

Abstract
Background
Ketone bodies are known to substitute for glucose as brain fuel when glucose availability is low. Ketogenic diets have been described as neuroprotective. Similar data have been reported for triheptanoin, a fatty oil and anaplerotic compound. In this study, we monitored the changes of energy metabolites in liver, blood, and brain after transient brain ischemia to test for ketone body formation induced by experimental stroke.
Methods and Results
Mice were fed a standard carbohydrate‐rich diet or 2 fat‐rich diets, 1 enriched in triheptanoin and 1 in soybean oil. Stroke was induced in mice by middle cerebral artery occlusion for 90 minutes, followed by reperfusion. Mice were sacrificed, and blood plasma and liver and brain homogenates were obtained. In 1 experiment, microdialysis was performed. Metabolites (eg glucose, β‐hydroxybutyrate, citrate, succinate) were determined by gas chromatography–mass spectrometry. After 90 minutes of brain ischemia, β‐hydroxybutyrate levels were dramatically increased in liver, blood, and brain microdialysate and brain homogenate, but only in mice fed fat‐rich diets. Glucose levels were changed in the opposite manner in blood and brain. Reperfusion decreased β‐hydroxybutyrate and increased glucose within 60 minutes. Stroke‐induced ketogenesis was blocked by propranolol, a β‐receptor antagonist. Citrate and succinate were moderately increased by fat‐rich diets and unchanged after stroke.
Conclusions
We conclude that brain ischemia induces the formation of β‐hydroxybutyrate (ketogenesis) in the liver and the consumption of β‐hydroxybutyrate in the brain. This effect seems to be mediated by β‐adrenergic receptors.
Keywords: cerebral ischemia, citrate, glucose, ketone bodies, lactate, middle cerebral artery occlusion, mouse, succinate, β‐hydroxybutyrate
Subject Categories: Metabolism, Ischemia, Cerebrovascular Disease/Stroke, Ischemic Stroke
Introduction
Stroke is a leading cause of death and disability,1, 2 a situation partly caused by a scarcity of successful treatments such as pharmacological interventions with recombinant tissue plasminogen activator or a mechanical thrombectomy.3, 4 Because of a high number of unsuccessful clinical studies with neuroprotective agents,5, 6 dietary approaches to reduce the severity of brain ischemia have gained popularity in the past decade. Although glucose is the main fuel of brain metabolism,3 ketone bodies such as acetoacetate and β‐hydroxybutyrate (BHB) can replace up to 50% of glucose to deliver the necessary metabolic energy for brain function. This contribution is particularly important during early life and during starvation.4 In brain ischemia, hyperglycemia is known to be detrimental, whereas ketone bodies are associated with significant benefits. In fact, fat‐rich and ketogenic diets have been proposed as neuroprotectants,5 and animal data indicate beneficial effects of ketogenic diets in experimental stroke studies.6 Moreover, BHB administration has been found to prevent neuronal cell death in cell cultures and in animal models of neurological disease.7, 8, 9
An alternative to the ketogenic diet may be triheptanoin, a triglyceride containing heptanoate, an odd‐numbered C7‐fatty acid. Triheptanoin is postulated to act as an anaplerotic compound replenishing substrates of the citric acid cycle such as succinate.10 Triheptanoin is beneficial in long‐chain mitochondrial β‐oxidation disorders.11 Recent work has also shown that triheptanoin is beneficial in epilepsy, where the ketogenic diet has long had a place in treatment.12 We reported previously that a triheptanoin‐rich diet reduced neuronal cell death after transient brain ischemia in mice.13 Because preliminary data from our laboratory showed that triheptanoin also induced ketogenesis, we initiated the current study in which we compared a standard diet, a triheptanoin‐rich diet, and a soybean oil diet for their effects on glucose and BHB levels in liver, blood, and brain. We found a dramatic, stroke‐induced ketogenesis in mice on fat‐rich diets that was accompanied by opposite changes of glucose levels and that was prevented by blockade of adrenergic β‐receptors.
Methods
Experimental Groups
Female CD‐1 mice (29–32 g; Charles River Laboratories) were used for the experiments. They were kept in standard cages, under 60% humidity, 22°C temperature, and a 12 hour‐light/dark cycle. Food and water were available ad libitum. The study was registered with the local animal committee (Regierungspräsidium Darmstadt). In accordance with GV‐Solas guidelines, all procedures were designed to minimize the suffering of the experimental animals.
Mice were randomized to study groups using a computer program for random number generation. In total, 173 mice were used for this study. Overall, 24 experiments could not be followed through because of surgical problems (insufficient blockade of the middle cerebral artery, continuous bleeding during reperfusion), and the mice had to be sacrificed. In 17 experiments, analytical problems caused a failure to obtain data (lack of perfusion in the microdialysis probe, problems during sample workup and gas chromatography–mass spectrometry measurements). The results shown in Figures 1, 2, 3, 4, 5 through 6 were obtained from 132 successful experiments (with an average of 8 experiments per group). Separate experiments were performed for the generation of figures, except that results in Figures 3 and 5 were from the same group of animals. Analytical measurements by gas chromatography–mass spectrometry were done by 3 different investigators, with identical results.
Figure 1.
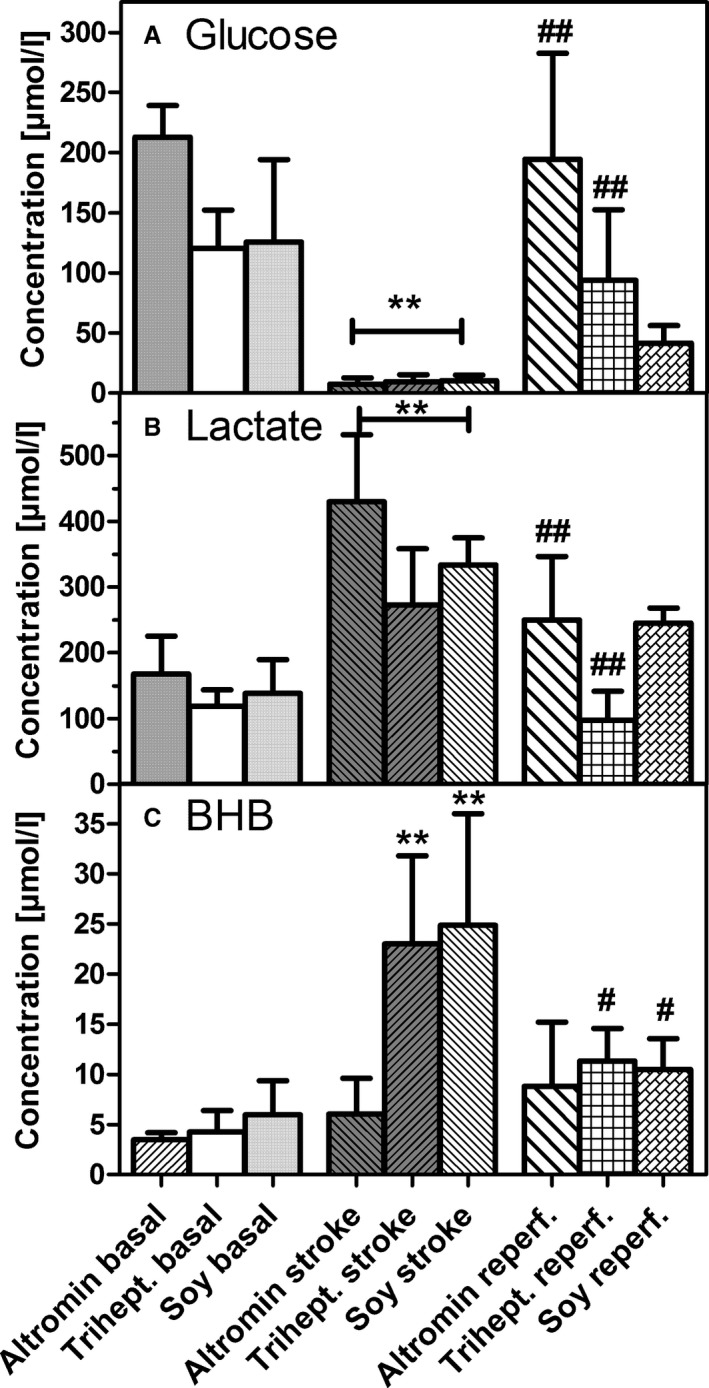
Changes in metabolite levels in microdialysates before (basal), during (stroke), and after (reperf.) transient (90 minutes) middle cerebral artery occlusion: (A) glucose, (B) lactate, (C) β‐hydroxybutyrate (BHB). Mice were kept on diets that were rich in carbohydrates (standard diet; Altromin), in triheptanoin (Trihept.), or in soybean oil (Soy) for 3 weeks. Data are concentrations that were calculated from 6 samples taken before, during, or after stroke and are mean±SD of 6 experiments. Statistics were calculated by 1‐way ANOVA and Tukey post‐test; **P<0.01 vs basal; # P<0.05; ## P<0.01 vs stroke.
Figure 2.
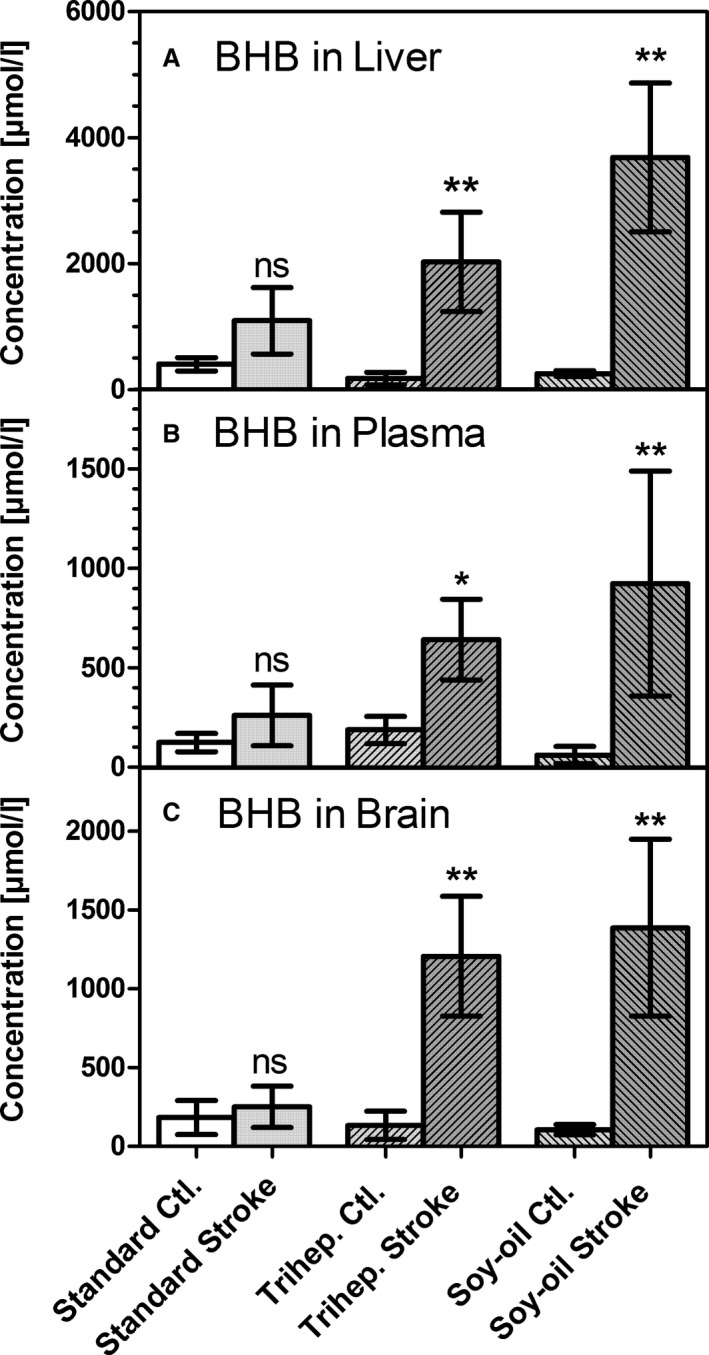
Concentrations of β‐hydroxybutyrate (BHB) in (A) liver homogenate, (B) blood plasma, and (C) brain homogenate (ischemic hemisphere). Samples were taken either from untreated animals (Ctl.) or immediately after 90 minutes of transient middle cerebral artery occlusion (stroke) in mice kept on a standard diet (Standard) or on a diet rich in soybean oil (Soy‐oil). Data are given as mean±SD (n=6). Statistics were calculated by 1‐way ANOVA and Tukey post‐test; *P<0.05; **P<0.01 vs Ctl. ns indicates not significant.
Figure 3.
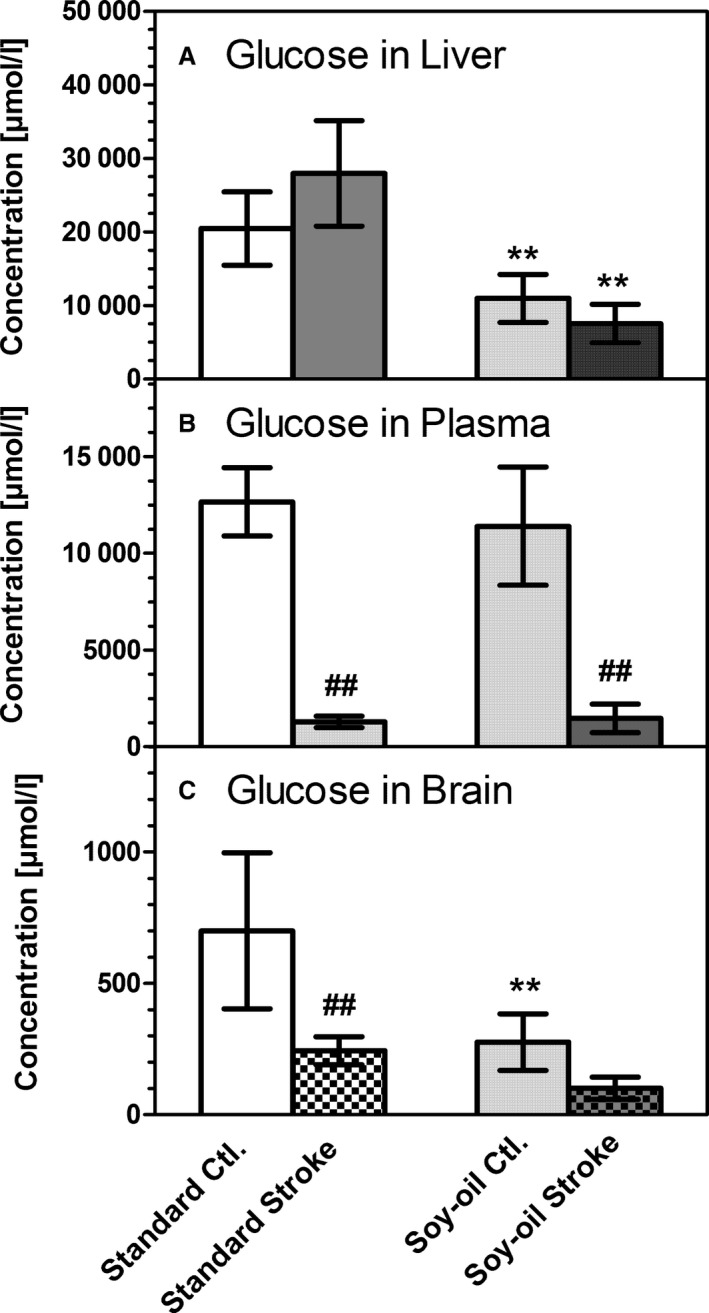
Concentrations of glucose in (A) liver homogenate, (B) blood plasma, and (C) brain homogenate (ischemic hemisphere). Samples were taken either from untreated animals (Ctl.) or immediately after 90 minutes of transient middle cerebral artery occlusion (stroke) in mice kept on a standard diet (Standard) or on a diet rich in soybean oil (Soy‐oil). Data are given as mean±SD (n=6). Statistics were calculated by 1‐way ANOVA and Tukey post‐test; **P<0.01 vs standard diet; ## P<0.01 vs Ctl.
Figure 4.
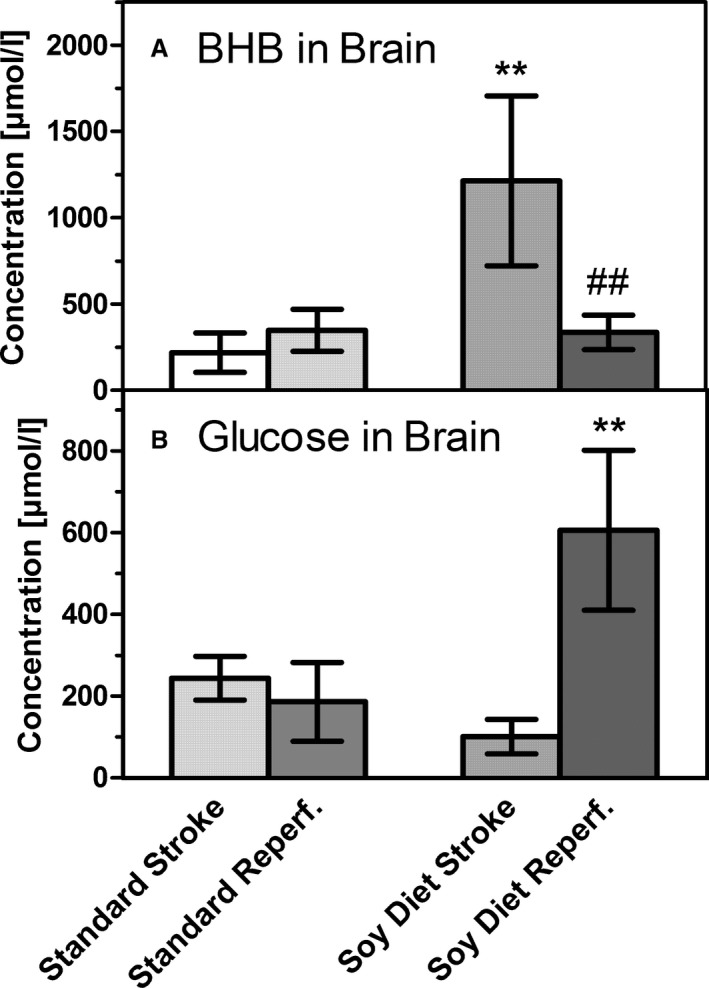
Changes of (A) β‐hydroxybutyrate (BHB) and (B) glucose after 60 minutes of reperfusion, measured in the left (ischemic) brain hemispheres. Samples were taken either immediately after the end of 90 minutes of transient middle cerebral artery occlusion (stroke) or after 60 minutes of reperfusion (Reperf.) in mice on a standard diet (Standard) or on a diet rich in soybean oil (Soy Diet). Data are given as mean±SD (n=6). Statistics were calculated by 1‐way ANOVA and Tukey post‐test; **P<0.01 vs standard diet; ## P<0.01 vs stroke.
Figure 5.
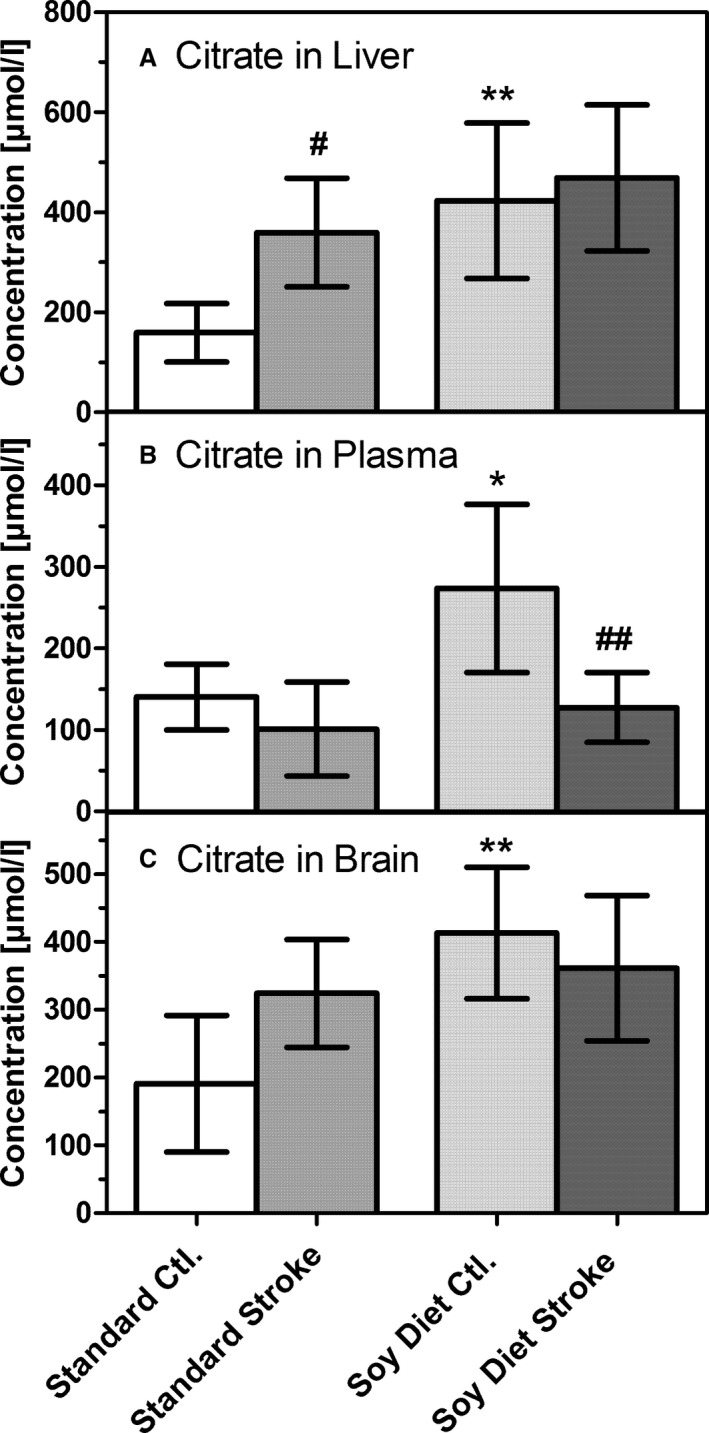
Concentrations of citrate in (A) liver homogenate, (B) blood plasma, and (C) brain homogenate (ischemic hemisphere). Samples were taken either from untreated animals (Ctl.) or after 90 minutes of transient middle cerebral artery occlusion (stroke) in mice on a standard diet (Standard) or on a diet rich in soybean oil (Soy Diet). Data are given as mean±SD (n=6). Statistics were calculated by 1‐way ANOVA and Tukey post‐test; *P<0.05; **P<0.01 vs standard diet; # P<0.05, ## P<0.01 vs Ctl.
Figure 6.
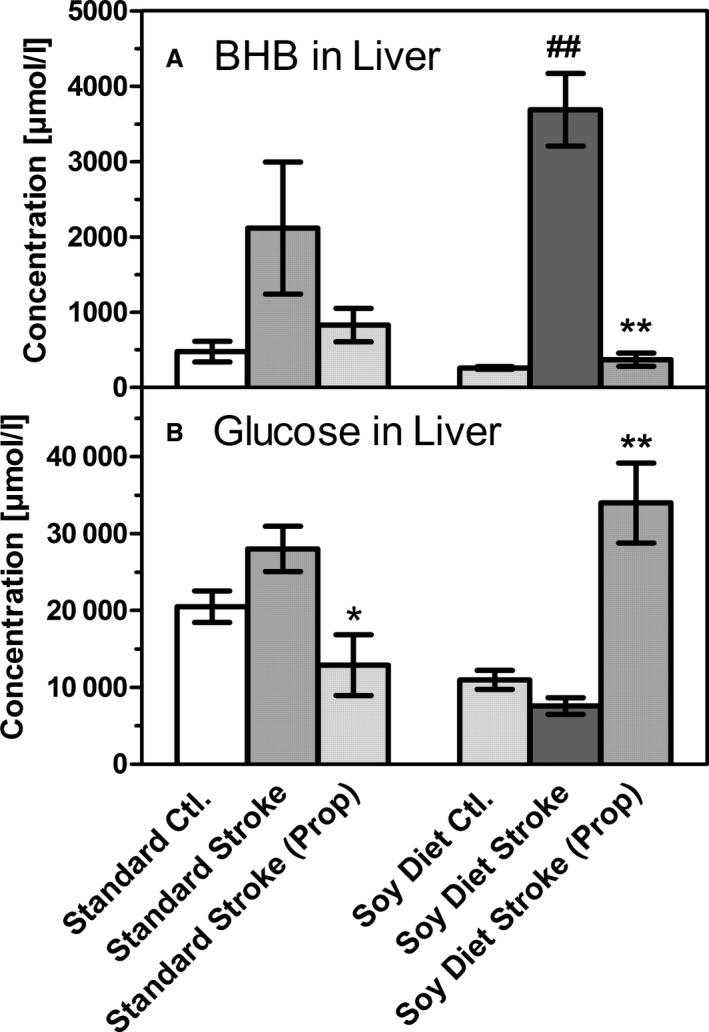
Changes of (A) β‐hydroxybutyrate (BHB) and (B) glucose in liver homogenates, taken from untreated mice (Ctl.), after 90 minutes of transient middle cerebral artery occlusion in untreated mice (stroke) or in mice that had received propranolol (prop; 2 mg/kg IP) prior to stroke induction. Mice had been fed a standard diet (Standard) or a diet rich in soybean oil (Soy Diet) for 3 weeks. Data are given as mean±SD (n=6). Statistics were calculated by 1‐way ANOVA and Tukey post‐test; *P<0.05; **P<0.01 vs standard diet; ## P<0.01 vs stroke.
Three dietary groups were used: a standard carbohydrate‐rich diet, a triheptanoin‐rich diet, and a soybean oil–rich diet. Diets were isocaloric and were fed for 3 weeks, with food and water available ad libitum. The standard diet (Altromin 1326 for mice; Altromin Co) contained 64% carbohydrate, 24% protein, and 12% fat (calculated as percentages of calories). The triheptanoin diet (Ssniff Co) contained 14% triheptanoin (wt/wt), which represents 35% of total caloric intake (carbohydrates 48%, protein 17%). The fat‐rich diet consisted of the same caloric amount of soybean oil (35%) with 48% carbohydrates and 17% protein and was also prepared by Ssniff Co. The amounts of fiber (4%) and ash (5.5%) were identical in the 3 diets, as were the food additives, which included vitamins A, C, D3, E, K3, and copper.
The study consisted of 2 parts. In the first part (Figures 1 and 2), the triheptanoin‐rich diet was compared with the soybean oil diet, similar to previous studies.12, 13 Both diets had the same composition, except for the fat constituent (see above). In this experiment, mice were implanted with microdialysis probes before stroke induction, and metabolite levels were measured in dialysates. In addition, the concentrations of BHB and some energy metabolites (citrate, succinate, fumarate, malate) were measured by gas chromatography–mass spectrometry in plasma and in liver and brain homogenates.
In the second part of the study (Figures 2, 3, 4, 5–6), mice were fed either the soybean oil–based fat‐rich diet or the carbohydrate‐rich standard diet (Altromin 1326). In this experiment, glucose, BHB, and energy metabolites were measured by gas chromatography–mass spectrometry in liver, blood, and brain homogenates. In some experiments, 2 mg/kg of (±)‐propranolol hydrochloride (99% pure, dissolved in saline; Sigma‐Aldrich) was given by intraperitoneal injection 15 minutes before the induction of stroke.
Transient Middle Cerebral Artery Occlusion
The procedure was performed as described previously.14, 15 Briefly, mice were anesthetized using isoflurane (2% in synthetic air), their body temperature was kept constant using a thermostatic device, and buprenorphine (0.1 mg/kg IP) was injected as an analgesic 15 minutes before performing surgery (this injection was repeated 8 hours later). After a paratracheal incision, a silicone suture (size 6‐0; Doccol) was inserted inside the left Arteria communis and advanced into the middle cerebral artery, where the silicone top of the filament blocked perfusion of the vessel. Cerebral blood flow was monitored with a laser Doppler monitoring device (Moor Instruments) to ascertain ischemia (<15% of blood flow versus basal). After 90 minutes, the suture was removed to allow reperfusion. Mice were sacrificed under isoflurane anesthesia either immediately after ischemia or after 60 minutes of reperfusion.
Microdialysis
At 24 hours before inducing stroke, we implanted a self‐built microdialysis probe into the left striatum, as described previously.14, 15 The probe was implanted under isoflurane anesthesia with the following coordinates (from bregma): anteroposterior +0.5 mm; lateral −2.2 mm; and dorsal ventral −3.8 mm.16 After surgery, local anesthetic was applied to the surgical site, and mice recovered for 24 hours in their home cages. On the day of surgery, probes were perfused with artificial cerebrospinal fluid (147 mmol/L NaCl, 4 mmol/L KCl, 1.2 mmol/L CaCl2, and 1.2 mmol/L MgCl2) at 2 μL/min. Fractions were collected in 30‐minute intervals.
Gas Chromatography–Mass Spectrometry Measurements
Blood plasma, liver, and brain samples were harvested immediately after decapitation of mice, frozen in liquid nitrogen, and stored at −80°C until measurement. Brain and liver homogenates were extracted using a biphasic Folch′s procedure,17 the aqueous supernatant was dried under a stream of nitrogen, and the dry residues were derivatized with BSTFA (N,O‐bis[trimethylsilyl]trifluoroacetamide) and TMCS (trimethylchlorosilane; 99:1). In plasma samples, proteins were precipitated by addition of methanol:water (9:1) and centrifuged, and the supernatants were treated as described earlier. Microdialysate samples were simply dried and derivatized directly with BSTFA/TMCS (99:1).
Samples were measured on an HP‐6890 Series GC System (Hewlett Packard) coupled to an Agilent Mass Selective Detector 5973 (Agilent) and an Agilent Autosampler 7683. We used a VF‐5MS capillary column (30 m×0.25 mm inner diameter; Varian Technologies) with a silylated precolumn (5 m). After the qualitative analysis of the metabolites (spectra adjusted to the NIST database), we established single ion monitoring parameters and used them for quantification of glucose, BHB, citrate, succinate, fumarate, and malate. The calculations were performed with internal and external standard methods.
Statistical Analysis
Values are expressed as mean±SD of n experiments. Statistical significance between treatment groups was evaluated by 1‐way ANOVA with Tukey post‐test, using GraphPad Prism software version 5.0.
Results
Effects of Fat‐Rich Diets on Energy Metabolites and Ketone Body Formation During Brain Ischemia
In the first experiment of our study, we compared 2 fat‐rich diets, the triheptanoin‐rich diet and a soybean oil diet, with the standard diet. Although fat content in the 2 fat‐rich diets was identical in terms of calories, it should be noted that soybean oil has a high amount of C16/18 fatty acids, whereas triheptanoin consists of glycerol esterified to C7‐heptanoic acid. Microdialysis was performed before, during, and after ischemia. The data (Figure 1) demonstrate that, as described previously,14 brain ischemia resulted in a rapid drop in brain glucose and a significant increase in lactate in all 3 dietary groups. Glucose and lactate levels normalized during reperfusion, with mice on the soybean oil diet showing less rapid recovery for lactate than the other 2 dietary groups (Figure 1A and 1B). The opposite was observed for glucose (Figure 1A and 1B). Interestingly, however, a significant ≈5‐fold rise in BHB was observed in the 2 dietary groups on fat‐rich diets (Figure 1C) and also normalized during reperfusion. No significant increase of BHB was seen in mice on the standard diet (Figure 1C).
In the following experiments, metabolite levels were measured in liver, in blood plasma, and in homogenates of the brain ischemic hemisphere. We found that ischemic stroke induces massive formation of ketone bodies in the liver (Figure 2A), the organ mainly responsible for ketogenesis. In the liver of triheptanoin‐fed mice, BHB rose from a basal level of 181±97 to 2031±787 μmol/L during brain ischemia, an 11‐fold increase. In the soybean oil–fed group, BHB rose from 258±46 to 3.688±1179 μmol/L, a 14‐fold increase. In comparison, this huge increase was not seen in mice fed the standard diet. Although the hepatic BHB level rose >2‐fold (from 406±108 to 1095±525 μmol/L) in these mice, this increase was not statistically significant (Figure 2A).
Concomitant with ketone body formation in the liver, plasma concentrations of BHB rose from 187±69 to 641±203 μmol/L in triheptanoin‐fed mice and from 61±42 to 924±565 μmol/L in soybean oil–fed mice, respectively; again, mice on the standard diet showed a much less prominent increase (from 124±46 to 261±152 μmol/L) after transient middle cerebral artery occlusion. Finally, increases in BHB were also reflected in brain homogenates: basal levels were 134±90 μmol/L in triheptanoin‐fed and 105±33 μmol/L in soybean oil–fed mice, whereas during ischemia, values increased to 1207±380 and 1387±561 μmol/L, respectively. BHB levels were not significantly changed in mice on the standard diet (Figure 2). These data demonstrate that ischemia in the brain causes a dramatic ketogenesis in the liver, which in turn leads to a several‐fold increase of BHB in blood plasma and the brain. This ketogenesis occurs only in mice that were previously fed the triheptanoin diet or the soybean oil diet, not the standard carbohydrate‐based diet.
In addition to BHB, we measured a range of metabolites in the brain to test for triheptanoin's anaplerotic effects. In particular, we measured succinate, a metabolite of the citric acid cycle that was postulated to be formed from propionyl‐CoA, a metabolite of C7‐heptanoic acid in triheptanoin.10, 11, 12 Succinate levels under basal conditions were 696±151 μmol/L after the standard diet and were higher in triheptanoin‐fed mice (1010±387 μmol/L; P value not significant versus standard diet) and in the soybean oil group (1256±324 μmol/L; P<0.01 versus standard diet). However, succinate levels were only slightly, and not significantly, increased in either of the diets after ischemia. Consequently, in this study, we found no direct evidence that triheptanoin acted as an anaplerotic substrate supplying succinate into the citric acid cycle. However, formation of propionate or succinate, possibly in specialized compartments such as mitochondria, may have occurred and may explain the neuroprotective activity of triheptanoin observed in our previous study.13 Because triheptanoin's metabolic actions in the present study strongly resembled those in the soybean oil‐rich diet, and because we failed to obtain additional triheptanoin from manufacturers or commercial sources, we continued our study with a comparison of the standard diet and the soybean oil diet.
Influence of Brain Ischemia on Energy Metabolism: Comparison of the Standard Diet and the Soybean Oil Diet
Figure 3 summarizes the data on glucose levels in liver, plasma, and brain. We found that the concentration of glucose in the liver was 2‐fold higher after the standard diet compared with the soybean oil diet group (P<0.01) (Figure 3A). No significant changes in liver glucose were observed after stroke. Basal plasma glucose levels were identical in the dietary groups (Figure 3B). Remarkably, plasma glucose levels decreased strongly during brain ischemia in both groups of mice, reaching levels of only 10% to 15% of basal levels corresponding to hypoglycemia (1.3–1.5 mmol/L) (Figure 3B). In the brain, as in liver, glucose concentrations were 2‐fold higher in mice on the standard diet (Figure 3C). Ischemia caused a reduction in glucose levels to 30% to 35% of basal levels in both dietary groups. Taken together, these data show that glucose levels, at least in blood and brain, changed in opposite ways to BHB, namely, they fell when BHB levels went up.
Next, we wanted to know how brain fuels glucose and BHB changed when perfusion was reinstated after brain ischemia. In mice on the standard diet, 60 minutes of reperfusion did not cause major changes in glucose or BHB in the brain (Figure 4). In mice on the soybean oil diet, however, the high concentration of BHB observed after stroke was much reduced after reperfusion; concomitantly, glucose levels were much increased, even beyond basal levels (Figure 4). In mice on the soybean oil diet, reperfusion was accompanied by a fall in BHB levels in the brain and reactive hyperglycemia.
Because BHB is a precursor of acetyl‐CoA and acetyl‐CoA is a precursor of citrate in the citric acid cycle, concentrations of citrate may serve as an indicator of cellular energy production. We measured the concentrations of citrate and other metabolites of the citric acid cycle and found that citrate concentrations were higher in liver, blood plasma, and brain in mice fed the soybean oil diet (P<0.01) (Figure 5). Citrate levels in these mice, however, were unchanged in liver and brain (and decreased in plasma) after stroke. In contrast, in mice fed the standard diet, citrate levels increased after stroke, reaching levels equal with the soybean oil diet group. Other metabolites (ie succinate, fumarate, and malate) did not show significant differences under either basal of ischemic conditions (data not shown).
Effects of Propranolol on Stroke‐Induced Ketogenesis
The sympathetic system is known to play a major role as a regulator of metabolism; therefore, in the final experiment, we chose to test whether β‐adrenergic receptors were involved in stroke‐induced ketogenesis. We applied propranolol (2 mg/kg IP), a nonselective β‐blocker, immediately before stroke and observed a dramatic shift of the glucose/BHB ratio in the liver (Figure 6). In mice on the soybean oil diet, the stroke‐induced manifold increase of BHB was completely prevented in the presence of propranolol, but glucose levels were increased by 3‐fold. In comparison, much less effect was seen in mice on the standard diet, in which hepatic levels of BHB and glucose were slightly decreased (Figure 6). These results clearly demonstrate a role for the adrenergic system in the balance of glucose and/or BHB formation in the liver.
Discussion
Glucose is the major fuel of the brain, but BHB can substitute for glucose under conditions of hypoglycemia.18, 19, 20 Ketogenesis (BHB formation) occurs largely in the liver and is a physiological response induced, for instance, by starving.21 The main finding of our study is that brain ischemia, induced by middle cerebral artery occlusion in mice, causes massive formation of ketone bodies in the liver, followed by an increase of BHB in the brain. This effect is dependent on diet and occurs to a great extent only in mice fed a fat‐rich diet.
In the first part of the study, we compared the standard diet with 2 fat‐rich diets, a soybean oil diet and a triheptanoin‐rich diet; in both cases, the fat content represented 35% of total calories. In contrast to our previous findings with a strictly ketogenic diet,22 these diets did not cause major ketosis by themselves (Figures 1 and 2), but they facilitated systemic ketosis when brain ischemia was induced. Our microdialysis data show that, with fat‐rich diets, brain ischemia leads to a several‐fold increase of BHB in the brain extracellular space, whereas no increase is seen after the standard diet. BHB is known to be brain permeable and to enter the brain dependent on its plasma levels.23 A more detailed analysis of BHB levels demonstrated a >10‐fold increased BHB concentration (reaching 2–4 mmol/L) in the liver of mice on fat‐rich diets, measured after 90 minutes of transient brain ischemia. Mice on the standard diet responded with minor ≈2‐fold BHB formation (Figure 2). Concomitantly, with fat‐rich diets, BHB levels were high in both blood plasma and brain homogenate (≈1 mmol/L).
It is generally accepted that the liver is the predominant site of ketone body production, whereas other organs, including glial cells in the brain, may be able to form small amounts of BHB.4, 24 Ketone body formation is typically seen at low glucose levels when alternative substrates are needed for fuel consumption. In our hands, BHB and glucose levels were changed in an opposite manner after stroke (Figures 2 and 3). After stroke, plasma glucose levels were strongly decreased, whereas BHB was high. Liver glucose levels remained high, but this may partly be due to enhanced glycogen breakdown, which was not measured in our study. Similarly, brain glucose levels dropped when BHB levels rose. When reperfusion was allowed, BHB levels in the brain reached normal levels within 1 hour, whereas glucose concentrations increased above normal levels (reactive hyperglycemia) (Figure 4). Taken together, stroke induced a hypoglycemic response that was accompanied by hepatic BHB production and uptake of BHB in the brain, reflected by increased extra‐ and intracellular concentrations of BHB.
It is remarkable that stroke‐induced ketogenesis was almost completely blocked when the β‐blocker propranolol was administered before stroke induction (Figure 6). At the same time, glucose levels in mice on the soybean oil diet increased under propranolol. This effect of propranolol is a preliminary finding, but it deserves further study. Brain ischemia is known to activate sympathetic outflow, which causes the release of adrenaline from the adrenal medulla. Adrenaline is expected to release glucose from the liver, an action that is mediated by β2‐adrenergic receptors. This may explain the reduction of plasma glucose in mice on the standard diet (Figure 6). Sympathetic activation in the liver may also occur; however, previous work demonstrated that this is not a signal for ketogenesis.25 Adrenaline as well as sympathetic outflow is also known to increase free fatty acids in the blood, an effect that favors ketone body formation.26, 27 Propranolol may reduce BHB formation indirectly by lowering free fatty acids, but this idea was not tested in the present study. Alternatively, propranolol may also have central effects on metabolic signaling,28 or it may influence plasma levels of insulin and glucagon, which are also known to regulate ketogenesis.29 Once formed, BHB may exert a positive influence on sympathetic activity mediated by hydroxycarboxylic acid (HCA2) receptors or free fatty acid (FFA3) receptors receptors.30, 31 Clearly, additional studies are required to identify the role of β‐receptors in stroke‐induced ketogenesis.
Finally, we also measured intermediate metabolites of the citric acid cycle for several reasons. First, succinate has raised a lot of interest as a receptor agonist and a potential mediator of antioxidant responses,32, 33 and succinate was also postulated to be a product of triheptanoin metabolism expected to replenish succinate for the citric acid cycle.10, 11, 12, 13 In this study, however, succinate was low in plasma and microdialysate (<1 μmol/L; data not shown) and, more important, was not significantly changed in either liver or brain by diets or stroke. Because triheptanoin caused strong ketogenesis but only limited succinate formation, we conclude that triheptanoin's major action in the present study was not as an anaplerotic compound; rather, it served as a source of ketone bodies under ischemic conditions (Figures 1 and 2). Concentrations of fumarate and malate, downstream products of succinate in the citric acid cycle, were unchanged by diets or stroke (data not shown). Citrate is the first metabolite of the citric acid cycle and is formed from oxalacetate and acetyl‐CoA. To test whether increased acetyl‐CoA and citrate formation played a role in energy metabolism under ischemic conditions, we measured citrate levels (Figure 5). We found citrate levels to be higher in mice on the soybean oil diet, a finding that may be due to more acetyl‐CoA for citrate synthesis being formed from fatty acids in the diet. Brain ischemia caused an increase of citrate levels in liver and brain of mice on the standard diet and equalized citrate levels between the 2 groups in all 3 compartments; therefore, an increase of citrate by the soybean oil diet was seen under basal conditions in liver and brain but not after stroke.
In summary, we described massive hepatic ketogenesis induced by transient brain ischemia in mice fed fat‐rich diets. The mechanism of ketone body formation involves β‐adrenergic receptors, and ketogenesis is accompanied by low glucose concentrations as long as ischemia persists and BHB remains high. The formation of BHB during brain ischemia may be beneficial because BHB can be used as a brain fuel, avoiding the use of glucose and potentially dangerous lactate formation. Because BHB is considered neuroprotective, fat‐rich diets may have neuroprotective properties in stroke‐prone patients.
Sources of Funding
This study was funded by internal funds from Goethe University to Klein.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e005556 DOI: 10.1161/JAHA.117.005556.)28381467
References
- 1. Strong K, Mathers C, Bonita R. Preventing stroke: saving lives around the world. Lancet Neurol. 2007;6:182–187. [DOI] [PubMed] [Google Scholar]
- 2. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker‐Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez‐Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez‐Ruiz F, Perico N, Phillips D, Pierce K, Pope CA III, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De León FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui‐Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study. Lancet. 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robinson M, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60:143–187. [DOI] [PubMed] [Google Scholar]
- 5. Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gibson CL, Murphy AN, Murphy SP. Stroke outcome in the ketogenic state—a systematic review of the animal data. J Neurochem. 2012;123:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech LR. D‐β‐hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci USA. 2000;97:5440–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson‐Lewis V, Ramasamy R, Przedborski S. D‐β‐hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puchowicz MA, Zechel JL, Valerio J, Emancipator DS, Xu K, Pundik S, LaManna JC, Lust WD. Neuroprotection in diet‐induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab. 2008;28:1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borges K, Sonnewald U. Triheptanoin—a medium chain triglyceride with odd chain fatty acids: a new anaplerotic anticonvulsant treatment?. Epilepsy Res. 2012;100:239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roe CR, Brunengraber H. Anaplerotic treatment of long‐chain fat oxidation disorders with triheptanoin: review of 15 years experience. Mol Genet Metab. 2015;116:260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Willis S, Stoll J, Sweetman L, Borges K. Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol Dis. 2010;40:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarzkopf TM, Koch K, Klein J. Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with anaplerotic substance triheptanoin. Neuroscience. 2015;300:201–209. [DOI] [PubMed] [Google Scholar]
- 14. Kiewert C, Mdzinarishvili A, Hartmann J, Bickel U, Klein J. Metabolic and transmitter changes in core and penumbra after middle cerebral artery occlusion in mice. Brain Res. 2010;1312:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwarzkopf T, Horn T, Lang D, Klein J. Blood gases and energy metabolites in mouse blood before and after cerebral ischemia: effect of anesthetics. Exp Biol Med. 2013;238:84–89. [DOI] [PubMed] [Google Scholar]
- 16. Franklin KB, Paxinos G. A Stereotaxic Atlas of the Mouse Brain. San Diego: Academic Press; 1997. [Google Scholar]
- 17. Folch J, Lees M, Sloane‐Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 18. Zhang Y, Kuang Y, Xu K, Harris D, Lee Z, LaManna J, Puchowicz MA. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab. 2013;33:1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chowdhury GM, Jiang L, Rothman DL, Behar KL. The contribution of ketone bodies to basal and activity‐dependent neuronal oxidation in vivo. J Cereb Blood Flow Metab. 2014;34:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunnane SC, Courchesne‐Loyer A, St‐Pierre V, Vandenberghe C, Pierotti T, Fortier M, Croteau E, Castellano CA. Can ketones compensate for deteriorating brain glucose uptake during aging? Ann NY Acad Sci. 2016;1367:12–20. [DOI] [PubMed] [Google Scholar]
- 21. Cahill GF Jr. Fuel metabolism in starvation. Ann Rev Nutr. 2006;26:1–22. [DOI] [PubMed] [Google Scholar]
- 22. Samala R, Klein J, Borges K. The ketogenic diet changes metabolite levels in hippocampal extracellular fluid. Neurochem Int. 2011;58:5–8. [DOI] [PubMed] [Google Scholar]
- 23. Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL. [2,4‐13C2]‐β‐hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab. 2002;22:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guzman M, Blazquez C. Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids. 2004;70:287–292. [DOI] [PubMed] [Google Scholar]
- 25. Gardemann A, Püschel GP, Jungermann K. Nervous control of liver metabolism and hemodynamics. Eur J Biochem. 1992;207:399–411. [DOI] [PubMed] [Google Scholar]
- 26. Keller U, Weiss M, Stauffacher W. Contribution of alpha‐ and beta‐receptors to ketogenic and lipolytic effects of norepinephrine in humans. Diabetes. 1989;38:454–459. [DOI] [PubMed] [Google Scholar]
- 27. Krentz AJ, Freedman D, Greene R, McKinley M, Boyle PJ, Schade DS. Differential effects of physiological vs. pathophysiological plasma concentrations of epinephrine and norepinephrine on ketone body metabolism and hepatic portal blood flow in man. Metabolism. 1996;45:1214–1220. [DOI] [PubMed] [Google Scholar]
- 28. Sakaguchi T, Arase K, Bray GA. Effect of intrahypothalamic hydroxybutyrate on sympathetic firing rate. Metabolism. 1988;37:732–735. [DOI] [PubMed] [Google Scholar]
- 29. Beylot M, Picard S, Chambrier C, Vidal H, Laville M, Cohen R, Cotisson A, Mornex R. Effect of physiological concentrations of insulin and glucagon on the relationship between nonesterified fatty acids availability and ketone body production in humans. Metabolism. 1991;40:1138–1146. [DOI] [PubMed] [Google Scholar]
- 30. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S. Short‐chain fatty acids and ketones directly regulate sympathetic nervous system via G protein‐coupled receptors. Proc Natl Acad Sci USA. 2011;108:8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Won YJ, Lu VB, Puhl HL, Ikeda SR. β‐Hydroxybutyrate modulates N‐type calcium channels in rat sympathetic neurons by acting as an agonist for the G‐protein‐coupled receptor FFA3. J Neurosci. 2013;33:19314–19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ariza AC, Deen PM, Robben JH. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress‐related conditions. Front Endocrinol. 2012;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb‐Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]