTGAATTTGCA | −1.63728 | 0.002 |
| 4 | ENSG00000252917_st | CCCAGCTGTAGGCAGCTTTCTAGGTTGCCTTGTACCTAGGCAAGCATTACACTTCTGGGAGAACAGTAGCCAATAGCTGATTGGCATTCTGGCCTGGTTCATGCCACCTCTATGTTGACTACACCAG | −1.64902 | 0.008 |
| 5 | ENSG00000252969_st | CTAGAAAAATTGAAGATTTGGCCACTCTGGAAACTGCTGAGGGCCCCATGTGTAATGGCTGCAAACAGCTTCCTTTGTTGTGTAGACAGCCTGAGTTTTTCTCTAAGGGACTATGGAGACAGCC | −1.66622 | 0.035 |
| 6 | hp_hsa‐mir‐4727_st | AAUCUGCCAGCUUCCACAGUGGCAGAUUUUCCCAUAGUGGGAAGCUGGCAGAUUC | −1.53796 | 0.005 |
| 7 | hsa‐miR‐151b_st | UCGAGGAGCUCACAGUCU | −1.50781 | 0.031 |
| 8 | ENSG00000238437_st | ATCCTTTTGTGGTTCAAAAGCATGATTGGGCTTTCATGCTTATGCATGAGTTGTGACTCCCTCAAACCTTATTAGGATGTCGGCGCATTACCCATCTGACA | −1.51200 | 0.010 |
| 9 | HBII‐52‐23_st | GGGTCAATGATGAGAACCCTATATTGTGTTGAAGAGAGGTGATGACTTAAAATTACCATGCTCAATGATTACGCTGAGGCCC | −1.51715 | 0.007 |
| 10 | hp_hsa‐mir‐214_st | GGCCUGGCUGGACAGAGUUGUCAUGUGUCUGCCUGUCUACACUUGCUGUGCAGAACAUCCGCUCACCUGUACAGCAGGCACAGACAGGCAGUCACAUGACAACCCAGCCU | −1.51789 | 0.028 |
| 11 | hp_hsa‐mir‐3678_st | GAAUCCGGUCCGUACAAACUCUGCUGUGUUGAAUGAUUGGUGAGUUUGUUUGCUCAUUGAUUGAAUCACUGCAGAGUUUGUACGGACCGGAUUC | −1.52620 | 0.033 |
| 12 | ENSG00000252138_x_st | AGCACTTGTATGTTTTAACATGTGGACATTTATAGACAGGTTCAAAAAGTCCTCTTGCAACCCAGAACTCGTTGTTCAGTATGAGTCTTGATACGTGTAAGAAGGGATTTG | −1.52812 | 0.009 |
Coexpression of miR‐486 Antagomir and miR‐204 Mimic Suppresses the Pro‐Osteogenic Activity of AVICs of Calcified Valves
We infected AVICs from calcified valves with lentivirus that expresses miR‐486 antagomir, miR‐204 mimic, or both and examined calcium deposit formation. The results in Figure 2 show that lentiviral expression of miR‐486 antagomir alone or miR‐204 mimic alone reduced calcium deposit formation in AVICs of calcified valves by 31.6% and 28.9%, respectively (P<0.05; ANOVA and nonparametric Kruskal–Wallis H test), whereas expression of irrelevant miR had no effect. Moreover, a combination of miR‐486 antagomir and miR‐204 mimic reduced the levels of calcium deposit formation close to baseline levels in AVICs of normal valves. Thus, both increased levels of miR‐486 and decreased levels of miR‐204 are involved in the mechanism underlying the elevated pro‐osteogenic activity in AVICs of calcified valves.
Figure 2.
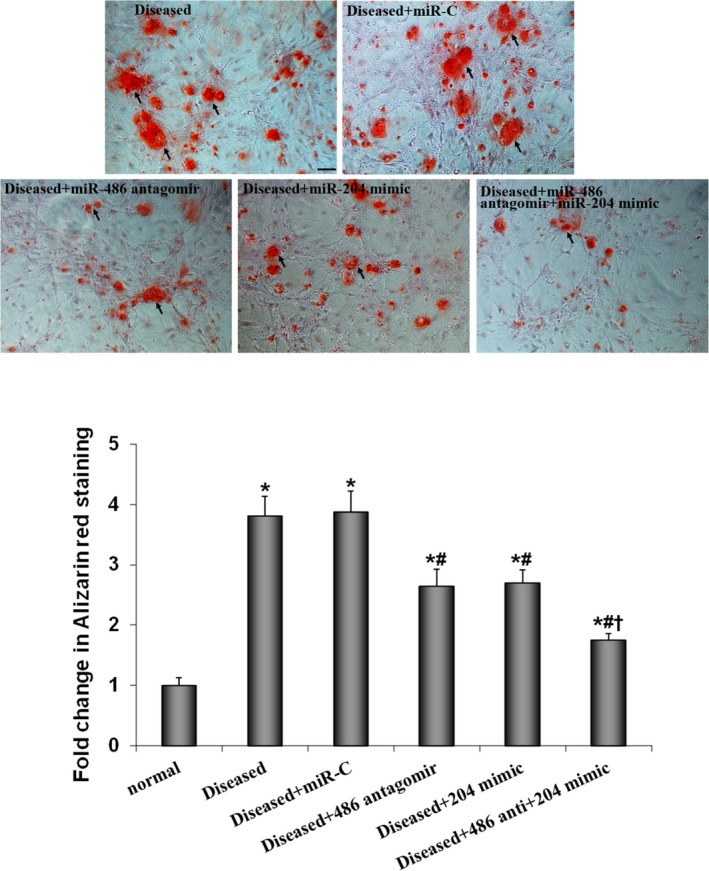
Both miR‐486 and miR‐204 are involved in the mechanism underlying calcium‐deposit formation in aortic valve interstitial cells (AVICs) of calcified valves. AVICs of calcified valves were untreated or infected with lentivirus that expresses irrelevant oligonucleotide (miR‐C), miR‐486 antagomir, miR‐204 mimic, or a combination of miR‐486 antagomir and miR‐204 mimic. Next, cells were incubated for 14 days in the conditioning medium (growth medium supplemented with 10 mmol/L β‐glycerophosphate, 10 nmol/L vitamin D3, 10 nmol/L dexamethasone, and 8 mmol/L CaCl2). Compared with cells treated with lentiviral miR‐C, cells treated with lentiviral miR‐486 antagomir or lentiviral miR‐204 mimic exhibited decreased calcium deposit formation. Coexpression of miR‐486 antagomir and miR‐204 mimic has a greater effect in suppressing calcium‐deposit formation. Data are expressed as mean±SE. n=7 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs normal AVICs incubated in the conditioning medium for 14 days; # P<0.05 vs AVICs of calcified valves treated with lentiviral miR‐C; † P<0.05 vs AVICs of calcified valves plus miR‐486 antagomir or AVICs of calcified valves plus miR‐204 mimic; scale bar=200 μm.
MiR‐486 Upregulates α‐SMA Expression in AVICs Through the AKT Signaling Pathway
The results in Figure 3A show that α‐SMA levels in AVICs of calcified valves were reduced by transfection of miR‐486 antagomir but were not affected by transfection of miR‐204 mimic. In addition, miR‐486 mimic upregulated α‐SMA expression in AVICs of normal valves in a time‐dependent manner (Figure 3B). The data of immunofluorescence analysis confirmed the effect of miR‐486 mimic on α‐SMA levels in AVICs of normal valves (Figure 3B). It appears that miR‐486 upregulates α‐SMA expression in human AVICs, and the increased levels of miR‐486 contribute to the mechanism underlying enhanced α‐SMA expression in AVICs of calcified valves.
Figure 3.
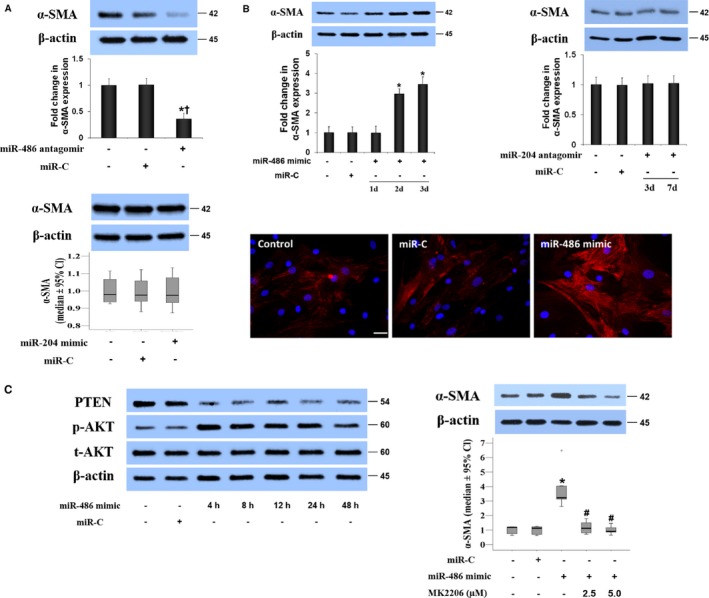
MiR‐486 upregulates α–smooth muscle actin (α‐SMA) expression in aortic valve interstitial cells (AVICs) through the AKT signaling pathway. A, Representative immunoblots and quantitative analysis show that transfection of AVICs from calcified valves with miR‐486 antagomir reduces α‐SMA levels at day 3 after transfection, whereas transfection with miR‐204 mimic or control microRNA (miR‐C) has no effect. Data are expressed as mean±SE or median and 95% CI (α‐SMA levels in diseased cells treated with miR‐204 mimic or control miR). n=6 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs untreated AVICs of calcified valves; † P<0.05 vs AVICs of calcified valves plus miR‐C. B, AVICs of normal valves were transfected with miR‐486 mimic or control microRNA (miR‐C). Representative immunoblots of 5 separated experiments and quantitative analysis show that miR‐486 mimic increases α‐SMA levels in normal AVICs, whereas miR‐204 antagomir has no effect. Representative immunofluorescence images show the effect of miR‐486 mimic on α‐SMA expression. Scale bar=100 μm. C, Representative immunoblots show that miR‐486 mimic reduces cellular PTEN levels at 4 to 48 hours and induces AKT phosphorylation in normal AVICs. Inhibition of AKT with MK2206 abrogates the effects of miR‐486 mimic on α‐SMA expression. Data are expressed as mean±SE or median and 95% CI (α‐SMA levels). n=7 in separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs control normal AVICs or normal AVICs plus miR‐C; # P<0.05 vs normal AVICs plus miR‐486 mimic.
We observed that the expression of miR‐486 mimic caused a reduction in cellular PTEN levels that is accompanied by AKT phosphorylation (Figure 3C). The effect of miR‐486 mimic on α‐SMA levels was essentially lost when the AKT signaling pathway was inhibited with a specific inhibitor (Figure 3C). Furthermore, overexpression of PTEN suppressed AKT phosphorylation and reduced α‐SMA levels in normal AVICs treated with miR‐486 mimic (Figure S2). The results suggest that the PTEN–AKT signaling pathway mediates miR‐486–induced α‐SMA expression in human AVICs.
MiR‐204 Regulates Runx2 and Osx Expression in AVICs Through Posttranscriptional Modulation
We transfected AVICs with miR‐204 antagomir or miR‐204 mimic to determine the role of miR‐204 in the upregulation of Runx2 and Osx. The miR‐204 mimic reduced the levels of Runx2 and Osx in AVICs of calcified valves (Figure 4A). Conversely, miR‐204 antagomir caused a marked increase in the levels of Runx2 and Osx in AVICs of normal valves (Figure 4B). Therefore, miR‐204 negatively regulates the expression of both Runx2 and Osx in human AVICs, and overexpression of these 2 osteogenic transcription factors in AVICs of calcified valves is due to a deficiency of miR‐204.
Figure 4.
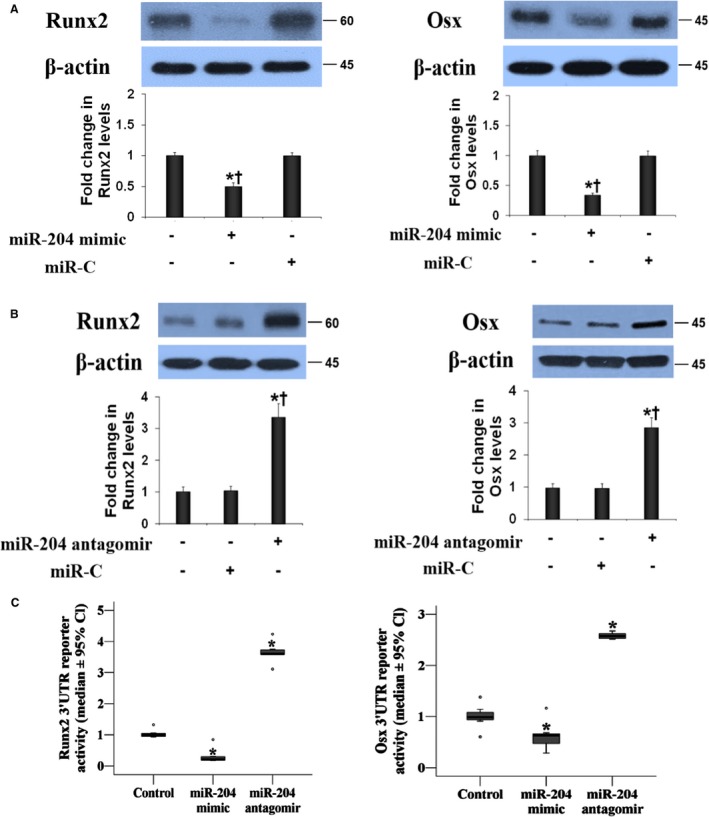
MiR‐204 modulates the expression of both runt‐related transcription factor 2 (Runx2) and osterix (Osx) in human aortic valve interstitial cells (AVICs). A, Representative immunoblots and quantitative analysis show that transfection of AVICs of calcified valves with miR‐204 mimic markedly reduces the levels of Runx2 and Osx examined at 72 hours after transfection. B, MiR‐204 antagomir upregulates the levels of Runx2 and Osx at 72 hours after transfection in AVICs of normal valves. Data are expressed as mean±SE. n=8 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs untreated control; † P<0.05 vs miR‐C control. C, Normal AVICs were infected with lentiviral luciferase reporter of Runx2 3′ untranslated region (UTR) or Osx 3′ UTR together with irrelevant oligonucleotide (miR‐C), miR‐204 mimic, or miR‐204 antagomir. Luciferase assays were performed 72 hours after infection. Quantitative analysis of 3′ UTR luciferase activities. Activities of Runx2 and Osx 3′ UTR reporters are decreased by miR‐204 mimic and increased by miR‐204 antagomir. Data are expressed as median and 95% CI (Runx2 and Osx reporter activity data). n=8 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs control (miR‐C).
We introduced luciferase reporters with insertion of human Runx2 3′ UTR or human Osx 3′ UTR to AVICs to examine the impact of miR‐204 on reporter activity. The reporter activity data are presented in Figure 4C. The miR‐204 mimic repressed and miR‐204 antagomir increased the luciferase reporter activities of Runx2 and Osx 3′ UTR. These results indicate a role for miR‐204 in posttranscriptionally repressing the expression of Runx2 and Osx proteins.
MiR‐486 Mimic and miR‐204 Antagomir Have a Synergistic Effect on the Upregulation of Valvular Pro‐Osteogenic Activity
We observed that miR‐486 mimic also upregulated the expression of Runx2 and Osx in AVICs of normal valves, although the effect of miR‐486 mimic was moderate in comparison to that of miR‐204 antagomir (Figure 5A). Interestingly, cells produced greater levels of Runx2 and Osx when they were cotransfected with miR‐486 mimic and miR‐204 antagomir (Figure 5A). Furthermore, AVICs of normal valves formed significantly more calcium deposits when they were subjected to a conditioning medium following lentiviral coexpression of miR‐486 mimic and miR‐204 antagomir (Figure 5B). More importantly, lentiviral coexpression of miR‐486 mimic and miR‐204 antagomir resulted in greater calcium deposition in human aortic valve tissue (Figure 5C). Thus, miR‐486 elevation and miR‐204 deficiency synergize in promotion of Runx2 and Osx expression as well as pro‐osteogenic activity in human aortic valves.
Figure 5.
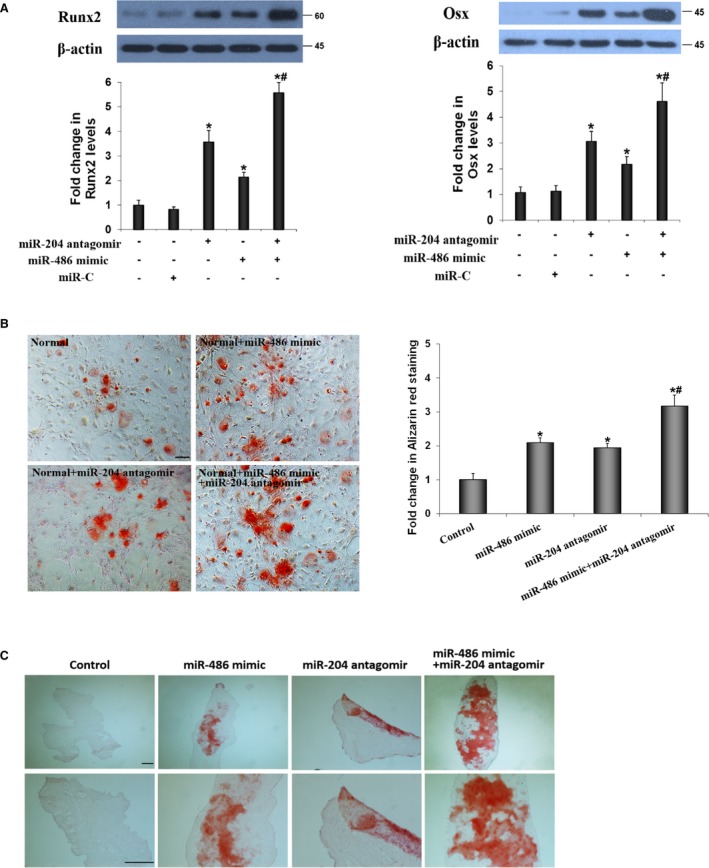
MiR‐486 mimic and miR‐204 antagomir have a synergistic effect in the upregulation of runt‐related transcription factor 2 (Runx2) and osterix (Osx). A, Lentiviral expression of miR‐486 mimic and miR‐204 antagomir synergistically upregulates Runx2 and Osx levels in normal aortic valve interstitial cells (AVICs). B, Lentiviral expression of both miR‐486 mimic and miR‐204 antagomir caused greater calcium deposition in normal AVICs. Scale bar=200 μm. Data are expressed as mean±SE. n=6 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs untreated control; # P<0.05 vs miR‐486 mimic or miR‐204 antagomir. C, Representative images show that infection of human aortic valve tissue with lenti–miR‐486 mimic and lenti–miR‐204 antagomir causes greater calcium deposition after the tissue is cultured in conditioning medium for 14 days. Scale bar=100 μm.
Neither Runx2 nor Osx could be a direct target of miR‐486. Because miR‐486 upregulates α‐SMA expression (a biomarker of myofibroblastic transition), it is likely that miR‐486 overexpression induces myofibroblastic transition in AVICs to enhance pro‐osteogenic activity. To verify a link between myofibroblastic transition and enhanced pro‐osteogenic activity, we applied lentiviral expression of α‐SMA shRNA to block the myofibroblastic transition induced by miR‐486 mimic in normal AVICs. The immunoblotting results in Figure 6A show that lentiviral α‐SMA shRNA reduced α‐SMA levels in AVICs of normal valves at day 3 after infection in the absence of stimulation. The immunofluorescence images show that lentiviral α‐SMA shRNA abolished the effect of miR‐486 mimic on α‐SMA expression (Figure 6A). Inhibition of α‐SMA expression essentially abrogated the effect of miR‐486 mimic on Runx2 and Osx levels (Figure 6B). Conversely, overexpression of α‐SMA in normal AVICs enhanced the expression of Runx2 and Osx (Figure 6C). Consequently, upregulation of α‐SMA expression appears to be the mechanism underlying the effect of miR‐486 on pro‐osteogenic activity in human AVICs.
Figure 6.
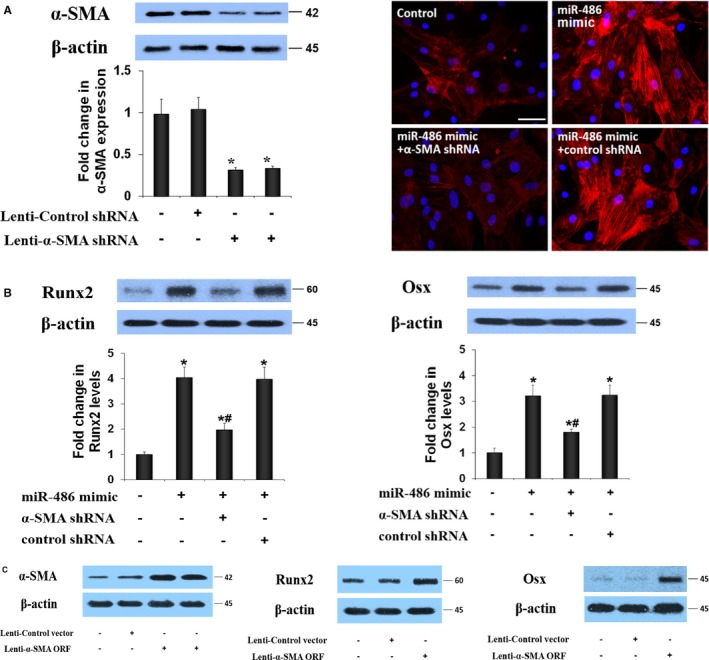
MiR‐486 induces myofibroblastic transition to upregulate the pro‐osteogenic activity in human aortic valve interstitial cells (AVICs). A, Representative immunoblots and quantitative analysis show that treatment with short‐hairpin RNA (shRNA) reduces cellular α–smooth muscle actin (α‐SMA) levels in AVICs of calcified valves. Representative immunofluorescence images show that knockdown of α‐SMA abolishes miR‐486–induced overexpression of α‐SMA in normal AVICs. Data are expressed as mean±SE. n=6 separate experiments using distinct cell isolates; ANOVA and nonparametric Kruskal–Wallis H test, *P<0.05 vs untreated control or control shRNA; scale bar=200 μm. B, Representative immunoblots and quantitative analysis show that knockdown of α‐SMA markedly reduces the effects of miR‐486 mimic on the expression of runt‐related transcription factor 2 (Runx2) and osterix (Osx). Data are expressed as mean±SE. n=6 separate experiments using distinct isolates; ANOVA and nonparametric Kruskal–Wallis H test; *P<0.05 vs untreated control; # P<0.05 vs miR‐486 mimic or miR‐486 mimic plus control shRNA. C, Representative immunoblots of 6 separate experiments show that treatment with an α‐SMA–ORF vector increases cellular α‐SMA levels in normal AVICs. Overexpression of α‐SMA elevates Runx2 and Osx levels in normal AVICs. ORF indicates open reading frame.
Discussion
CAVD is a leading cardiovascular disease in older adults. Progressive calcification of aortic valve leaflets eventually results in valvular dysfunction. Although aortic valve replacement surgery is an available therapy, the replaced valve can fail, and valve replacement surgery is associated with several complications.39 Pharmacological intervention for limitation of this disease progression is desperately needed and relies on identification of therapeutic targets. AVICs of calcified valves undergo profibrogenic and pro‐osteogenic transitions and thus play an important role in CAVD progression17, 40; however, the mechanism underlying AVIC phenotypic transition remains poorly understood. Using a systematic approach, we found that the expression of miR‐486 and miR‐204 is markedly altered in AVIC isolates from noncalcified tissue of calcified human aortic valves, and altered expression of these 2 miRs is responsible for the AVIC pro‐osteogenic phenotype. Additional novel findings of the present study include that miR‐486 upregulates α‐SMA expression through the PTEN–AKT pathway, and miR‐204 negatively regulates the expression of both Runx2 and Osx through posttranscriptional modulation. Moreover, miR‐486 elevates AVIC pro‐osteogenic activity (expression of Runx2 and Osx and formation of calcium deposits) by inducing myofibroblastic transition (overexpression of α‐SMA). These findings provide insights into the mechanism underlying CAVD pathogenesis and may help identify therapeutic targets for suppression of CAVD progression.
Both miR‐486 and miR‐204 Are Involved in the Mechanism Underlying the Pro‐Osteogenic Activity in AVICs of Calcified Valves
CAVD is now recognized as an active pathobiological process involving AVIC reprogramming. In normal aortic valves, AVICs are fibroblast‐like cells; however, AVICs in calcified valves display an osteoblast‐like phenotype.15, 41 Activated AVICs express high levels of α‐SMA, a biomarker of myofibroblast. It is believed that these cells can undergo osteoblast‐like differentiation, expressing chondrogenic and osteogenic biomarkers and forming calcific nodules.15, 41 Such phenotypic change is also observed in AVICs isolated from the areas surrounding calcification nodules in calcified aortic valves.8 AVICs form normal aortic valves do not have spontaneous pro‐osteogenic activity, whereas cells from calcified aortic valves display elevated pro‐osteogenic activity. In the present study, we confirmed that AVICs isolated from noncalcified areas of calcified aortic valves have higher levels of α‐SMA, Runx2 and Osx, and form greater amounts of calcium deposits in vitro in the presence of a conditioning medium that contains higher levels of calcium and phosphate. Increased levels of miR‐486 and decreased levels of miR‐204 are prominent in the list of altered miRs in AVICs of calcified valves.
miR‐204 has been found to suppress osteoblastic differentiation in C2C12 mesenchymal progenitor cells and stem cells by negative regulation of Runx2.25, 42 In addition, miR‐486 can upregulate AKT activity through negative regulation of PTEN expression.43 AKT signaling is important in endothelial‐to‐mesenchymal transition and myofibroblastic transformation in fibroblasts from the heart and lungs.32, 33 Consequently, it is likely that both increased expression of miR‐486 and decreased expression of miR‐204 in AVICs of calcified valves contribute to the mechanism underlying the pro‐osteogenic phenotype in human AVICs. The results of experiments using the lentiviral expression approach support this notion because the expression of miR‐486 antagomir alone or miR‐204 mimic alone results in a reduction of calcium deposition in AVICs of calcified valves. Moreover, coexpression of miR‐486 antagomir and miR‐204 mimic essentially abolishes calcium deposition in AVICs of calcified valves. The results of experiments using the miR‐486 loss‐of‐function and miR‐204 gain‐of‐function approaches demonstrate that both miR‐486 and miR‐204 are involved in upregulating the pro‐osteogenic activity of AVICs of calcified valves and that miR‐486 is a positive regulator and miR‐204 is a negative regulator of AVIC pro‐osteogenic activity. Increased levels of miR‐486 and decreased levels of miR‐204 may exert an effect on aortic valve pro‐osteogenic activity.
Our microarray data also show altered expression of miR‐602 (≈2.1‐fold increase) and miR‐411 (≈1.9‐fold decrease) in AVICs of calcified valves. These 2 miRs have been found to modulate the expression of mediators involved in the degradation of extracellular matrix proteins in cartilage and to play a role in protection against osteoarthritis through downregulation of inflammatory responses.44, 45 Because proinflammatory mechanisms also play a role in CAVD progression, miR‐602 and miR‐411 may contribute to the aortic valve pathobiology associated with CAVD. Their potential roles in aortic valve inflammation and calcification need to be determined in future studies.
It remains unknown from the present study what is responsible for the altered expression of miR‐486 and miR‐204 in AVICs of calcified valves. The levels of TGF‐β1 and BMP‐2 are significantly increased in calcified aortic valves, and these 2 growth factors are potent in elevation of AVIC pro‐osteogenic activity.22 It is possible that TGF‐β1 and BMP‐2 are involved in the mechanism underlying the altered expression of miRs in AVICs of calcified valves. In addition, multiple pro‐inflammatory mediators have been identified in calcified human aortic valves.46 Our previous studies demonstrate that exogenous and endogenous ligands of Toll‐like receptors are capable of elevating the pro‐osteogenic activity in human AVICs.8, 17, 29, 47, 48, 49 It is noteworthy that activation of certain Toll‐like receptors in human AVICs by pro‐inflammatory stimuli leads to overproduction of TGF‐β1 and/or BMP‐2.8, 17, 29, 47, 48, 49 Consequently, it is likely that TGF‐β1 and BMP‐2 are also involved in alteration of miR expression by Toll‐like receptor ligands. The potential central role of TGF‐β1 and BMP‐2 in modulation of AVIC miR expression awaits future investigation.
MiR‐204 Negatively Regulates the Expression of Runx2 and Osx in Human AVICs via Posttranscriptional Modulation
miR‐204 is known to negatively regulate Runx2 expression in stem cells and to suppress osteoblastic differentiation.25 In addition, miR‐204 has been found to negatively modulate Runx2 expression to antagonize osteoblastic differentiation in AVICs stimulated by BMP‐2.50 The results of the present study show that markedly downregulated miR‐204 levels in AVICs of calcified valves correlate with elevated levels of Runx2 and Osx. Analysis of miR‐204 targets using mirSVR and TargetScanS indicates both Runx2 and Osx are the targets of miR‐204. The role of miR‐204 in regulating Osx expression, however, has not been determined. The results obtained from experiments using AVICs of calcified valves show that miR‐204 mimic not only reduces Runx2 levels but also markedly decreases Osx levels. This observation suggests that miR‐204 negatively regulates both Runx2 and Osx in human AVICs. Additional results obtained from experiments using AVICs of normal valves show that miR‐204 antagomir increases cellular levels of Runx2 and Osx. Together, our findings highlight the role of miR‐204 as a negative regulator of both Runx2 and Osx in human AVICs.
To understand the mechanism by which miR‐204 modulates Runx2 and Osx expression, we utilized luciferase reporters with insertion of human Runx2 or Osx 3′ UTR and examined the effect of miR‐204 mimic and antagomir on reporter activity. The miR‐204 mimic repressed and miR‐204 antagomir increased the activities of both Runx2 and Osx reporters. These results revealed a role for miR‐204 in posttranscriptionally repressing the expression of both Runx2 and Osx. Runx2 and Osx are master osteogenic transcription factors, and neither Runx2‐null mice nor Osx‐null mice form mature osteoblasts.51, 52 The results of the present study suggest that downregulation of miR‐204 in human AVICs causes pro‐osteogenic reprogramming by enhancement of expression of both Runx2 and Osx.
MiR‐486 Induces Myofibroblastic Transition in Human AVICs to Promote Pro‐Osteogenic Activity
Fibroblast is the most abundant cell type within normal heart valves, whereas myofibroblasts constitute a small fraction. Our previous studies show that myofibroblasts are abundant in calcified aortic valves.8 In the present study, we found miR‐486 is increased in AVICs of calcified valves. In determining the role of miR‐486 in AVIC myofibroblastic transition, we found that miR‐486 mimic upregulates the expression of α‐SMA, a biomarker of myofibroblasts, although it does not induce myofibroblastic transition uniformly in AVIC culture, likely due to cell heterogeneity. Conversely, inhibition of miR‐486 suppresses α‐SMA expression in AVICs of calcified valves. Consequently, miR‐486 plays a role in myofibroblastic transition in human AVICs.
AKT signaling is important in endothelial‐to‐mesenchymal transition and myofibroblastic transformation in fibroblasts from the heart and lungs.32, 33 miR‐486 may upregulate AKT activity through negative regulation of PTEN expression.43 The phosphoinositide phosphatase PTEN converts PIP3 to PIP2 and thereby negatively modulates AKT activity,31 thus downregulation of PTEN can upregulate AKT phosphorylation.43, 53 We evaluated the role of the AKT pathway in mediating α‐SMA expression induced by miR‐486 mimic. miR‐486 decreases PTEN expression and induces AKT phosphorylation in human AVICs. Importantly, inhibition of AKT with a specific inhibitor abolishes α‐SMA expression induced by miR‐486 mimic. The results suggest that the PTEN–AKT pathway is involved in the upregulation of α‐SMA expression and myofibroblastic transition mediated by miR‐486 in human AVICs. These findings are consistent with previous reports that the AKT signaling pathway modulates α‐SMA expression in lung fibroblasts and porcine AVICs.33, 54
It should be noted that overexpression of miR‐204 downregulates AKT signaling in cancer cells and leads to inhibition of cellular migration and invasion.55 Nevertheless, in our preliminary experiments, we found no change in AKT phosphorylation in human AVICs treated with miR‐204 mimic or antagomir; therefore, miR‐204 is unlikely to be involved in the modulation of AKT signaling in human AVICs.
Interestingly, we observed that normal AVICs that coexpress miR‐486 mimic and miR‐204 antagomir produce markedly higher levels of Runx2 and Osx and exhibit greater calcium deposition than cells that express miR‐486 mimic alone or miR‐204 antagomir alone. Our ex vivo data demonstrate that coexpression of miR‐486 mimic and miR‐204 antagomir also results in greater calcium deposition in normal human aortic valve tissue. It seems that increased levels of miR‐486 and decreased levels of miR‐204 have a synergistic effect in upregulating valvular pro‐osteogenic activity.
When evaluating the effect of miR‐486 alone on the pro‐osteogenic activity, we noted that miR‐486 mimic can cause moderate increases in Runx2 and Osx levels as well as calcium deposition. A question is how miR‐486 upregulates AVIC pro‐osteogenic activity and synergizes with miR‐204 deficiency in upregulation of the pro‐osteogenic activity in AVICs and valvular tissue. In further experiments, we determined the potential role of myofibroblastic transition, reflected by overexpression of α‐SMA. Interestingly, the effect of miR‐486 mimic on pro‐osteogenic activity is abolished by inhibition of the expression of α‐SMA with shRNA. Furthermore, overexpression of α‐SMA promotes the expression of Runx2 and Osx in normal AVICs in the absence of miR‐486 mimic expression. Our results suggest that miR‐486 upregulates pro‐osteogenic activity in human AVICs through inducing myofibroblastic transition. In this regard, several studies indicate that myofibroblasts in the aortic valve may have elevated pro‐osteogenic potential.18, 56 Our findings provide evidence that human AVICs that overexpress α‐SMA have elevated pro‐osteogenic activity.
It appears that a synergistic effect of miR‐486 overexpression and miR‐204 deficiency could exaggerate pro‐osteogenic activity in AVICs and valvular tissue; however, it is unclear from this study whether myofibroblasts and “osteoblast‐like cells” in the aortic valve are the same and how α‐SMA overexpression leads to the upregulation of Runx2 and Osx. Nevertheless, our loss‐of‐function and gain‐of‐function experiments provide evidence that miR‐486 and miR‐204 synergistically upregulate Runx2 and Osx expression. Further study will address the impact of α‐SMA overexpression or myofibroblastic transition on the levels of these 2 miRs and on the activities of Runx2 and Osx genes.
Conclusion
Increased levels of miR‐486 and decreased levels of miR‐204 are characteristics of AVICs from calcified human aortic valves and are responsible for the enhanced pro‐osteogenic activity observed in such cells. Normal AVICs that overexpress miR‐486 exhibit a myofibroblastic phenotype, whereas normal AVICs that overexpress miR‐204 antagomir become pro‐osteogenic. miR‐486 promotes the pro‐osteogenic activity through induction of myofibroblastic transition. A combination of increased miR‐486 and decreased miR‐204 synergistically upregulates the pro‐osteogenic activity in human AVICs and aortic valve tissue (Figure 7). These novel findings suggest that altered expression of miR‐486 and miR‐204 plays a significant role in promoting valvular pro‐osteogenic activity and that modulation of the expression of these 2 miRs may have therapeutic potential for suppression of CAVD progression.
Figure 7.
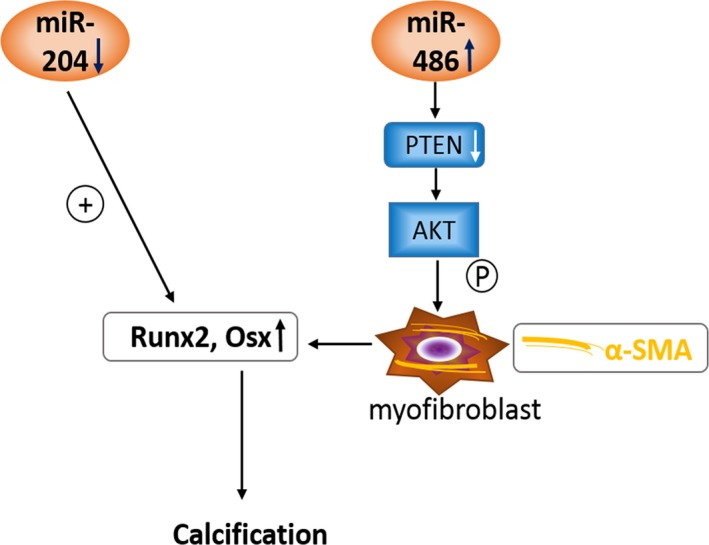
Schematic diagram depicting the mechanism underlying aortic valve calcification. miR‐204 functions as a negative regulator of runt‐related transcription factor 2 (Runx2) and osterix (Osx). miR‐486 positively regulates α–smooth muscle actin (α‐SMA) expression through the PTEN–AKT pathway and promotes aortic valve interstitial cell pro‐osteogenic activity by inducing myofibroblastic transition. Upregulated expression of miR‐486 and downregulated expression of miR‐204 synergistically enhances valvular pro‐osteogenic activity.
Sources of Funding
Research reported in this manuscript was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award no. R01HL106582. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
Supporting information
Table S1. Patient Demographics
Figure S1. Vimentin levels in aortic valve interstitial cells of normal and calcified aortic valves.
Figure S2. PTEN modulates AKT activation and α–smooth muscle actin expression in human aortic valve interstitial cells induced by miR‐486 mimic.
(J Am Heart Assoc. 2017;6:e005364 DOI: 10.1161/JAHA.116.005364.)28438736
A part of this work was presented at the American Heart Association Scientific Sessions, November 7–11, 2015, in Orlando, FL.
References
- 1. Mohler ER III. Mechanisms of aortic valve calcification. Am J Cardiol. 2004;94:1396–1402, A1396. [DOI] [PubMed] [Google Scholar]
- 2. Messika‐Zeitoun D, Bielak LF, Peyser PA, Sheedy PF, Turner ST, Nkomo VT, Breen JF, Maalouf J, Scott C, Tajik AJ, Enriquez‐Sarano M. Aortic valve calcification: determinants and progression in the population. Arterioscler Thromb Vasc Biol. 2007;27:642–648. [DOI] [PubMed] [Google Scholar]
- 3. Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111:3316–3326. [DOI] [PubMed] [Google Scholar]
- 4. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez‐Sarano M. Burden of valvular heart diseases: a population‐based study. Lancet. 2006;368:1005–1011. [DOI] [PubMed] [Google Scholar]
- 5. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hafizi S, Taylor PM, Chester AH, Allen SP, Yacoub MH. Mitogenic and secretory responses of human valve interstitial cells to vasoactive agents. J Heart Valve Dis. 2000;9:454–458. [PubMed] [Google Scholar]
- 7. Chester AH, Taylor PM. Molecular and functional characteristics of heart‐valve interstitial cells. Philos Trans R Soc Lond B Biol Sci. 2007;362:1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song R, Zeng Q, Ao L, Yu JA, Cleveland JC, Zhao KS, Fullerton DA, Meng X. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll‐like receptor‐2. Arterioscler Thromb Vasc Biol. 2012;32:2711–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, Frangogiannis NG. Fibroblasts in post‐infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schroer AK, Merryman WD. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J Cell Sci. 2015;128:1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Leinwand LA, Anseth KS. Roles of transforming growth factor‐beta1 and OB‐cadherin in porcine cardiac valve myofibroblast differentiation. FASEB J. 2014;28:4551–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yanagawa B, Lovren F, Pan Y, Garg V, Quan A, Tang G, Singh KK, Shukla PC, Kalra NP, Peterson MD, Verma S. miRNA‐141 is a novel regulator of BMP‐2‐mediated calcification in aortic stenosis. J Thorac Cardiovasc Surg. 2012;144:256–262. [DOI] [PubMed] [Google Scholar]
- 13. Osman L, Chester AH, Amrani M, Yacoub MH, Smolenski RT. A novel role of extracellular nucleotides in valve calcification: a potential target for atorvastatin. Circulation. 2006;114:I566–I572. [DOI] [PubMed] [Google Scholar]
- 14. Yip CY, Blaser MC, Mirzaei Z, Zhong X, Simmons CA. Inhibition of pathological differentiation of valvular interstitial cells by C‐type natriuretic peptide. Arterioscler Thromb Vasc Biol. 2011;31:1881–1889. [DOI] [PubMed] [Google Scholar]
- 15. New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114:I547–I552. [DOI] [PubMed] [Google Scholar]
- 17. Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Meng X. Pro‐osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll‐like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. [DOI] [PubMed] [Google Scholar]
- 18. Chen JH, Simmons CA. Cell‐matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. [DOI] [PubMed] [Google Scholar]
- 19. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 20. He X, Jing Z, Cheng G. MicroRNAs: new regulators of Toll‐like receptor signalling pathways. Biomed Res Int. 2014;2014:945169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR‐9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song R, Fullerton DA, Ao L, Zheng D, Zhao KS, Meng X. BMP‐2 and TGF‐beta1 mediate biglycan‐induced pro‐osteogenic reprogramming in aortic valve interstitial cells. J Mol Med (Berl). 2015;93:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–1784. [DOI] [PubMed] [Google Scholar]
- 24. Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang J, Zhao L, Xing L, Chen D. MicroRNA‐204 regulates Runx2 protein expression and mesenchymal progenitor cell differentiation. Stem Cells. 2010;28:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ, Stein GS. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci USA. 2011;108:9863–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang M, Liu X, Zhang X, Song Z, Han L, He Y, Xu Z. MicroRNA‐30b is a multifunctional regulator of aortic valve interstitial cells. J Thorac Cardiovasc Surg. 2014;147:1073–1080.e1072. [DOI] [PubMed] [Google Scholar]
- 28. Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, Mohamed SA. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–465. [PMC free article] [PubMed] [Google Scholar]
- 29. Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC Jr, Fullerton DA. Expression of functional Toll‐like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–C35. [DOI] [PubMed] [Google Scholar]
- 30. Zeng Q, Jin C, Ao L, Cleveland JC Jr, Song R, Xu D, Fullerton DA, Meng X. Cross‐talk between the Toll‐like receptor 4 and Notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation. 2012;126:S222–S230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naik UP. PTEN: not just a tumor suppressor. Blood. 2010;116:2404–2405. [DOI] [PubMed] [Google Scholar]
- 32. Meadows KN, Iyer S, Stevens MV, Wang D, Shechter S, Perruzzi C, Camenisch TD, Benjamin LE. Akt promotes endocardial‐mesenchyme transition. J Angiogenes Res. 2009;1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nyp MF, Navarro A, Rezaiekhaligh MH, Perez RE, Mabry SM, Ekekezie II. TRIP‐1 via AKT modulation drives lung fibroblast/myofibroblast trans‐differentiation. Respir Res. 2014;15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 35. Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. [DOI] [PubMed] [Google Scholar]
- 36. Ao L, Zou N, Cleveland JCJ, Fullerton DA, Meng X. Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP‐1 expression induced by extracellular HSC70. Am J Physiol Heart Circ Physiol. 2009;297:H21–H28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cowan CM, Zhang X, James AW, Mari Kim T, Sun N, Wu B, Ting K, Soo C. NELL‐1 increases pre‐osteoblast mineralization using both phosphate transporter Pit1 and Pit2. Biochem Biophys Res Commun. 2012;422:351–357. [DOI] [PubMed] [Google Scholar]
- 38. Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huygens SA, Mokhles MM, Hanif M, Bekkers JA, Bogers AJ, Rutten‐van Molken MP, Takkenberg JJ. Contemporary outcomes after surgical aortic valve replacement with bioprostheses and allografts: a systematic review and meta‐analysis. Eur J Cardiothorac Surg. 2016;50:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Can J Cardiol. 1996;12:231–236. [PubMed] [Google Scholar]
- 41. Rajamannan NM, Evans FJ, Aikawa E, Grande‐Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease‐2011 update. Circulation. 2011;124:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Z, Hassan MQ, Volinia S, van Wijnen AJ, Stein JL, Croce CM, Lian JB, Stein GS. A microRNA signature for a BMP2‐induced osteoblast lineage commitment program. Proc Natl Acad Sci USA. 2008;105:13906–13911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Small EM, O'Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, Richardson JA, Olson EN. Regulation of PI3‐kinase/Akt signaling by muscle‐enriched microRNA‐486. Proc Natl Acad Sci USA. 2012;107:4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akhtar N, Makki MS, Haqqi TM. MicroRNA‐602 and microRNA‐608 regulate sonic hedgehog expression via target sites in the coding region in human chondrocytes. Arthritis Rheumatol. 2015;67:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang G, Zhang Y, Zhao X, Meng C, Ma L, Kong Y. MicroRNA‐411 inhibited matrix metalloproteinase 13 expression in human chondrocytes. Am J Transl Res. 2015;7:2000–2006. [PMC free article] [PubMed] [Google Scholar]
- 46. Rajamannan NM, Bonow RO, Rahimtoola SH. Calcific aortic stenosis: an update. Nat Clin Pract Cardiovasc Med. 2007;4:254–262. [DOI] [PubMed] [Google Scholar]
- 47. Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu D, Fullerton DA, Meng X. Notch1 promotes the pro‐osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor‐kappaB activation. Arterioscler Thromb Vasc Biol. 2013;33:1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhan Q, Song R, Zeng Q, Yao Q, Ao L, Xu D, Fullerton DA, Meng X. Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF‐kappaB and ERK1/2 pathways. Int J Biol Sci. 2015;11:482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zeng Q, Song R, Fullerton DA, Ao L, Zhai Y, Li S, Ballak DB, Cleveland JC Jr, Reece TB, McKinsey TA, Xu D, Dinarello CA, Meng X. Interleukin‐37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci USA. 2017;114:1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y, Chen S, Deng C, Li F, Wang Y, Hu X, Shi F, Dong N. MicroRNA‐204 targets Runx2 to attenuate BMP‐2‐induced osteoblast differentiation of human aortic valve interstitial cells. J Cardiovasc Pharmacol. 2015;66:63–71. [DOI] [PubMed] [Google Scholar]
- 51. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. [DOI] [PubMed] [Google Scholar]
- 52. Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger‐containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. [DOI] [PubMed] [Google Scholar]
- 53. Korkaya H, Paulson A, Charafe‐Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta‐catenin signaling. PLoS Biol. 2009;7:e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McFall‐Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet‐Loso T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA. 2013;110:3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Imam JS, Plyler JR, Bansal H, Prajapati S, Bansal S, Rebeles J, Chen HI, Chang YF, Panneerdoss S, Zoghi B, Buddavarapu KC, Broaddus R, Hornsby P, Tomlinson G, Dome J, Vadlamudi RK, Pertsemlidis A, Chen Y, Rao MK. Genomic loss of tumor suppressor miRNA‐204 promotes cancer cell migration and invasion by activating AKT/mTOR/Rac1 signaling and actin reorganization. PLoS One. 2012;7:e52397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early‐stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Patient Demographics
Figure S1. Vimentin levels in aortic valve interstitial cells of normal and calcified aortic valves.
Figure S2. PTEN modulates AKT activation and α–smooth muscle actin expression in human aortic valve interstitial cells induced by miR‐486 mimic.