

Abstract
Background
End points and adverse events (AEs) are collected separately in clinical trials, yet regulatory requirements for serious AE reporting vary across regions, so classifying end points according to seriousness criteria can be useful in global trials.
Methods and Results
In the Apixaban for Prevention of Acute Ischemic Events 2 (APPRAISE‐2) trial, patients with a recent acute coronary syndrome were randomized to apixaban or placebo for the prevention of recurrent ischemic events. Suspected end points (myocardial infarction, stroke, or bleeding) were adjudicated by an independent clinical events classification committee. Safety criteria were collected for suspected end points and AEs. Patient‐level event rates per 100 patient‐days of follow‐up, modeled using Poisson regression, explored the influence of region and patient characteristics on event reporting. Overall, 13 909 events were reported by 858 sites in 39 countries; 8.4% (n=1166) were suspected end points, and 91.6% (n=12 743) were AEs. Overall, 66.0% of suspected end points were confirmed by the clinical events classification committee. Most clinical events classification committee‐confirmed end points met criteria to be classified as serious (94.0%); many clinical events classification committee‐negated end points also did (63.2%), but fewer AEs met seriousness criteria (17.9%). The most common seriousness criterion was hospitalization (79.9%, n=2594). Region explained 28.7% of end point‐ and 26.4% of serious AE‐reporting variation, and patient characteristics explained an additional 25.4% of end point and 13.4% of serious AE variation. Nonserious AE‐reporting variation was not explained by adjustment.
Conclusions
An integrated collection of end points and serious AEs is feasible in a multinational trial and illustrates the shared characteristics of events. Tailoring event collection to fit the phase and purpose of the trial is achievable and informative.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00831441.
Keywords: acute coronary syndrome, clinical end points, clinical events classification, safety, serious adverse events
Subject Categories: Clinical Studies, Cardiovascular Disease, Acute Coronary Syndromes, Myocardial Infarction
Introduction
Collection of trial end point and adverse event (AE) data in clinical trials is vital to determine both the efficacy and safety of the study treatment. Trial end points are established early during the trial design with specific definitions and form the basis for event‐driven trial completion and regulatory approval.1 AEs, commonly reported by study participants during trial follow‐up, follow a regulatory path if they meet criteria for seriousness and represent a key element of the product label.2, 3 Traditionally, these events are captured with unique data elements and criteria, but overlap exists. It may be particularly relevant to understand which end point events also meet serious AE criteria to meet regulatory reporting requirements in a global trial. The regulatory environment varies in different countries. For example, some countries do not require end points to be reported as serious AEs, but others require all serious AEs to be reported. In the Apixaban for Prevention of Acute Ischemic Events 2 (APPRAISE‐2) trial,4 data collection was designed to capture safety criteria for trial end points, which provides a unique opportunity to describe events by seriousness criteria regardless of reporting criteria or adjudication outcome. Limited data are available to reflect on overall event collection, variation across sites, and shared aspects of end points and AEs in multinational trials. The generalizability of study results from international clinical trials may be influenced by regional differences in health care and event reporting. A better understanding of site reporting variation may provide insights into optimizing event collection to suit the study phase and objectives. Therefore, we describe both trial end points and AEs by seriousness, explore site variation in reporting by event type, and discuss observations from a trial with integrated collection of end point and safety events.
Methods
Participants and Study Design
APPRAISE‐2 was a double‐blind randomized controlled trial that enrolled high‐risk acute coronary syndrome patients and included 7392 participants at 858 sites in 39 countries. The design and main results of APPRAISE‐2 have been previously published.4 Study participants were randomized in a 1:1 fashion to receive either apixaban (5 mg twice daily) or placebo on top of standard antiplatelet therapy. A reduced dose of apixaban (2.5 mg twice daily) was given to participants with a creatinine clearance lower than 40 mL/min. Key exclusion criteria were severe renal impairment (creatinine clearance <20 mL/min), advanced heart failure, high risk of bleeding, previous intracranial hemorrhage, ischemic stroke within the last 7 days, and current use of anticoagulants. The trial was stopped early after the enrollment of 7392 participants due to an increased rate of bleeding events with apixaban not accompanied by a reduction in ischemic end points (cardiovascular death, myocardial infarction [MI], or ischemic stroke). The period between the first dose of study drug and 2 days after the last dose was used for safety analyses. Institutional review board approval was obtained at all sites. All participating patients gave written informed consent.
Clinical Events
All events reported by site investigators were collected on case report forms as either suspected trial end points or AEs. Suspected trial end points were collected on the dedicated pages for MI or unstable angina, cerebrovascular event, or bleeding. The definitions of end points in the APPRAISE‐2 trial are shown in Table S1. A clinical events classification committee (CEC) blinded to study drug assignment adjudicated the suspected end points according to trial definitions. When the suspected trial end point was negatively adjudicated, no further action was taken. All suspected end points were sent to the CEC with the exception of site‐reported bleeding classified as Thrombolysis in Myocardial Infarction (TIMI) minimal, which was sent to be reviewed by a coordinator. For this analysis, TIMI minimal bleeds sent to coordinator review are grouped with AEs. Prespecified AEs were listed and included heart failure, pneumonia, urinary tract infection, atrial fibrillation, hepatotoxicity, hypertension, headache, dizziness, dyspnea, chest pain, and syncope. Other AEs were reported as free text.
Suspected trial end points and AEs were assessed by a site investigator for seriousness based on regulatory criteria. This included those that resulted in death or were life threatening, led to hospitalization (or prolonged current hospitalization), caused persistent or significant disability or a congenital anomaly, or were thought to be an important medical event, based on clinical judgment. All clinical data from APPRAISE‐2 were collected centrally in a database at the Duke Clinical Research Institute.
Statistical Analysis
Clinical events are presented overall and by event type (suspected trial end points or AEs). Events reported as both suspected trial end points and AEs (n=185) were classified as suspected trial end points if they represented the same event. Continuous variables are presented as median (25th, 75th percentiles), and categorical variables as number (percentage). Continuous variables were compared using Wilcoxon rank‐sum tests, and categorical variables were compared using chi‐square tests.
Site‐level analyses excluded 371 sites that enrolled fewer than 5 patients and 246 sites that enrolled 5 to 9 patients, leaving the 241 sites that enrolled 10 or more patients. Sites were divided into tertiles of event reporting volume per 100 patient‐days of follow‐up (high, middle, and low). Patient‐level event rates per 100 patient‐days of follow‐up were modeled using Poisson regression with site as a random intercept to explore the influence of region and patient characteristics on reporting variation. Models were separately fit for trial end points, serious AEs, and nonserious AEs. The random effect variance was estimated for each model interpreted as the variation in log event rate attributable to between‐site differences. Proportional change in variance was calculated for pairs of nested models: proportional change in variance =(V0−V1)/V0, where V0 was variance of the initial model and V1 was variance of the model with additional covariates. The log event rate was assumed to be normally distributed with mean equal to the intercept parameter of the model and variance equal to the random effect variance. Region (Asia Pacific, North America, South America, Western Europe, and Eastern Europe) and patient characteristics (age, sex, and comorbidities [hypertension, diabetes mellitus, depression, heart failure, peripheral vascular disease, cardiovascular disease, atrial fibrillation, renal dysfunction]) were added as independent variables to an intercept‐only model. Parameter estimates from the models were used to estimate the parameters of the normal distribution. The relative reduction in variability of site reporting was plotted as a probability density function of event rates on a log scale, separately for each event type, overlaying the intercept‐only model, the model adding region, and the full model. The same models were used to explore the association of geographic region and patient characteristic with event reporting for each type of event (reported as relative risk with 95% confidence intervals and F values).
Event rates for end points, serious AEs, and nonserious AEs were plotted by site with smoothing splines where sites were sorted according to the rate of any type of event. Data were analyzed using SAS Version 9.4 (SAS Institute Inc, Cary, NC). P<0.05 are considered statistically significant.
Results
Clinical Events Distribution
A total of 13 909 events were reported by 858 sites, of which 8.4% (n=1166) were suspected trial end points and 91.6% (n=12 743) were AEs (Figure 1). Among all AEs, 33.6% were prespecified (n=4278). The most common prespecified AEs were minimal bleeding, chest pain, heart failure, and hypertension.
Figure 1.
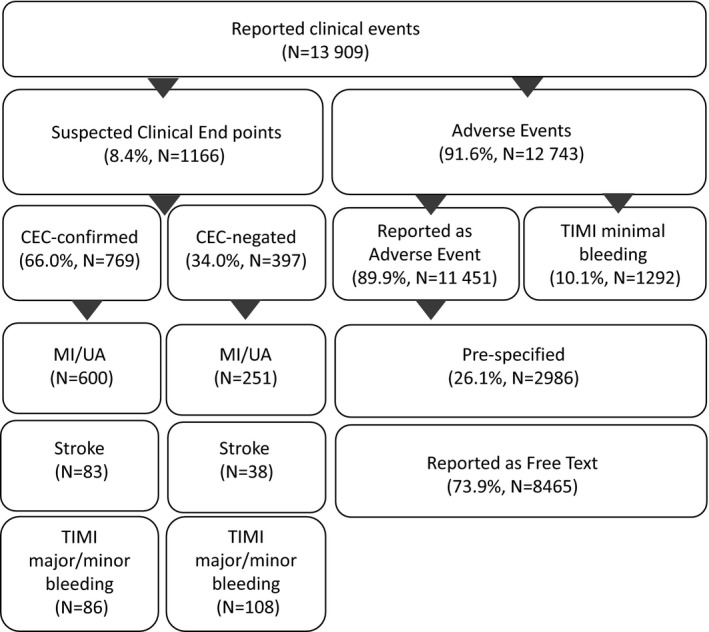
Site‐reported clinical events distribution. CEC indicates clinical events classification committee; MI, myocardial infarction; TIMI, Thrombolysis in Myocardial Infarction; UA, unstable angina.
Among suspected end points forwarded to the CEC, 66.0% (n=769) were confirmed: 70.5% (n=600) MI or unstable angina, 68.6% (n=83) stroke, and 44.3% (n=86) bleeding. Among bleeding events, 13.1% (n=194) were sent for CEC review. The other 86.9% (n=1292) were TIMI minimal bleeding events reviewed by a coordinator and grouped with AEs.
Seriousness of Clinical Events
Seriousness criteria were met for 17.9% (2276/12 743) of reported AEs and 83.6% (974/1166) of all suspected clinical end points. Of CEC‐confirmed end points, 94.0% (723/769) met seriousness criteria: 98% of MIs, 95% of unstable anginas, 94% of strokes, and 73% of bleedings. Of CEC‐negated events, 63.2% (251/397) met seriousness criteria: 72.4% of negated MIs or unstable anginas, 81.6% of negated strokes, and 36.1% of negated bleeding. Hospitalization or prolongation of hospitalization was the most common seriousness criterion for end points and AEs (79.9%, 2594/3250), followed by death (9.9%, 321/3250) (Table 1). Rates of serious events by prespecified AEs and end points are also shown in Table S2.
Table 1.
Seriousness Criteria of Clinical Events
| Clinical Event | Overall, n | Serious, n (% of Overall) | Hospitalization/Prolongation, n (% of Serious Events) | Death, n (% of Serious Events) | Life Threatening, n (% of Serious Events) | Important Medical Event, n (% of Serious Events) | Disability, n (% of Serious Events) |
|---|---|---|---|---|---|---|---|
| Overall | 13 909 | 3250 (23.4) | 2594 (79.9) | 321 (9.9) | 173 (5.3) | 93 (2.9) | 38 (1.2) |
| AEsa | 12 743 | 2276 (17.9) | 1839 (80.9) | 221 (9.7) | 89 (3.9) | 78 (3.4) | 19 (0.8) |
| Suspected clinical end points | 1166 | 974 (83.6) | 755 (77.6) | 100 (10.3) | 84 (8.6) | 15 (1.5) | 19 (2.0) |
| CEC‐confirmed | 769 | 723 (94.0) | 563 (78.0) | 69 (9.6) | 70 (9.7) | 5 (0.7) | 15 (2.1) |
| MI | 416 | 407 (97.8) | 309 (76.1) | 46 (11.3) | 42 (10.3) | 1 (0.2) | 8 (2.0) |
| UA | 184 | 175 (95.1) | 160 (91.4) | 2 (1.1) | 9 (5.1) | 1 (0.6) | 3 (1.7) |
| Stroke | 83 | 78 (94.0) | 49 (62.8) | 16 (20.5) | 8 (10.3) | 1 (1.3) | 4 (5.1) |
| TIMI bleeding, major or minor | 86 | 63 (73.3) | 45 (71.4) | 5 (7.9) | 11 (17.5) | 2 (3.2) | 0 (0) |
| CEC‐negated | 397 | 251 (63.2) | 192 (76.5) | 31 (12.4) | 14 (5.6) | 10 (4.0) | 4 (1.6) |
| MI or UA | 251 | 181 (72.4) | 143 (79.0) | 25 (13.8) | 7 (3.9) | 4 (2.2) | 2 (1.1) |
| Stroke | 38 | 31 (81.6) | 18 (58.1) | 4 (12.9) | 4 (12.9) | 3 (9.7) | 2 (6.5) |
| TIMI bleeding, major or minor | 108 | 39 (36.1) | 31 (79.5) | 2 (5.1) | 3 (7.7) | 3 (7.7) | 0 (0) |
AE indicates adverse event; CEC, clinical events classification committee; MI, myocardial infarction; TIMI, Thrombolysis in Myocardial Infarction; UA, unstable angina.
Includes the bleeds that were sent only for coordinator‐level review. Three serious AEs and 1 serious end point did not provide a cause. Twenty‐two serious AEs were cancer.
Site‐Level Patterns of Clinical Event Reporting
Of the 858 sites, the 371 sites (43%) that enrolled fewer than 5 patients as well as the 246 sites (29%) that enrolled between 5 and 9 patients were excluded from site‐level analysis. This left 241 sites (28%) that enrolled ≥10 patients. The rates of event‐reporting per 100 patient‐days of follow‐up in these 241 sites are presented in Figure S1. Median rates for nonserious AEs were 1.15 events per 100 patient‐days in the highest‐reporting tertile, 0.52 events per 100 patient‐days in the middle tertile, and 0.14 events per 100 patient‐days in the lowest‐reporting tertile; median rates for serious AEs were 0.20 events per 100 patient‐days in the highest‐reporting tertile, 0.13 events per 100 patient‐days in the middle tertile, and 0.04 events per 100 patient‐days in the lowest‐reporting tertile. Finally, median rates for suspected trial end points were 0.09 events per 100 patient‐days in the highest‐reporting tertile, 0.06 events per 100 patient‐days in the middle tertile, and 0.04 events per 100 patient‐days in the lowest‐reporting tertile.
Patient characteristics across tertiles of reporting (patient n=4831) are shown in Table 2. Participants were older in the high‐ and middle‐reporting sites than in low‐reporting sites. Hypertension, diabetes mellitus, impaired renal function, depression, and cerebrovascular disease were more prevalent in high‐reporting sites. North American sites were more often in the high‐reporting tertile, whereas sites in Asia and Eastern Europe were more often in the low‐reporting tertile.
Table 2.
Region and Patient Characteristics by Site‐Tertile of Reporting Clinical Events for Sites With ≥10 Patients
| Characteristic | High‐Reporting Tertile (80 Sites, 1320 Patients) | Middle‐Reporting Tertile (81 Sites, 1608 Patients) | Low‐Reporting Tertile 80 Sites, 1903 Patients) | P Value |
|---|---|---|---|---|
| Age, y, median (25th, 75th percentile) | 67.0 (58.4, 73.4) | 67.4 (59.6, 73.2) | 63.5 (55.0, 71.3) | <0.0001 |
| Sex, women | 460 (34.8) | 526 (32.7) | 605 (31.8) | 0.1873 |
| Hypertension | 1088 (82.5) | 1259 (78.3) | 1401 (73.6) | <0.0001 |
| Diabetes mellitus | 645 (48.9) | 701 (43.6) | 779 (40.9) | <0.0001 |
| Depression before index ACS event | 127 (9.6) | 41 (2.5) | 37 (1.9) | <0.0001 |
| Peripheral vascular disease | 264 (20.0) | 256 (15.9) | 367 (19.3) | 0.0070 |
| Heart failure or LVEF <40% | 559 (42.4) | 709 (44.1) | 798 (41.9) | 0.4120 |
| Impaired renal function | 403 (32.0) | 506 (33.0) | 464 (26.0) | <0.0001 |
| Atrial fibrillation | 76 (5.8) | 80 (5.0) | 96 (5.0) | 0.5779 |
| Cerebrovascular disease | 146 (11.1) | 154 (9.6) | 132 (6.9) | 0.0002 |
| 2 or more chronic conditions other than hypertension | 570 (43.2) | 562 (35.0) | 602 (31.6) | <0.0001 |
| 3 or more chronic conditions other than hypertension | 159 (12.0) | 119 (7.4) | 129 (6.8) | <0.0001 |
| Region | <0.0001 | |||
| Asia Pacific | 109 (8.3) | 361 (22.5) | 441 (23.2) | |
| Eastern Europe | 349 (26.4) | 738 (45.9) | 811 (42.6) | |
| North America | 418 (31.7) | 129 (8.0) | 233 (12.2) | |
| South America | 169 (12.8) | 69 (4.3) | 180 (9.5) | |
| Western Europe | 275 (20.8) | 311 (19.3) | 238 (12.5) |
ACS indicates acute coronary syndrome; LVEF, left ventricular ejection fraction.
Categorical variables are expressed as n (%). Data on renal function were missing for 5% of patients.
The rate of CEC confirmation of site‐reported trial end points was similar across the tertiles (Table 3). The exception was less CEC confirmation of stroke in the highest‐reporting tertile compared with the middle and low‐reporting tertiles (52.2%, 80.0%, and 81.8%, respectively).
Table 3.
Ratio of CEC‐Confirmed End Points to Suspected End Points Across Tertiles by Type of End Points
| End Points | High‐Reporting Tertile (80 Sites, 1320 Patients) | Middle‐Reporting Tertile (81 Sites, 1608 Patients) | Low‐Reporting Tertile 80 Sites, 1903 Patients) | P Value |
|---|---|---|---|---|
| Overall | 179/286 (62.6) | 183/264 (69.3) | 117/164 (71.3) | 0.102 |
| MI/UA | 148/220 (67.3) | 132/179 (73.7) | 88/117 (75.2) | 0.208 |
| Stroke | 12/23 (52.2) | 28/35 (80.0) | 18/22 (81.8) | 0.035 |
| TIMI bleeding | 19/43 (44.2) | 23/50 (46.0) | 11/25 (44.0) | 0.979 |
CEC indicates clinical events classification committee; MI, myocardial infarction; TIMI, Thrombolysis in Myocardial Infarction; UA, unstable angina.
Influence of Geographic Region and Patient Characteristics in Between‐Site Variation in Event Reporting
Geographic region explained 28.7% of site variation in trial end point reporting and 26.4% of site variation for serious AE reporting but had little impact on nonserious AE reporting (6.7%) (Figure 2). In the model, geographic regions (specifically Eastern Europe and Asia Pacific) were less likely to report clinical end point and serious AE events (Table S3). Patient characteristics further reduced site variation in end point (25.4%) and serious AE (13.4%) reporting but also had virtually no impact on nonserious AE (2.2%) reporting. Older age, diabetes mellitus, and heart failure were associated with more reported end points, and diabetes mellitus, heart failure, depression, atrial fibrillation, renal dysfunction, and peripheral vascular disease were associated with more reported serious AEs. Female sex and depression were more strongly associated with more reported nonserious AEs.
Figure 2.
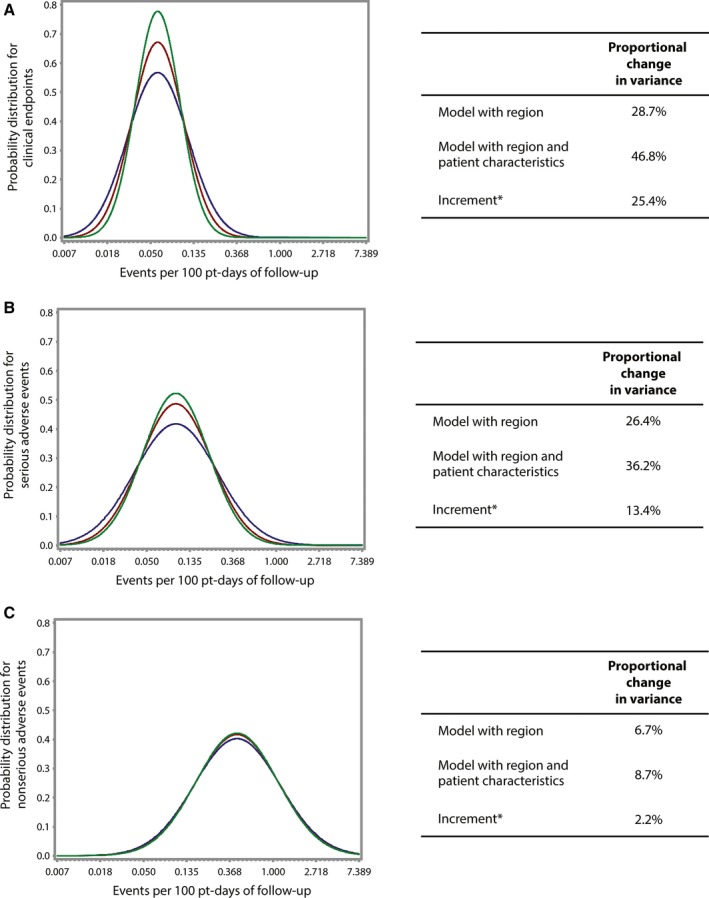
Impact of geographic region and patient characteristics on site variation in event reporting (A, clinical end points; B, serious adverse events; C, nonserious adverse events) per 100 patient‐days of follow‐up. Blue represents the unadjusted model. Red represents the adjustment for geographic region. Green represents the adjustment for geographic region and patient characteristics. *Incremental to the previous model.
Discussion
In this multinational acute coronary syndrome trial including 7392 participants, ≈14 000 clinical events were reported by site investigators, the majority of which were nonserious AEs. Although most CEC‐confirmed trial end points and two thirds of CEC‐negated trial end points met seriousness criteria, only 18% of site‐reported AEs did. Serious AEs are collected with specific regulatory guidelines, and the exemption of trial end points from regulatory reporting is an important step in simplifying trial burden. However, gathering seriousness criteria for study end points may be warranted in some cases to enable analysis of all serious events, including end points, for global reporting needs. There is also site variation in the reporting of suspected trial end points and serious AEs, which was partly explained by region and participant characteristics. Importantly, the rate of CEC‐confirmed end points did not vary by reporting tertile, suggesting that sites reported similar types of events, just different rates of them. This study demonstrates that collection of trial end points and safety events can be tailored to suit the purpose of the trial and that end points and serious AEs share site variation patterns.
Site reporting variation for end points and serious AEs was influenced by geographic region and patient characteristics but persisted after adjustment. Patient characteristics such as age, comorbidities, and renal function have been shown to be independently associated with serious AE reporting in acute coronary syndrome trials.5 In this analysis we confirmed that baseline characteristics such as heart failure, diabetes mellitus, peripheral vascular disease, atrial fibrillation, depression, and renal dysfunction were associated with serious AE reporting. Furthermore, female sex and depression were most strongly associated with nonserious AE reporting. Moreover, trial end points and serious AEs were influenced by geographic region, whereas nonserious AEs were not. Nonserious AE reporting, of unclear relevance in later‐phase studies and the majority of events reported in this phase 3 trial, was weakly associated with patient characteristics or region. This suggests a more random nature to the reporting of nonserious AEs in clinical trials driven by factors other than patient characteristics or site.
The CEC process provides independent, blinded, and systematic adjudication of events which determines trial results.6, 7, 8 Approximately two thirds of the suspected trial end points met definitions and were confirmed by CEC. A similar rate of site‐reported to confirmed trial end points was seen in the Platelet Inhibition and Patient Outcomes trial.9 In our study the proportion of CEC‐confirmed to site‐reported trial end points was similar across tertiles, with the exception of stroke. This suggests that sites in low‐reporting regions are reporting the same types of events, just fewer of them. Differences in population or health care across regions, such as hospital access and use of troponin, may contribute to variability in event reporting.10 This variability is not unexpected, but notable geographic variation is worth further exploration.
Seriousness criteria were collected for AEs and suspected trial end points by design. As expected, almost all (94.0%) suspected trial end points confirmed by the CEC process met seriousness criteria. Over 60% of CEC‐negated end points also met seriousness criteria. The most common seriousness criterion for these events was hospitalization (76.5%) followed by death (12.4%) and life‐threatening condition (5.6%). There have been recent concerns expressed by regulatory agencies that CEC‐negated trial end points are a potential source for missed serious AEs.11, 12 AEs are required to be reported to regulatory agencies if serious, unexpected, and potentially caused by the investigational drug. Although end points are exempt from serious AE reporting, negatively adjudicated trial end points have been a topic of concern. Often these events are similar in causality and pathophysiology to the suspected trial end point event, but with insufficient elements to meet trial definitions. In this case, leaving them as negatively adjudicated end points exempt from reporting as serious AEs makes sense. However, among CEC‐negated end points that also meet seriousness criteria, review for missed serious AEs is important. Therefore, classification of the seriousness status of end points may also focus attention on those events most likely to contain other serious AEs. There is no single way to collect event data, but integrating the CEC and safety processes provides support for sorting and filtering all clinical events without limitations of standard classification schemes.
Our results should be interpreted in light of some limitations. For the site‐level analysis, we included only sites with ≥10 patients, thus excluding two thirds of the sites. However, including sites with only a few patients enrolled would add more uncertainty to observations. Additionally, stratification of sites in tertiles of reporting is driven by the rates of nonserious AEs because these events were more prevalent. Our results were derived from a single acute coronary syndrome trial, which may impair generalizability to other scenarios. Nevertheless, the unique way that seriousness of clinical events was collected in this study allowed us to demonstrate the importance of an integrated process when assessing clinical and safety end points.
Conclusion
An integrated collection of trial end points and serious AEs demonstrates how these clinical events share similar characteristics and reporting trends. Tailoring event collection to fit the phase and purpose of the trial is both feasible and informative.
Author Contributions
Dr Guimarães, Dr Lopes, and Dr K. Alexander had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analyses.
Sources of Funding
This analysis was funded internally by the Duke Clinical Research Institute, Durham, NC.
Disclosures
Dr Lopes reported receiving research support from Bristol‐Myers Squibb and GlaxoSmithKline and personal fees from Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, GlaxoSmithKline, Pfizer, and Portola. Dr J. Alexander reported receiving research support from Boehringer Ingelheim, Bristol‐Myers Squibb, CSL Behring, Pfizer, and Tenax and personal fees from Bristol‐Myers Squibb, CSL Behring, Pfizer, and Portola. No other disclosures were reported.
Supporting information
Table S1. APPRAISE‐2 Clinical Endpoint Definitions
Table S2. Rates of Serious and Nonserious Clinical Events
Table S3. Associations Between Region and Patient Characteristics and Event Reporting
Figure S1. Rates of site‐reported clinical endpoints, serious adverse events, and nonserious adverse events per 100 patient‐days of follow‐up at site level (sites with 10 or more patients, n=241).
Acknowledgments
Dr Guimarães thanks CNPq–Conselho Nacional de Desenvolvimento Cientifico e Tecnologico, of Ministry of Science, Technology and Innovation of Brazil for her fellowship funding.
(J Am Heart Assoc. 2017;6:e005490 DOI: 10.1161/JAHA.117.005490.)28438739
References
- 1. Pocock SJ, Clayton TC, Stone GW. Design of major randomized trials: part 3 of a 4‐part series on statistics for clinical trials. J Am Coll Cardiol. 2015;66:2757–2766. [DOI] [PubMed] [Google Scholar]
- 2. Sherman RB, Woodcock J, Norden J, Grandinetti C, Temple RJ. New FDA regulation to improve safety reporting in clinical trials. N Engl J Med. 2011;365:3–5. [DOI] [PubMed] [Google Scholar]
- 3. Food and Drug Administration, Department of Health and Human Services . Investigational new drug safety reporting requirements for human drug and biological products and safety reporting requirements for bioavailability and bioequivalence studies in humans. Final rule. Fed Regist. 2010;75:59935–59963. [PubMed] [Google Scholar]
- 4. Alexander JH, Lopes RD, James S, Kilaru R, He Y, Mohan P, Bhatt DL, Goodman S, Verheugt FW, Flather M, Huber K, Liaw D, Husted SE, Lopez‐Sendon J, De Caterina R, Jansky P, Darius H, Vinereanu D, Cornel JH, Cools F, Atar D, Leiva‐Pons JL, Keltai M, Ogawa H, Pais P, Parkhomenko A, Ruzyllo W, Diaz R, White H, Ruda M, Geraldes M, Lawrence J, Harrington RA, Wallentin L; APPRAISE‐2 Investigators . Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365:699–708. [DOI] [PubMed] [Google Scholar]
- 5. Zimerman A, Lopes RD, Stebbins AL, Guimarães PO, Haque G, Melloni C, Trollinger K, James SK, Alexander JH, Tricoci P, Roe MT, Ohman EM, Mahaffey KW, Held C, Tinga B, Pieper KS, Alexander KP. Pooled analysis of adverse event collection from 4 acute coronary syndrome trials. Am Heart J. 2016;174:60–67. [DOI] [PubMed] [Google Scholar]
- 6. Mahaffey KW, Harrington RA, Akkerhuis M, Kleiman NS, Berdan LG, Crenshaw BS, Tardiff BE, Granger CB, DeJong I, Bhapkar M, Widimsky P, Corbalon R, Lee KL, Deckers JW, Simoons ML, Topol EJ, Califf RM; for the PURSUIT Investigators . Systematic adjudication of myocardial infarction end‐points in an international clinical trial. Curr Control Trials Cardiovasc Med. 2001;2:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granger CB, Vogel V, Cummings SR, Held P, Fiedorek F, Lawrence M, Neal B, Reidies H, Santarelli L, Schroyer R, Stockbridge NL, Zhao F. Do we need to adjudicate major clinical events? Clin Trials. 2008;5:56–60. [DOI] [PubMed] [Google Scholar]
- 8. Dechartres A, Boutron I, Roy C, Ravaud P. Inadequate planning and reporting of adjudication committees in clinical trials: recommendation proposal. J Clin Epidemiol. 2009;62:695–702. [DOI] [PubMed] [Google Scholar]
- 9. Mahaffey KW, Held C, Wojdyla DM, James SK, Katus HA, Husted S, Steg PG, Cannon CP, Becker RC, Storey RF, Khurmi NS, Nicolau JC, Yu CM, Ardissino D, Budaj A, Morais J, Montgomery D, Himmelmann A, Harrington RA, Wallentin L; PLATO Investigators . Ticagrelor effects on myocardial infarction and the impact of event adjudication in the PLATO (Platelet Inhibition and Patient Outcomes) trial. J Am Coll Cardiol. 2014;63:1493–1499. [DOI] [PubMed] [Google Scholar]
- 10. Akkerhuis KM, Deckers JW, Boersma E, Harrington RA, Stepinska J, Mahaffey KW, Wilcox RG, Lincoff AM, Keltai M, Topol EJ, Califf RM, Simoons ML. Geographic variability in outcomes within an international trial of glycoprotein IIb/IIIa inhibition in patients with acute coronary syndromes. Results from PURSUIT. Eur Heart J. 2000;21:371–381. [DOI] [PubMed] [Google Scholar]
- 11. Lopes RD, Dickerson S, Hafley G, Burns S, Tourt‐Uhlig S, White J, Newby LK, Komajda M, McMurray J, Bigelow R, Home PD, Mahaffey KW. Methodology of a reevaluation of cardiovascular outcomes in the RECORD trial: study design and conduct. Am Heart J. 2013;166:208–216.e228. [DOI] [PubMed] [Google Scholar]
- 12. Mahaffey KW, Hafley G, Dickerson S, Burns S, Tourt‐Uhlig S, White J, Newby LK, Komajda M, McMurray J, Bigelow R, Home PD, Lopes RD. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J. 2013;166:240–249.e241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. APPRAISE‐2 Clinical Endpoint Definitions
Table S2. Rates of Serious and Nonserious Clinical Events
Table S3. Associations Between Region and Patient Characteristics and Event Reporting
Figure S1. Rates of site‐reported clinical endpoints, serious adverse events, and nonserious adverse events per 100 patient‐days of follow‐up at site level (sites with 10 or more patients, n=241).