

Abstract
Background
Particulate matter (PM; PM 2.5 [PM with diameters of <2.5 μm]) exposure during development is strongly associated with adverse cardiovascular outcomes at adulthood. In the present study, we tested the hypothesis that in utero PM 2.5 exposure alone could alter cardiac structure and function at adulthood.
Methods and Results
Female FVB mice were exposed either to filtered air or PM 2.5 at an average concentration of 73.61 μg/m3 for 6 h/day, 7 days/week throughout pregnancy. After birth, animals were analyzed at 12 weeks of age. Echocardiographic (n=9–10 mice/group) and pressure‐volume loop analyses (n=5 mice/group) revealed reduced fractional shortening, increased left ventricular end‐systolic and ‐diastolic diameters, reduced left ventricular posterior wall thickness, end‐systolic elastance, contractile reserve (dP/dtmax/end‐systolic volume), frequency‐dependent acceleration of relaxation), and blunted contractile response to β‐adrenergic stimulation in PM 2.5‐exposed mice. Isolated cardiomyocyte (n=4–5 mice/group) function illustrated reduced peak shortening, ±dL/dT, and prolonged action potential duration at 90% repolarization. Histological left ventricular analyses (n=3 mice/group) showed increased collagen deposition in in utero PM 2.5‐exposed mice at adulthood. Cardiac interleukin (IL)‐6, IL‐1ß, collagen‐1, matrix metalloproteinase (MMP) 9, and MMP13 gene expressions were increased at birth in in utero PM 2.5‐exposed mice (n=4 mice/group). In adult hearts (n=5 mice/group), gene expressions of sirtuin (Sirt) 1 and Sirt2 were decreased, DNA methyltransferase (Dnmt) 1, Dnmt3a, and Dnmt3b were increased, and protein expression (n=6 mice/group) of Ca2+‐ATPase, phosphorylated phospholamban, and Na+/Ca2+ exchanger were decreased.
Conclusions
In utero PM 2.5 exposure triggers an acute inflammatory response, chronic matrix remodeling, and alterations in Ca2+ handling proteins, resulting in global adult cardiac dysfunction. These results also highlight the potential involvement of epigenetics in priming of adult cardiac disease.
Keywords: air pollution, calcium signaling, cardiac, cardiovascular function, heart failure, in utero, myocardial, myocyte, particulate matter
Subject Categories: Animal Models of Human Disease, Calcium Cycling/Excitation-Contraction Coupling, Contractile function
Introduction
Particulate matter (PM) air pollution has been ranked as the ninth cause of overall disease burden attracting significant attention as a major health concern globally.1 Various clinical and epidemiological investigations have demonstrated that exposure to fine PM air pollution (ambient particles with diameters of <2.5 μm; PM2.5) increases the risk for cardiovascular disease2 (CVD) and correlates with arrhythmias, hypertension, myocardial infarction, and cardiac remodeling, resulting in heart failure.3, 4 Air pollution exposure not only exacerbates preexisting heart conditions, but also plays a pivotal role in the development of CVD, particularly if exposed during the developmental period.
The perinatal period is particularly susceptible to developmental defects attributed to the highly plastic nature of the developing organs. Animal studies have demonstrated that exposure to air pollution during the pre‐ and early postnatal periods results in fetal inflammation, promoting adult disease susceptibility in offspring.5, 6 Furthermore, clinical studies have reported that exposure to high levels of PM2.5 during pregnancy impairs fetal development, resulting in reduced birth weight and preterm birth.7, 8, 9 Other reports showed that human exposure to PM2.5 during pregnancy decreases placental mitochondrial DNA10 as well as causes placental DNA hypomethylation.11 These effects are thought to be attributed to epigenetic reprogramming, where changes in DNA methylation and histone modification have been observed to promote long‐lasting effects on gene expression that alter physiological and cellular function. Here, we hypothesized that air pollution exposure during the crucial intrauterine period of developmental plasticity may have important health implications given that it is ubiquitous and can persistently alter the structure and function of developing organs, leading to adult‐onset disease.
In our previous work, we demonstrated the effects of PM2.5 exposure during the combined in utero and postnatal developmental periods (from gestation until weaning at 4 weeks of age) on adult cardiac dysfunction.12 This prior work demonstrated the importance of the developmental period in influencing an adult disease phenotype; however, it did not determine whether PM2.5 exposure during the in utero or postnatal period alone causes detrimental alterations on adult cardiac function. Therefore, in the present study, we sought to investigate the impact of in utero PM2.5 exposure on adult epigenetic changes and cardiovascular function and potential mechanisms that could define the developmental diseases paradigm as it relates to air pollution exposure and may have strong clinical implications benefitting public health.
Materials and Methods
Animals and Exposure
All animal procedures were conducted according to National Institutes of Health guidelines under an Institutional Animal Care and Use Committee protocol approved at The Ohio State University (Columbus, OH). FVB male and female mice were housed for at least 1 week in our facility before breeding. Dams were paired overnight and exposure was begun the day after a plug was observed. Pregnancy was confirmed and time dated with the presence of a vaginal plug. Pregnant dams were exposed to either PM2.5 or filtered air (FA) for 6 h/day, 7 days/week throughout pregnancy. We used the aerosol concentration system located at the Ohio State University4 for the concentrated PM2.5 exposure from the Columbus, Ohio, region. The average PM2.5 concentration that the dams were exposed to was 73.61 μg/m3. For FA treatment, an identical system was used, except that a high‐efficiency particulate arrestance filter at the inlet to the system was used to remove all ambient particles. After birth, pups were nursed and raised in room air until experiments were conducted at 12 weeks of age. Echocardiography (n=9–10 mice/group), cardiomyocyte function (n=4–5 mice/group), and protein expression (n=6 mice/group) were assessed in both male and female offspring, whereas pressure‐volume (PV) loops (n=5 mice/group), quantitative polymerase chain reaction (qPCR; n=4; 1‐day‐old pups/group; n=5 adult mice/group), and assessment of collagen deposition (n=3) was performed only in male offspring as described below.
Echocardiographic Assessment
At 12 weeks of age, echocardiographic assessments were performed using a 40‐MHz transducer (Vevo 2100; Visualsonics, Toronto, Ontario, Canada). Mice were anesthetized with 1% isoflurane in 100% O2 through a nose cone, and internal body temperature was maintained at 37°C throughout the assessment. Following induction of anesthesia, when the heart rate of the animal returned to normal (500–550 beats per minute [bpm]), parasternal short‐axis views were obtained using a 15‐MHz probe. Cine loops collected from M mode views were analyzed for left ventricular (LV) systolic and diastolic internal dimensions (LVESd and LVEDd) and systolic and diastolic posterior wall thickness (PWTs and PWTd). Percent fractional shortening (%FS) was calculated using the equation: %FS=[(LVEDd−LVESd)/LVEDd×100]. Data were averaged from at least 3 cardiac cycles per mouse. Echocardiographic assessments and analyses were performed according to the American Heart Association leading‐edge technique by an investigator blinded to group assignment.
Pressure‐Volume Loop Analyses
Cardiac hemodynamic measurements were assessed by closed chest using a 1.4 French Millar PV catheter. Mice were anesthetized with ketamine (55 mg/kg) plus xylazine (15 mg/kg) and placed in a supine position on a heating pad. Following a midline neck incision, the underlying muscles were dissected to expose the carotid artery. Using a 4‐0 suture, the artery was tied and the PV catheter was advanced through the artery into the left ventricle of the heart. After 5 to 10 minutes of stabilization, values at baseline and stimulation at varying frequencies (4–10 Hz) were recorded as described previously.13 PV loops were also obtained at varying preloads by inferior vena cava occlusions to obtain Ees. To measure the beta‐adrenergic response, 5 mg/kg of dobutamine was injected intraperitoneally. All of the measurements and analyses were evaluated using LabChart7 (AD Instruments, Colorado Springs, CO).
In Vitro Assessment of Cardiomyocyte Function
Cardiomyocytes were isolated using a standard protocol as described previously.4 Cells were stimulated (1 Hz, 3‐ms duration) with a Myopacer Field‐Stimulator system (IonOptix, Milton, MA) and functional properties of the cells were evaluated using the Sarclen Sarcomere Length Acquisition Module with the Myocam‐S Digital charge‐coupled device camera video imaging system (IonOptix, Milton, MA). Sarcomere percent peak shortening (normalized to baseline length, %PS; cellular corollary of %FS), sarcomere maximal departure and return velocities (+dL/dT, −dL/dT), and sarcomere time to 90% peak shortening (TPS90) and time to 90% relengthening (TR90) were measured to assess inotropic and lusitropic function.
Electrophysiological Recordings
Amphotericin‐B perforated patch clamp technique with a bath temperature of 36±0.5°C was used to assess myocyte electrophysiology. Action potentials (APs) were recorded from isolated cardiomyocytes in a train of 25 traces at 3, 4, and 5 Hz. The average of the last 10 traces (ie, from trace 18 to 27) was used to calculate the action potential duration (APD). APD was calculated at 50% and 90% of repolarization (APD50 and APD90). For current recordings, only recordings with an access resistance <20 MΩ were included in the analyses. Transient outward potassium current (Ito) was elicited as described previously.14
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted from LV tissue using the RNeasy kit (Qiagen, Hilden, Germany), according to the manufacturer's protocols. For qPCR, an aliquot of reverse‐transcription product representing 1 ng of total RNA was amplified using the iQ SYBR Green Supermix kit on a CFX Thermocycler (BioRad, Hercules, CA). Primers for target genes were used at a final concentration of 0.25 to 0.5 μmol/L and normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression. Relative gene expression levels were quantified using the formula 2‐ΔΔCt. Gene‐specific primer sequences are presented in Table. An initial denaturation step at 95°C for 10 minutes was followed by 45 cycles of denaturation (95°C, 1 second), annealing (65°C, 10 seconds), and extension (72°C, 20 seconds).
Table 1.
Primer Sequences Used for PCR Amplification
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Gapdh | CTCACTCAAGATTGTCAGCAATG | GAGGGAGATGCTCAGTGTTGG |
| IL‐6 | CCAAGAGGTGAGTGCTTCCC | CTGTTGTTCAGACTCTCTCCCT |
| IL‐1B | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG |
| Col‐1 | GCTAACGTGGTTCGTGACCGTG | GGTCAGCTGGATAGCGACATC |
| MMP‐9 | CAGGAGTCTGGATAAGTTGGGTC | GGTACTGGAAGATGTCGTGTGAG |
| MMP‐13 | GACAGATTCTTCTGGCGCCT | CATAACTCCACACGTGGTTCTCAG |
| Dnmt1 | ATCCTGTGAAAGAGAACCCTGT | CCGATGCGATAGGGCTCTG |
| Dnmt3a | CTGTCAGTCTGTCAACCTCAC | GTGGAAACCACCGAGAACAC |
| Dnmt3b | AGCGGGTATGAGGAGTGCAT | GGGAGCATCCTTCGTGTCTG |
| Sirt1 | TGATTGGCACCGATCCTCG | CCACAGCGTCATATCATCCAG |
| Sirt2 | GCGGGTATCCCTGACTTCC | CGTGTCTATGTTCTGCGTGTAG |
Col‐1 indicates collagen‐1; Dnmt, DNA methyltransferase; Gapdh, glyceraldehyde 3‐phosphate dehydrogenase; IL, interleukin; MMP, matrix metalloproteinase; PCR, polymerase chain reaction; Sirt, sirtuin.
Qualitative and Quantitative Assessment of Collagen
Morphology of interstitial and perivascular collagen was assessed from the picrosirius red–stained sections using bright‐field microscopy.15
Western Blotting
Protein extracts (20 μg/sample) from LV tissues were analyzed by SDS‐PAGE and subjected to western blotting analyses using specific antibodies and the enhanced chemiluminescence method (SuperSignal West Pico chemiluminescent substrate; Pierce, Rockford, IL). The primary and secondary antibodies used in our experiments were the following: mouse anti‐Na+/Ca2+ exchanger (NCX) and anti‐sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA‐2A; MA3‐926 and MA3‐919, respectively; Thermo Fisher Scientific, Waltham, MA); goat antiphosphorylated phospholamban (p‐PLN; sc‐12963; Santa Cruz Biotechnology, Dallas, TX); rabbit anticalsequestrin (ab62662; Abcam, Cambridge, MA); and rabbit anti‐troponin‐I (TnI; 4004; Cell Signaling Technology, Danvers, MA). Films were scanned and analyzed using Image Lab software (version 4.1; Bio‐Rad). Band density of the protein of interest was normalized to beta‐actin (A1978; Sigma‐Aldrich, St. Louis, MO) or extracellular signal‐regulated kinase 1 and 2 (ERK 1&2; sc‐93 and sc‐154; Santa Cruz Biotechnology).
Statistical Analyses
All data are reported as mean±SEM. Data were analyzed using a Student's t test (2‐tailed), ANOVA for PV loops because multiple heart rates were assessed, and Mann–Whitney U test for parameters with samples size n<5 using Prism software (version 6; GraphPad Software Inc, San Diego, CA). Differences were considered statistically significant if P<0.05.
Results
In Utero PM2.5 Exposure Is Associated With Left Ventricular Remodeling at Adulthood
Twelve‐week‐old mice exposed to PM2.5 in utero demonstrated increased LVESd (2.01±0.07 mm of FA; 2.59±0.08 mm of PM2.5; P<0.001) and LVEDd (3.56±0.07 mm of FA; 3.79±0.05 mm PM2.5; P<0.05) dimensions and reduced PWTs (1.24±0.03 mm of FA; 0.98±0.041 mm of PM2.5; P<0.001) compared with FA‐exposed mice. PWTd was not significantly different (data not shown). Morphological alterations were associated with lower systolic function, as indicated by reduced %FS (43.63±2.05% FA; 33.16±1.63% PM2.5; P<0.001) in PM2.5‐exposed mice compared with FA‐exposed mice (Figure 1). There was no difference in %FS in aged‐matched female mice exposed in utero to FA or PM2.5 (Figure S1A).
Figure 1.
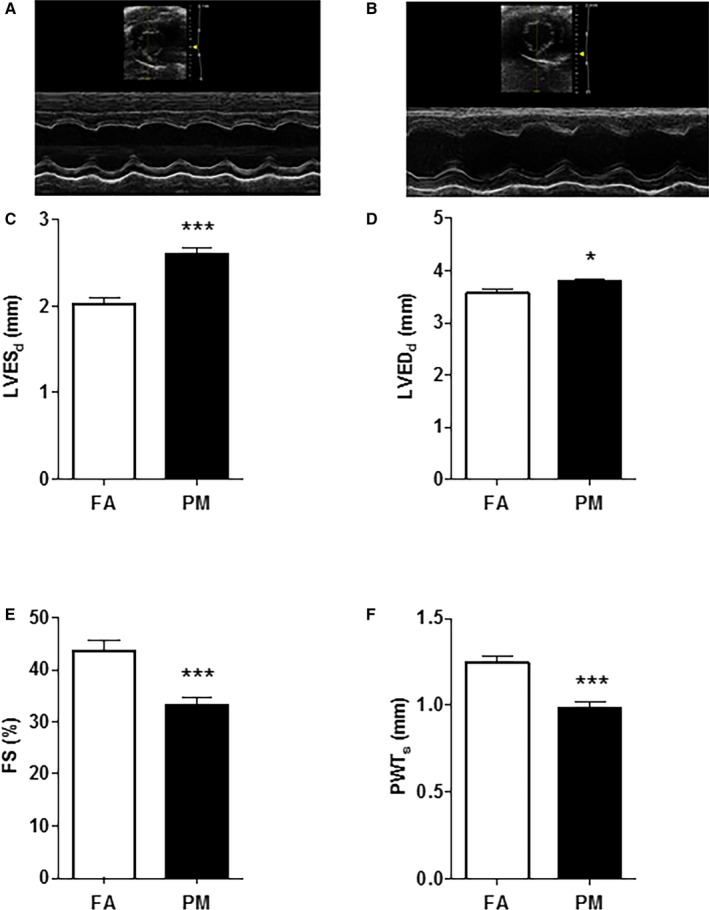
Echocardiographic parameters from adult mice exposed in utero to FA or PM 2.5. Representative M‐mode images obtained from (A) FA; (B) PM 2.5‐exposed mice; (C) left ventricular end‐systolic diameter (LVES d); (D) left ventricular end‐diastolic diameter (LVED d); (E) % fractional shortening (%FS); and (F) posterior wall thickness during systole (PWTs) from n=9 to 10 mice in each treatment group and no more than 2 mice per litter. Five beat cycles were captured and 3 loops averaged per assessment. Results are mean±SEM. *P<0.05; ***P<0.001 vs FA controls. FA indicates filtered air; PM, particulate matter; PM 2.5, particulate matter with diameters of <2.5 μm.
In Utero Exposure to PM2.5 Impairs In Vivo Hemodynamics at Adulthood
Twelve‐week‐old mice exposed to PM2.5 in utero demonstrated diminished contractility measured by end‐systolic elastance (Ees; Figure 2A). We further investigated contractility by increasing heart rate and observed a blunted contractile reserve (dP/dtmax/EDV [end‐diastolic volume]) in in utero PM2.5‐exposed mice (Figure 2B). Not only was contractility impaired, but the frequency‐dependent acceleration of relaxation was also blunted (Figure 2C). Consistent with a heart failure phenotype, mice exposed to PM2.5 in utero also had a blunted contractile response to β‐adrenergic stimulation (dobutamine; Figure 2D). These results suggest that adult mice exposed to PM2.5 in utero have substantial in vivo cardiac dysfunction and corroborate our echocardiography results.
Figure 2.
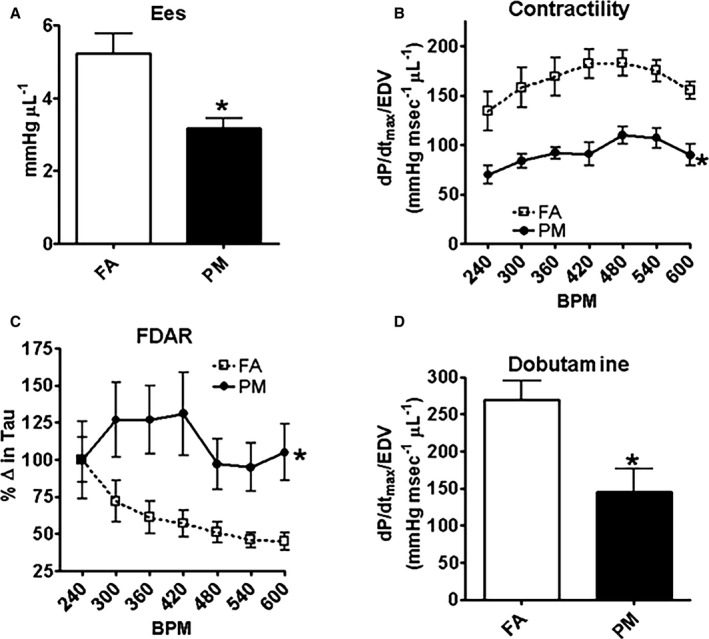
In vivo hemodynamic function from adult mice exposed in utero to FA or PM 2.5. A, End‐systolic elastance (Ees); (B) dP/dtmax/EDV; (C) frequency‐dependent acceleration of relaxation (FDAR); and (D) dP/dtmax/EDV (with β‐adrenergic stimulation). Results are mean±SEM from 5 mice each group. *P<0.05 vs FA controls. BPM indicates beats per minute; EDV, end‐systolic volume; FA, filtered air; PM, particulate matter; PM 2.5, particulate matter with diameters of <2.5 μm.
In Utero Exposure to PM2.5 Impairs Cardiomyocyte Function at Adulthood
Cardiomyocytes isolated from 12‐week‐old mice exposed to PM2.5 in utero showed significant decreases in %PS (12.06±0.6% FA; 8.59±0.41% PM; P<0.001; Figure 3A), −dL/dT (−4.62±0.31 μm/s FA; −3.13±0.46 μm/s PM; P<0.01; Figure 3E), and +dL/dT (4.26±0.25 μm/s FA; 2.87±0.26 μm/s PM; P<0.001; Figure 3D) compared with in utero FA‐exposed mice. TR90 and TPS90 were not different between groups (Figure 3B and 3C). These results corroborated with the echocardiography results, indicating that in vivo cardiac dysfunction is also evident at the cardiomyocyte level. There was no difference in these parameters in aged‐matched female mice exposed in utero to FA or PM2.5 (Figure S1B).
Figure 3.
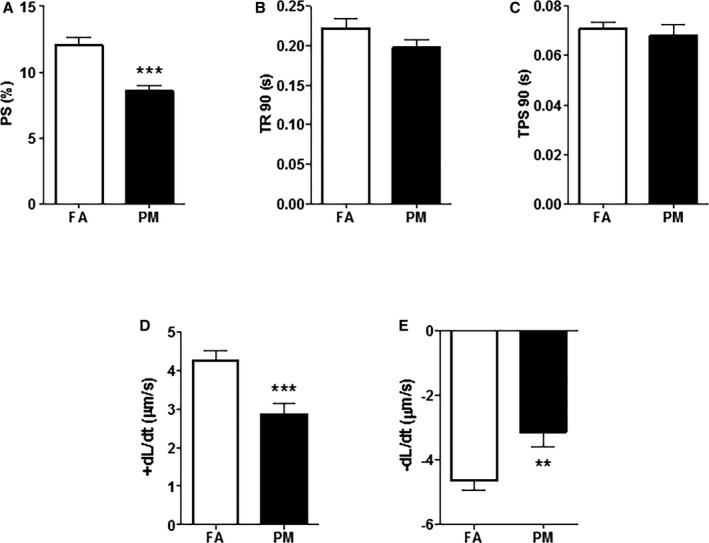
In vitro cardiomyocyte functional parameters obtained from adult mice exposed in utero to FA or PM 2.5. Graphs depicted are representative of (A) peak shortening amplitude (%PS); (B) time‐to‐90% relengthening (TR 90); (C) time‐to‐peak shortening (TPS 90); (D) positive velocity (+dL/dT); and (E) negative velocity (−dL/dT). Results are mean±SEM, n=8 to 10 cells/group from 4 to 5 mice each group. **P<0.01; ***P<0.001 vs FA controls. FA, filtered air; PM, particulate matter with diameters of <2.5 μm; PS, peak shortening.
In Utero PM2.5 Exposure Results in Prolonged Action Potential Duration at Adulthood
We observed significant prolongation in APD90 at 4 and 5 Hz in cardiomyocytes isolated from 12‐week‐old in utero PM2.5‐exposed mice compared with FA‐exposed mice (Figure 4). Analysis of current recordings demonstrated no difference in transient outward current (Ito) density and the corresponding slope conductance between cells obtained from PM2.5‐ and FA‐exposed mice (data not shown).
Figure 4.
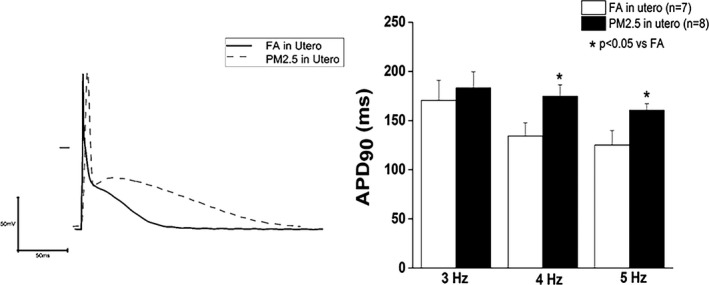
Representative action potential at 3, 4, and 5 Hz in cardiomyocytes obtained from either FA or PM 2.5 in utero exposed adult hearts. Results are mean±SEM, n=7 to 8 cells/group from 4 to 5 mice each group. *P<0.05 vs FA controls. ADP90 indicates 90% action potential duration; FA, filtered air; PM 2.5, particulate matter with diameters of <2.5 μm.
In Utero PM2.5 Exposure Results in an Inflammatory and Profibrotic Response in 1‐Day‐Old Neonates
We analyzed cardiac inflammation and fibrosis at birth and 12 weeks of age in mice exposed to FA or PM2.5 in utero. qPCR analysis of 1‐day‐old mouse hearts exposed to PM2.5 showed a significant increase in expression of interleukin‐6 and ‐1β (IL‐6, IL‐1ß; Figure 5A and 5B), collagen‐1 (Col‐1; Figure 5C), matrix metalloproteases 9 and 13 (MMP‐9, MMP‐13; Figure 5D and 5E). At adulthood, expression level of MMP‐13 remained significantly increased in the in utero PM2.5‐exposed group (2.69±0.64‐fold; P<0.05).
Figure 5.
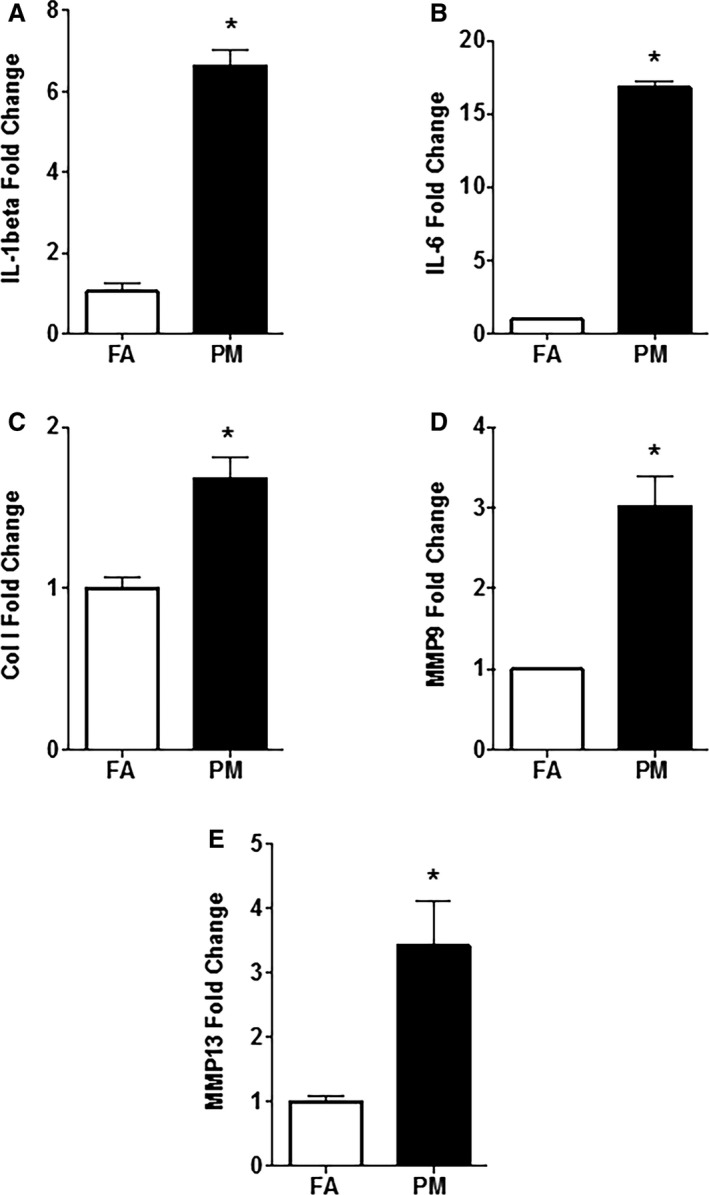
Quantitative polymerase chain reaction (qPCR) analysis of RNA samples obtained from newborn in utero FA‐ and PM 2.5‐exposed mice hearts (n=4 mice from each group). A, IL‐6; (B) IL‐1β; (C) Col‐1; (D) MMP‐9; (E) MMP‐13. Results are mean±SEM. *P<0.05 vs FA controls. Col‐1 indicates collagen‐1; FA, filtered air; IL, interleukin; MMP, matrix metalloproteinase; PM, particulate matter; PM 2.5, particulate matter with diameters of <2.5 μm.
In Utero PM2.5 Exposure Is Associated With Increased Cardiac Collagen Deposition in the Adult Heart
Morphometric analyses of picrosirius red–stained histological sections indicated an increase in collagen deposition in hearts of 12‐week‐old mice that were exposed to PM2.5 in utero compared with FA‐exposed mice (Figure 6).
Figure 6.
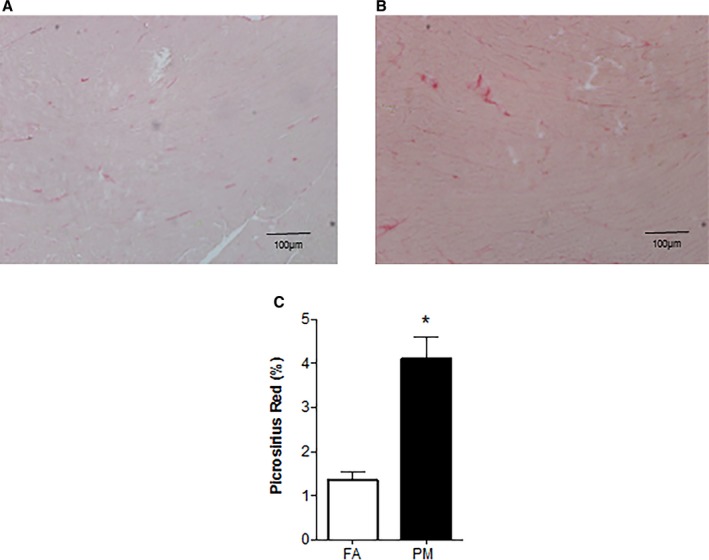
A, Collagen deposition from 12‐week‐old mice (n=3 mice from each group) that were exposed in utero to (A) FA; (B) PM 2.5; and (C) morphometric analyses of areas stained positive for picrosirius red. Original magnification ×10, Scale bar=100 μm. *P<0.05 vs FA controls. FA indicates filtered air; PM, particulate matter with diameters of <2.5 μm.
In Utero PM2.5 Exposure Alters Baseline Expression and Remodeling of Calcium Homeostatic Proteins in the Adult Heart
Cardiac SERCA‐2A (1.11±0.14 FA; 0.59±0.10 PM2.5; P<0.05), p‐PLN (0.39±0.05 FA; 0.22±0.03 PM2.5; P<0.05), and NCX (1.03±0.07 FA; 0.67±0.06 PM2.5; P<0.01) were significantly decreased in 12‐week‐old in utero PM2.5‐exposed mice, suggesting alterations in mechanisms that handle Ca2+ release and reuptake into the sarcoplasmic reticulum during in utero development (Figure 7). There was no difference in expression of calsequestrin and TnI between both groups (Figure S2). Interestingly, protein expression of SERCA‐2A, NCX, and p‐PLN was not different in female mice that were exposed to PM2.5 during in utero development (corroborating the in vivo and in vitro functional data; Figure S3).
Figure 7.
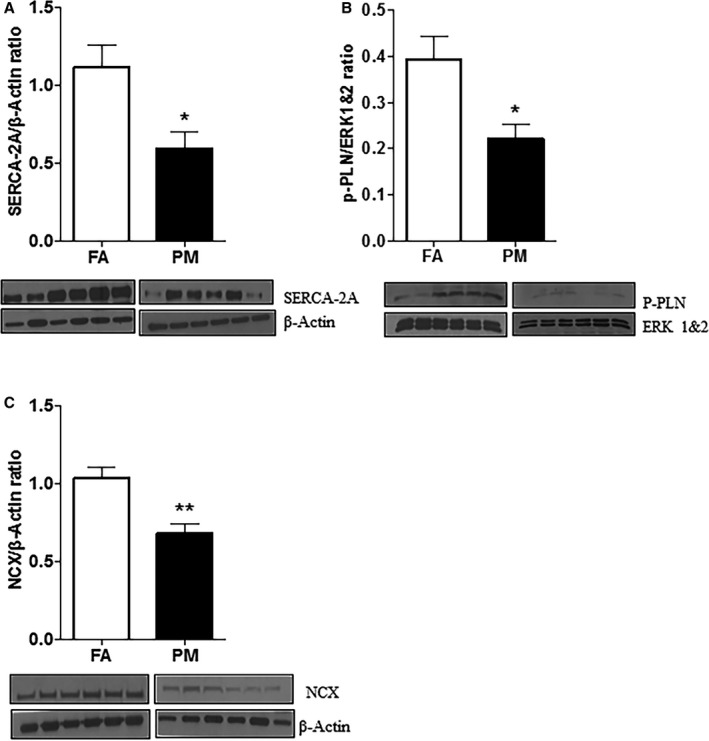
Protein analyses and representative western immune blots from whole adult heart homogenates of in utero FA‐ and PM 2.5‐exposed mice (n=6 mice each group). A, SERCA‐2A; (B) p‐PLN; and (C) NCX. Results are mean±SEM. *P<0.05, **P<0.01 vs FA controls. ERK 1&2 indicates extracellular signal‐regulated kinase 1 and 2; FA, filtered air; NCX, Na+/Ca2+ exchanger; p‐PLN, phosphorylated phospholamban; PM, particulate matter with diameters of <2.5 μm; SERCA‐2A, Ca2+‐ATPase.
In Utero PM2.5 Exposure Alters Epigenetic Mechanisms in Adult Heart
mRNA samples obtained from 12‐week‐old mouse hearts exposed to PM2.5 showed a significant decrease in expression of sirtuins (Sirt1 and Sirt2; Figure 8A and 8B) and increase in expression of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b; Figure 8C through 8E).
Figure 8.
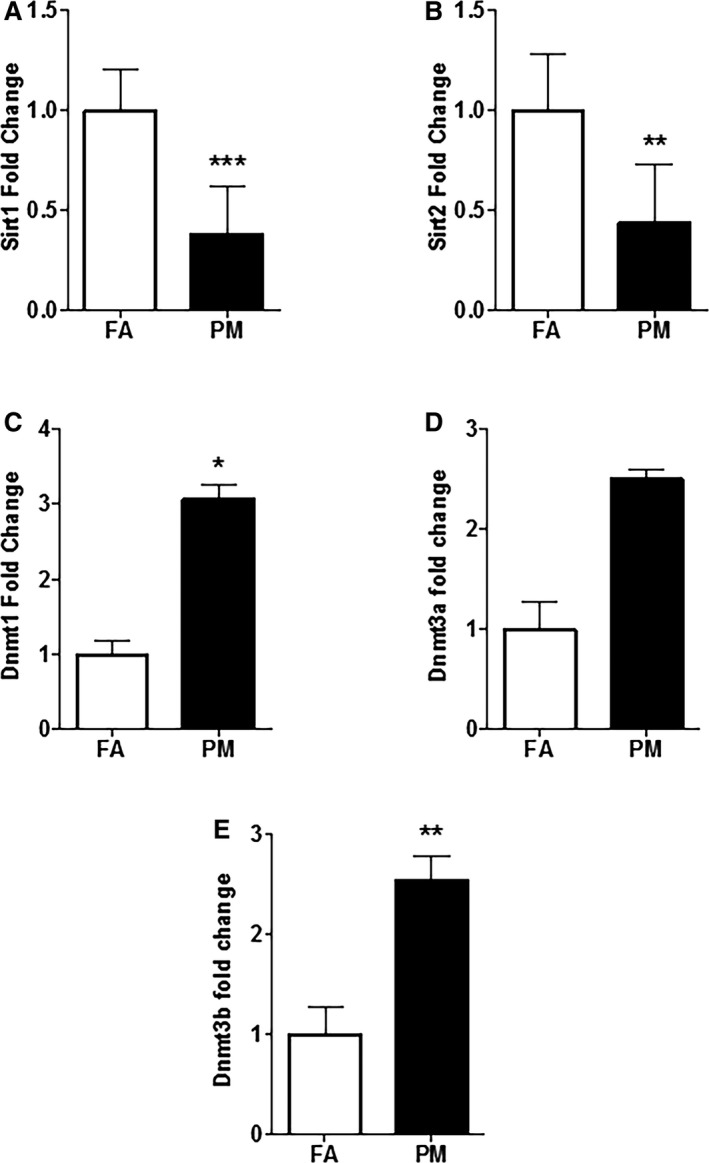
Quantitative polymerase chain reaction (qPCR) analysis of RNA samples obtained from in utero FA‐ and PM 2.5‐exposed adult mice hearts (n=5 mice from each group). A, Sirt1; (B) Sirt2; (C) Dnmt1; (D) Dnmt3a; and (E) Dnmt3b. Results are mean±SEM. *P<0.05; **P<0.01; ***P<0.001 vs FA controls. Dnmt indicates DNA methyltransferase; FA, filtered air; PM, particulate matter with diameters of <2.5 μm; Sirt, sirtuin.
Discussion
In the present study, we provide evidence that exposure of pregnant dams to PM2.5 (73.61 μg/m3) promotes significant cardiac dysfunction in male offspring at adulthood, which is manifested by in vivo LV remodeling and dysfunction, in vitro cardiomyocyte dysfunction, acute inflammation, chronic matrix remodeling, fibrosis, and alterations in calcium homeostasis. We have recently reported that combined in utero and perinatal exposure to PM2.5 caused cardiovascular dysfunction at adulthood.12 However, most important, the current study indicates that maternal PM2.5 exposure during intrauterine development alone results in an adult CVD phenotype. Taken together, our data expand the fetal origins of adult diseases (Barker) hypothesis to include exposure to air pollutants merely during pregnancy, a critical period of intrauterine development as a potential hazard resulting in adult susceptibility and onset of CVDs.
Changes in LV wall dimension and thickness affect chamber size, in a form of cardiac remodeling that alters demand‐supply ratio and impairs overall cardiac performance. In concordance with our previous observation,12 we observed cardiac remodeling in adult mice that were exposed to PM2.5 only during in utero development, as shown by decreased %FS and increased LVESd, LVEDd, and PWTs. PV loop analysis also corroborated the echocardiography data. Furthermore, cells isolated from in utero PM2.5‐exposed hearts manifested compromised contractile function at the cellular level, as indicated by decreased %PS and ±dL/dT, and electrical remodeling, as shown by prolonged APD90.
The mechanisms by which in utero exposure to PM2.5 promotes adult CVD are poorly understood. However, an understanding of the underlying molecular events may help to define this process. AP prolongation is consistently observed in human and animal models of dilated cardiomyopathy, hypertrophy, and heart failure.16, 17 In the present study, mice exposed to environmentally relevant concentrations of PM2.5 during the in utero period demonstrated significant prolongation in APD with no changes in Ito. The reductions in calcium regulatory proteins (discussed below) interact in complicated ways when considering [Ca2+]i and NCX as a driver of delayed repolarization. The increases in interleukins (as observed in our study) do raise an alternative explanation for the APD prolongation as both IL‐1 and IL‐6 have been implicated in repolarization prolongation through a number of different mechanisms.18, 19 Thus, the prolongation of APD90 is independent of a reduction in Ito and likely reflects reductions in other repolarizing currents. It is possible that the observed increased in cytokines and other inflammatory markers contribute to the delayed repolarization. A second mechanism may be mediated through cardiac inflammation that is regarded as an inciting stimulus to various cardiac pathophysiological events causing functional deterioration and matrix remodeling. Acute and chronic PM2.5 exposure impairs vascular reactivity and induces systemic inflammation,20, 21 contributing to various manifestations of CVD.22 In the present study, cardiac IL‐6 and IL‐1β mRNA was expressed ≈6‐ and 16‐fold, respectively, in 1‐day‐old pups exposed to in utero PM2.5. These results indicate that maternal PM2.5 exposure during in utero development results in an acute inflammatory response in fetal hearts during a critical phase of development, potentially reprogramming the fetal heart,23 resulting in the observed adult cardiac dysfunction.
The fetal inflammatory reaction following maternal pollutant exposure has been reported previously. Maternal diesel exhaust and air pollution exposure increased cytokine expression in placental tissue24 and the fetal heart, which can promote disease susceptibility in offspring later in life.6, 7 Our results are in concordance with these studies and indicate that PM2.5 is associated with increased inflammation in offspring through maternal exposure. The increased cytokine expression was transient; however, matrix remodeling is ongoing in adult mice that were exposed to PM2.5 during intrauterine development, as indicated by increased MMP expression. These results suggest that a fetal inflammatory response activates adult cardiac dysfunction by triggering remodeling pathways, including activation of extracellular matrix (ECM) proteins and MMPs.25
Cytokine expression precedes the increase in local collagen and MMP activity, a hallmark of fibrosis and contributes to cardiac remodeling.26 Increased mRNA expression of Col‐1 (1.6‐fold), MMP‐9 (3‐fold), and MMP‐13 (≈4‐fold) in newborn mice hearts exposed in utero to PM2.5 in our study suggest cardiac remodeling that was only modestly observed at adulthood, but resulted in increased collagen deposition. Although a profibrotic response has not yet been shown in air pollution studies, our results are in accord with experimental models of myocardial infarction and hypertension, where inflammatory response causes proliferation and differentiation of fibroblasts contributing to synthesis of ECM proteins.27 IL‐1β has also been shown to be upregulated and to activate MMPs that are initially responsible for collagen degradation and subsequent matrix deposition.28 Besides animal studies, clinical data also support the notion that MMPs, especially MMP‐9, are associated with increased LV diastolic dimensions and wall thickness, contributing to cardiac dysfunction.29
The third major finding was persistently altered expression of proteins regulating calcium homeostasis, a common finding in human and animal models of heart failure. In the present study, expression of SERCA‐2A, PLN, and NCX was significantly decreased in adult hearts exposed in utero to PM2.5. Reduced SERCA‐2A expression is correlated with cardiac dysfunction in various animal models30, 31 and human heart failure32 and likely accounts for the slower rate of Ca2+ reuptake by the sarcoplasmic reticulum, which means less Ca2+ is available for contraction (systolic dysfunction). A further consequence of the delay in cell relaxation (diastolic dysfunction) may result from an elevation of diastolic Ca2+, leading to further impairment in overall cardiac function. A decrease in SERCA‐2A also correlates with decreased or altered myocardial force‐frequency response.33
Phospholamban negatively regulates SERCA‐2A given that dephosphorylated PLN is an inhibitor of SERCA‐2A activity. When phosphorylated, inhibition of SERCA‐2A is alleviated and calcium flux into the sarcoplasmic reticulum is increased. We found decreased expression of p‐PLN in adult mice exposed to PM2.5 in utero, which explains the substantially inhibited SERCA‐2A, suggesting that both proteins regulate each other as previously reported.34 Similar findings have been shown previously in animal and human models of heart failure.35, 36
Consistent with previous studies, we also observed a decrease in NCX expression.37, 38 Differences in calcium signaling proteins that were evident at the protein level were not altered at the RNA level. This could be attributed to the fact that mRNA and protein synthesis or degradation can be altered in diseased conditions,39 and transient alterations in mRNA levels have a persistent impact on protein concentrations. No change in expression of calsequestrin and TnI suggests that there is no alteration in mechanisms of calcium binding or contractile machinery.
Last, the decrease in Sirt1 and Sirt2 and increase in Dnmt1, Dnmt3a, and Dnmt3b expression indicated altered epigenomic pathways in our study. Earlier findings demonstrated an increase in DNA deletion after transgenerational exposure of diesel exhaust particles in mice.40 It has also been reported that there is increased DNA methylation with abnormal SERCA‐2A expression in heart failure,41 leading to impaired calcium homeostasis and thus cardiac dysfunction. Furthermore, studies showed Sirt1 upregulation to be protective in heart failure. Sirt1 is shown to be upregulated in dog hearts undergoing pacing induced heart failure.42 Sirt2 is thought to be involved in regulating longevity43 and is also shown to suppress autophagy in neurons.44 Thus, our results suggested that long‐term effects of in utero PM2.5 exposure are also mediated through altered expression of sirtuins and DNA methyltransferases.
In our study, we did not observe development of heart failure features in female offspring at adulthood. Numerous studies documented earlier that prognosis of heart failure is much worse and features develop earlier in males than females45, 46 or females have better cardioprotective mechanisms than males.47, 48 Long‐term analysis of sex‐related differences could prove informative in our future studies. Furthermore, we did not investigate potential confounders like other environmental pollutants, which contribute to complexity in natural environment that may more closely mimic the clinical scenario. Additionally, attributed to practical reasons the mice in our experiment were cycled between 6‐hour bouts of higher PM2.5 concentrations and then FA. How this paradigm affected the results compared to constant, low‐level PM2.5 inhalation cannot be predicted.
In conclusion, this study describes potential molecular mechanisms responsible for in utero PM2.5‐exposure–induced adult cardiac dysfunction. Although in utero exposure of PM2.5 caused a modest alteration in cardiac function in vivo and in vitro, this could also serve as an important indicator that these animals are more susceptible to developing features of heart failure if subjected to conditions of increased myocardial demand. These results also indicate that PM2.5 can gestationally reprogram developing hearts and therefore provides evidence to study more in‐depth epigenetic mechanisms (global DNA methylation and gene‐specific methylation) responsible for the observed cardiac dysfunctions.
Sources of Funding
This work was supported by an American Heart Association Predoctoral Fellowship (16700011 to Gorr) and two National Institutes of Health grants (R01ES019923 and R01NR012618) to Wold.
Disclosures
None.
Supporting information
Figure S1. Echocardiographic and in vitro cardiomyocyte functional parameters obtained from adult female mice exposed in utero to FA or PM2.5. A, Fractional shortening (%FS) from n=5 to 7 mice in each treatment group and no more than 2 mice per litter; (B) peak shortening amplitude (%PS); (C) time‐to‐90% relengthening (TR90); (D) time‐to‐peak shortening (TPS90); (E) positive (+dL/dT); and (F) negative (−dL/dT). Results are mean±SEM, n=6 to 8 cells/group from 4 to 5 mice each group. FA indicates filtered air; PM2.5, particulate matter with diameters of <2.5 μm
Figure S2. Protein analyses and representative western immune blots from whole adult male heart homogenates of in utero FA‐ (n=4 mice; lanes 1–4) and PM2.5‐exposed mice (n=5 mice; lanes 5–9). A, TnI and (B) calsequestrin. ERK 1&2 was used as loading control. ERK 1&2 indicates extracellular signal‐regulated kinase 1 and 2; FA, filtered air; PM2.5, particulate matter with diameters of <2.5 μm; TnI, troponin‐1.
Figure S3. Protein analyses and representative western immune blots from whole adult female heart homogenates of in utero FA‐exposed (n=4 mice; lanes 1–4) and PM2.5‐exposed mice (n=5 mice; lanes 5–9). A, NCX and (B) SERCA‐2A and p‐PLN. β‐Actin was used as loading control. FA indicates filtered air; Na+/Ca2+ exchanger; p‐PLN, phosphorylated phospholamban; PM2.5, particulate matter with diameters of <2.5 μm; SERCA‐2A, Ca2+‐ATPase.
(J Am Heart Assoc. 2017;6:e005796 DOI: 10.1161/JAHA.117.005796.)28400369
References
- 1. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair‐Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker‐Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan‐Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD III, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA III, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez‐Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez‐Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stockl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brook RD, Rajagopalan S, Pope CA III, Brook JR, Bhatnagar A, Diez‐Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD; American Heart Association Council on E, Prevention CotKiCD, Council on Nutrition PA and Metabolism . Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. [DOI] [PubMed] [Google Scholar]
- 3. Rich DQ, Schwartz J, Mittleman MA, Link M, Luttmann‐Gibson H, Catalano PJ, Speizer FE, Dockery DW. Association of short‐term ambient air pollution concentrations and ventricular arrhythmias. Am J Epidemiol. 2005;161:1123–1132. [DOI] [PubMed] [Google Scholar]
- 4. Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, Youtz DJ, Wang A, Lucchesi PA, Sun Q, Rajagopalan S. Cardiovascular remodeling in response to long‐term exposure to fine particulate matter air pollution. Circ Heart Fail. 2012;5:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auten RL, Gilmour MI, Krantz QT, Potts EN, Mason SN, Foster WM. Maternal diesel inhalation increases airway hyperreactivity in ozone‐exposed offspring. Am J Respir Cell Mol Biol. 2012;46:454–460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex‐specific manner. FASEB J. 2012;26:4743–4754. [DOI] [PubMed] [Google Scholar]
- 7. Fleischer NL, Merialdi M, van Donkelaar A, Vadillo‐Ortega F, Martin RV, Betran AP, Souza JP. Outdoor air pollution, preterm birth, and low birth weight: analysis of the World Health Organization global survey on maternal and perinatal health. Environ Health Perspect. 2014;122:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Breton CV, Mack WJ, Yao J, Berhane K, Amadeus M, Lurmann F, Gilliland F, McConnell R, Hodis HN, Kunzli N, Avol E. Prenatal air pollution exposure and early cardiovascular phenotypes in young adults. PLoS One. 2016;11:e0150825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleisch AF, Rifas‐Shiman SL, Koutrakis P, Schwartz JD, Kloog I, Melly S, Coull BA, Zanobetti A, Gillman MW, Gold DR, Oken E. Prenatal exposure to traffic pollution: associations with reduced fetal growth and rapid infant weight gain. Epidemiology. 2015;26:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, Fierens F, Penders J, Vangronsveld J, Gyselaers W, Nawrot TS. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect. 2012;120:1346–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, Tarantini L, Schwartz J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119:977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gorr MW, Velten M, Nelin TD, Youtz DJ, Sun Q, Wold LE. Early life exposure to air pollution induces adult cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;307:H1353–H1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang B, Davis JP, Ziolo MT. Cardiac catheterization in mice to measure the pressure volume relationship: investigating the Bowditch effect. J Vis Exp. 2015;100:e52618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonilla IM, Vargas‐Pinto P, Nishijima Y, Pedraza‐Toscano A, Ho HT, Long VP III, Belevych AE, Glynn P, Houmsse M, Rhodes T, Weiss R, Hund TJ, Hamlin RL, Gyorke S, Carnes CA. Ibandronate and ventricular arrhythmia risk. J Cardiovasc Electrophysiol. 2014;25:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89:397–410. [DOI] [PubMed] [Google Scholar]
- 16. Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208–217. [DOI] [PubMed] [Google Scholar]
- 17. Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–H1041. [DOI] [PubMed] [Google Scholar]
- 18. Sordillo PP, Sordillo DC, Helson L. Review: the prolonged QT interval: role of pro‐inflammatory cytokines, reactive oxygen species and the ceramide and sphingosine‐1 phosphate pathways. In Vivo. 2015;29:619–636. [PubMed] [Google Scholar]
- 19. Lazzerini PE, Capecchi PL, Laghi‐Pasini F. Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med. 2015;2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruckerl R, Ibald‐Mulli A, Koenig W, Schneider A, Woelke G, Cyrys J, Heinrich J, Marder V, Frampton M, Wichmann HE, Peters A. Air pollution and markers of inflammation and coagulation in patients with coronary heart disease. Am J Respir Crit Care Med. 2006;173:432–441. [DOI] [PubMed] [Google Scholar]
- 21. Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, Gillen DL, Kleinman MT, Vaziri ND, Longhurst J, Zaldivar F, Sioutas C. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect. 2008;116:898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. [DOI] [PubMed] [Google Scholar]
- 23. Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4:611–624. [DOI] [PubMed] [Google Scholar]
- 24. Fujimoto A, Tsukue N, Watanabe M, Sugawara I, Yanagisawa R, Takano H, Yoshida S, Takeda K. Diesel exhaust affects immunological action in the placentas of mice. Environ Toxicol. 2005;20:431–440. [DOI] [PubMed] [Google Scholar]
- 25. Deardorff R, Spinale FG. Cytokines and matrix metalloproteinases as potential biomarkers in chronic heart failure. Biomark Med. 2009;3:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deten A, Volz HC, Briest W, Zimmer HG. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res. 2002;55:329–340. [DOI] [PubMed] [Google Scholar]
- 27. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. [DOI] [PubMed] [Google Scholar]
- 28. Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, Fishbein MC, Lehman TJ, Arditi M. Interleukin‐1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125:1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Galis ZS, Muszynski M, Sukhova GK, Simon‐Morrissey E, Libby P. Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Ann N Y Acad Sci. 1995;748:501–507. [DOI] [PubMed] [Google Scholar]
- 30. Zarain‐Herzberg A, Afzal N, Elimban V, Dhalla NS. Decreased expression of cardiac sarcoplasmic reticulum Ca(2+)‐pump ATPase in congestive heart failure due to myocardial infarction. Mol Cell Biochem. 1996;163–164:285–290. [DOI] [PubMed] [Google Scholar]
- 31. Kiss E, Ball NA, Kranias EG, Walsh RA. Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)‐ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure‐overload hypertrophy and congestive heart failure. Circ Res. 1995;77:759–764. [DOI] [PubMed] [Google Scholar]
- 32. Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)‐ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. [DOI] [PubMed] [Google Scholar]
- 33. Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force‐frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. [DOI] [PubMed] [Google Scholar]
- 34. Ji Y, Lalli MJ, Babu GJ, Xu Y, Kirkpatrick DL, Liu LH, Chiamvimonvat N, Walsh RA, Shull GE, Periasamy M. Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. J Biol Chem. 2000;275:38073–38080. [DOI] [PubMed] [Google Scholar]
- 35. Huang B, Wang S, Qin D, Boutjdir M, El‐Sherif N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of beta‐adrenergic pathway, G(i) protein, phosphodiesterase, and phosphatases. Circ Res. 1999;85:848–855. [DOI] [PubMed] [Google Scholar]
- 36. Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca(2+)‐sensitivity of SERCA 2a in failing human myocardium due to reduced serin‐16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. [DOI] [PubMed] [Google Scholar]
- 37. Dhalla NS, Dixon IM, Rupp H, Barwinsky J. Experimental congestive heart failure due to myocardial infarction: sarcolemmal receptors and cation transporters. Basic Res Cardiol. 1991;86(suppl 3):13–23. [DOI] [PubMed] [Google Scholar]
- 38. Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, Muelheims G, Ross J Jr, Barry WH. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing‐induced heart failure. Am J Physiol. 1998;275:H1441–H1448. [DOI] [PubMed] [Google Scholar]
- 39. Sainte Beuve C, Allen PD, Dambrin G, Rannou F, Marty I, Trouve P, Bors V, Pavie A, Gandgjbakch I, Charlemagne D. Cardiac calcium release channel (ryanodine receptor) in control and cardiomyopathic human hearts: mRNA and protein contents are differentially regulated. J Mol Cell Cardiol. 1997;29:1237–1246. [DOI] [PubMed] [Google Scholar]
- 40. Reliene R, Hlavacova A, Mahadevan B, Baird WM, Schiestl RH. Diesel exhaust particles cause increased levels of DNA deletions after transplacental exposure in mice. Mutat Res. 2005;570:245–252. [DOI] [PubMed] [Google Scholar]
- 41. Kao YH, Cheng CC, Chen YC, Chung CC, Lee TI, Chen SA, Chen YJ. Hydralazine‐induced promoter demethylation enhances sarcoplasmic reticulum Ca2+ ‐ATPase and calcium homeostasis in cardiac myocytes. Lab Invest. 2011;91:1291–1297. [DOI] [PubMed] [Google Scholar]
- 42. Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–980. [DOI] [PubMed] [Google Scholar]
- 43. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. [DOI] [PubMed] [Google Scholar]
- 44. Inoue T, Nakayama Y, Li Y, Matsumori H, Takahashi H, Kojima H, Wanibuchi H, Katoh M, Oshimura M. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 2014;281:2623–2637. [DOI] [PubMed] [Google Scholar]
- 45. Tamura T, Said S, Gerdes AM. Gender‐related differences in myocyte remodeling in progression to heart failure. Hypertension. 1999;33:676–680. [DOI] [PubMed] [Google Scholar]
- 46. Ranki HJ, Budas GR, Crawford RM, Jovanovic A. Gender‐specific difference in cardiac ATP‐sensitive K(+) channels. J Am Coll Cardiol. 2001;38:906–915. [DOI] [PubMed] [Google Scholar]
- 47. Bae S, Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J Pharmacol Exp Ther. 2005;315:1125–1135. [DOI] [PubMed] [Google Scholar]
- 48. Wang M, Baker L, Tsai BM, Meldrum KK, Meldrum DR. Sex differences in the myocardial inflammatory response to ischemia‐reperfusion injury. Am J Physiol Endocrinol Metab. 2005;288:E321–E326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Echocardiographic and in vitro cardiomyocyte functional parameters obtained from adult female mice exposed in utero to FA or PM2.5. A, Fractional shortening (%FS) from n=5 to 7 mice in each treatment group and no more than 2 mice per litter; (B) peak shortening amplitude (%PS); (C) time‐to‐90% relengthening (TR90); (D) time‐to‐peak shortening (TPS90); (E) positive (+dL/dT); and (F) negative (−dL/dT). Results are mean±SEM, n=6 to 8 cells/group from 4 to 5 mice each group. FA indicates filtered air; PM2.5, particulate matter with diameters of <2.5 μm
Figure S2. Protein analyses and representative western immune blots from whole adult male heart homogenates of in utero FA‐ (n=4 mice; lanes 1–4) and PM2.5‐exposed mice (n=5 mice; lanes 5–9). A, TnI and (B) calsequestrin. ERK 1&2 was used as loading control. ERK 1&2 indicates extracellular signal‐regulated kinase 1 and 2; FA, filtered air; PM2.5, particulate matter with diameters of <2.5 μm; TnI, troponin‐1.
Figure S3. Protein analyses and representative western immune blots from whole adult female heart homogenates of in utero FA‐exposed (n=4 mice; lanes 1–4) and PM2.5‐exposed mice (n=5 mice; lanes 5–9). A, NCX and (B) SERCA‐2A and p‐PLN. β‐Actin was used as loading control. FA indicates filtered air; Na+/Ca2+ exchanger; p‐PLN, phosphorylated phospholamban; PM2.5, particulate matter with diameters of <2.5 μm; SERCA‐2A, Ca2+‐ATPase.