

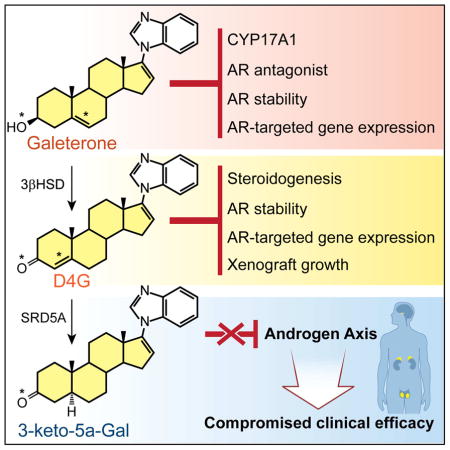
SUMMARY
Galeterone is a steroidal CYP17A1 inhibitor, androgen receptor (AR) antagonist and AR degrader, and was under evaluation in a phase III clinical trial for castration-resistant prostate cancer (CRPC). The A/B steroid ring (Δ5, 3β-hydroxyl) structure of galeterone is identical to cholesterol, which makes endogenous steroids with the same structure (e.g., DHEA and pregnenolone) substrates for the enzyme 3β-hydroxysteroid dehydrogenase (3βHSD). We found that galeterone is metabolized by 3βHSD to Δ4-galeterone (D4G), which is further converted by steroid-5α-reductase (SRD5A) to 3-keto-5α-galeterone (5αG), 3α-OH-5α-galeterone, 3β-OH-5α-galeterone, and in vivo is also converted to the 3 corresponding 5β-reduced metabolites. D4G inhibits steroidogenesis, suppresses AR protein stability, AR target gene expression and xenograft growth comparably to galeterone, and further conversion by SRD5A leads to loss of several activities that inhibit the androgen axis that may compromise clinical efficacy. Together, these findings define a critical metabolic class effect of steroidal drugs with a Δ5, 3β-hydroxyl structure.
eTOC Blurb
Alyamani et al. show that galeterone, a steroidal androgen signaling inhibitor, is converted by enzymes that normally process endogenous steroids to metabolites that have varying activities on androgen signaling pathway components.
INTRODUCTION
Despite an array of improved treatment options that have become available over the past decade, prostate cancer remains the second leading cause of cancer mortality in men in the United States(Siegel et al., 2016). Metastatic prostate cancer generally responds to androgen deprivation therapy (i.e., medical or surgical castration) but the tumor eventually progresses as castration-resistant prostate cancer (CRPC), which develops by way of tumor synthesis of testosterone and/or dihydrotestosterone (DHT) and other mechanisms that stimulate the androgen receptor (AR)(Attard et al., 2015; Dai, In press; Watson et al., 2015). Abiraterone and enzalutamide are 2 next-generation hormonal therapies that have a profound effect on metastatic disease, prolong survival and are FDA-approved for treatment of CRPC. Abiraterone inhibits the enzyme 17α-hydroxylase/17,20-lyase (CYP17A1) (de Bono et al., 2011; Ryan et al., 2012) and thus targets residual androgen synthesis, which suppresses AR signaling, whereas enzalutamide directly antagonizes the AR(Beer et al., 2014; Scher et al., 2012). Unfortunately, tumor resistance eventually develops against both of these agents and some evidence suggests that resistance to abiraterone is yet again engendered by a reinstatement of AR signaling (Attard et al., 2012; Carreira et al., 2014).
New agents and a clearer understanding of drug mechanisms are both urgently required to improve treatment outcomes. Currently under clinical development, Galeterone (Gal), 17-(1H-benzimidazol-1-yl) androsta-5,16-dien-3β-ol, is a steroidal 17-azole compound that inhibits CYP17A1, directly competes with androgens to bind and antagonize AR, promotes AR protein degradation and has clinical activity in a phase I/II clinical trial (Kwegyir-Afful et al., 2015; Montgomery et al., 2016; Njar and Brodie, 2015; Purushottamachar et al., 2013; Yu et al., 2014). Gal shares its Δ5, 3β-hydroxyl structure with abiraterone. The two drugs are distinguished by their C17 moieties – the benzamidazole ring of Gal – and the 3-pyridyl structure of abiraterone. These differences may explain why Gal has been reported to have more direct effects on AR. However, abiraterone is metabolized in vivo to Δ4-abiraterone (D4A), which more potently than abiraterone inhibits the androgen axis (Li et al., 2015), and 5α-abiraterone, which is in contrast an AR agonist that promotes tumor progression (Li et al., 2016). The differing activities of abiraterone and Gal may be due to their respective steroidal metabolites that interact with steroidogenic enzymes and AR that may lead to an incomplete accounting of the context and activities of the parent drugs. However, metabolites of Gal along steroidogenic pathways have not yet been identified. We hypothesized that steroidogenic enzymes act upon Gal and that steroidal metabolites of Gal may in part account for its effects and contribute to distinguishing its activities from abiraterone and its steroidal metabolites (Li et al., 2016; Li et al., 2015).
Here, we report that Gal undergoes metabolism by steroidogenic enzymes that are normally responsible for processing endogenous steroids, in vitro and in vivo, to multiple metabolites detectable by a liquid chromatography mass spectroscopy (LC-MS/MS) method that can distinguish diastereoisomers (Alyamani et al., 2016). Gal is sequentially metabolized by 3β-hydroxysteroid dehydrogenase (3βHSD) to Δ4-galeterone (D4G) and steroid-5α-reductase to 3 5α-reduced metabolites. D4G is also converted in vivo to 3 5β-reduced metabolites. Gal metabolites inhibit multiple androgen synthesis enzymes. D4G binds AR with higher affinity than Gal. However, this was insufficient for increasing the anti-tumor activity of D4G over Gal, which stands in contrast to the activities of D4A conferring more potent antitumor activity over abiraterone (Li et al., 2015). These results suggest that metabolism by steroidogenic enzymes is a common attribute that define a class effect of Δ5, 3β-hydroxyl steroidal drugs with important consequences on resultant downstream metabolites and mechanisms of drug response.
RESULTS
In vitro and in vivo metabolism of galeterone by steroidogenic enzymes
The structural identity in the steroid A and B rings between Gal and endogenous Δ5, 3β-hydroxyl steroids (e.g., DHEA; Fig. 1A), which is also shared with abiraterone, raises the possibility that Gal undergoes metabolism by steroidogenic enzymes that normally physiologically process endogenous steroids. We hypothesized that Gal would be first converted to D4G, which has a Δ4, 3-keto structure. D4G then would be modified by 5α-reductase and/or 5β-reductase activity to produce 3-keto-5α-galeterone (5αG) or 3-keto-5β-galeterone (5βG), respectively. 3-keto-reduction of 5αG or 5βG might reversibly convert 5αG and 5βG to their 3α-OH and 3β-OH congeners, making a total of 6 metabolites downstream of D4G (Fig. 1A Fig S1A and S1B). Two prostate cancer cell lines, LNCaP (which expresses mutant 3βHSD1 with high enzyme activity) and LAPC4 (which expresses wild-type 3βHSD1 with low enzyme activity) (Chang et al., 2013), were used to explore the metabolism of Gal in vitro (Fig. 1B and C). After treating LNCaP and LAPC4 cells with Gal for 24 or 48h, Gal metabolites including D4G, 5αG, 3α-OH-5αG and 3β-OH-5αG were detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Fig S1C). Metabolism of Gal to D4G and to subsequent 5α-reduced metabolites was more robust in LNCaP than LAPC4 cells, consistent with the known high 3βHSD enzyme activity in LNCaP cells. D4G was converted to 5αG, 3α-OH-5αG, and 3β-OH-5αG but not back to Gal, indicating that conversion from Gal to D4G is irreversible. 5αG treatment led to the production of 3α-OH-5αG and 3β-OH-5αG but not D4G or Gal. No 5β-reduced metabolites were detectable, consistent with the absence of 5β-reductase activity in prostate cancer cell lines. We next injected Gal and its metabolites into NSG mice to determine its metabolism in vivo (Fig. 1D). In addition to D4G and the three 5α-reduced metabolites, three 5β-reduced metabolites – 5βG, 3α-OH-5βG and 3β-OH-5βG – were detected in mouse serum after Gal injection. Furthermore, injecting any one 5αG-compound led to detection of all three 5α-reduced metabolites, which indicates that 5αG, 3α-OH-5α-G and 3β-OH-5α-G are interconvertible in vivo. In subsequent studies, we focused on D4G and the 5α-reduced metabolites because 5α-reduction leads to the formation of a planar structure, as occurs with the most potent endogenous androgen, DHT (Bruchovsky and Wilson, 1968; Mostaghel et al., 2010). On the other hand, 5β-reduction of 3-keto steroids introduces a 90° bend, which generally inactivates steroid hormones and facilitates their clearance.
Figure 1. Galeterone metabolism in prostate cancer cell lines and mice.

(A) Schema of galeterone (Gal) metabolism by steroidogenic enzymes. Gal is similar in structure to DHEA therefore it is converted by 3β-hydroxysteroid dehydrogenase (3βHSD) to 3-keto, Δ4 -galeterone (D4G). D4G can be 5α-reduced or 5β-reduced to yield 5α-galeterone (3-keto-5β-Gal) or 5β-galeterone (3-keto-5α-Gal), respectively. 5β-Gal or 5α-Gal are further metabolize by 3-keto-reduction to 3β-OH-5β-Gal, 3α-OH-5β-Gal, 3β-OH-5α-Gal and 3α-OH-5α-Gal. See Figure S1 for chemical synthesis and metabolites separation by Mass spec. (B, C) Gal metabolism in prostate cancer cell lines LNCaP (B) and LAPC4 (C) respectively. Cells were treated with 0.1 μM Gal or the indicated metabolite for 24 or 48 h. Metabolites from the medium were extracted and detected by liquid chromatography–tandem mass spectrometry (LC–MS/MS) in triplicate. (D) Gal metabolites detected in mice after treatment with Gal, D4G and all 3 5α-Gal metabolites. Treatment with Gal or D4G results in detection of all six 5(α/β)-reduced metabolites. Treatment with any of the three 5α-Gal metabolites results in detection of the other two 5α-reduced metabolites, demonstrating reversible interconversion by way of 3-keto reduction and 3-OH-oxidation. n = 3 mice per group. Error bars represent s.d. in B and C, D.
We hypothesized that steroidogenic enzymes directly metabolize Gal. Expression of 3βHSD1 in HEK-293 cells permitted the conversion from Gal to D4G (Fig. 2A). Similarly, expression of SRD5A1 or SRD5A2 resulted in the conversion of D4G to 5αG (Fig. 2B). In LAPC4 cells with endogenous SRD5A1 activity, the SRD5A inhibitor LY191704 or dutasteride blocked the conversion from D4G to 5αG (Fig. 2C). Stable SRD5A1 knockdown with short hairpin RNAs similarly ablated metabolism from D4G to 5αG (Fig. 2D). Expression of AKR1C2, the major enzyme that converts endogenous 3-keto steroids such as DHT to their 3α-OH metabolites, enabled the conversion from 5αG to 3α-OH-5α-Gal in a time- and concentration-dependent manner (Fig. 2E). Together, these data support our hypothesis that Gal undergoes direct metabolism by steroidogenic enzymes, similar to endogenous steroids and the structurally related drug, abiraterone.
Figure 2. Steroidogenic enzymes required for Gal metabolism.

(A) 3βHSD1 catalyzes the conversion from Gal to D4G. 293 cells transfected with the indicated amount of plasmid expressing 3βHSD1 or empty vector were incubated with 0.1 μM of Gal for 3 or 6 h. (B) SRD5A catalyzes the conversion from D4G to 3-keto-5α-Gal. 293 cells were transfected with indicated plasmids and treated with 0.1 μM D4G. (C) SRD5A inhibitors suppress conversion from D4G to 3-keto-5α-Gal. LAPC4 cells were treated with 0.1 μM D4G together with or without the SRD5A inhibitors dutasteride or LY191704. (D) Silencing SRD5A blocks 5α-reduction of D4G. LAPC4 cells stably expressing short hairpin RNAs targeting SRD5A1 or non-silencing control vector (shCtrl) were treated with D4G. (E) AKR1C2 catalyzes conversion of 3-keto-5α-Gal to 3α-OH-5α-Gal. HEK-293 cells were transfected with vector or AKR1C2-expressing plasmid and incubated with 0.1 μM 3-keto-5α-Gal. Metabolites in all experiments were detected by LC-MS/MS. Error bars represent s.d. for experiments performed in triplicate.
Effects of galeterone metabolites on steroidogenic enzyme activity
We further investigated the effects of Gal metabolites on the androgen pathway. D4A, the abiraterone metabolite that most potently inhibits steroidogenesis, was compared with Gal and its metabolites (Li et al., 2015). Although published studies on effects against CYP17A1 expressed in E. coli suggest that Gal may be a more potent CYP17A1 inhibitor than abiraterone (Njar and Brodie, 2015; Yin and Hu, 2014), the comparative effects of their respective metabolites on steroidogenic enzymes is not known. In HEK-293 cells stably expressing CYP17A1, D4A, at a concentration of 10 nM, completely blocked CYP17A1-catalyzed conversion of [3H]-pregnenolone to DHEA. Gal and its metabolites were approximately 100-fold weaker in blocking the production of DHEA (Fig. 3A). This result indicated that Gal and its metabolites are 100-fold less potent than D4A in blocking CYP17A1 activity. In LNCaP cells, which possess high 3βHSD enzyme activity, Gal inhibited the conversion of [3H]-DHEA to Δ4-androstenedione (AD), comparably to D4G and D4A (Fig. 3B). Conversion from D4G to 5αG slightly reduces the capacity to inhibit 3βHSD1 activity. D4G was more potent than Gal and comparable to D4A in inhibiting SRD5A activity, as assessed with the conversion of [3H]-AD to 5α-dione (Chang et al., 2011) in LAPC4 cells (Fig. 3C).
Figure 3. Effects of Gal and its metabolites on steroidogenic enzymes.

(A) Gal and its metabolites are less potent CYP17A1 inhibitors than D4A. HEK-293 cells overexpressing CYP17A1 were treated with [3H]-pregnenolone, in the presence of D4A (0.1, 1 and 10 nM), or Gal and its metabolites (1, 10 and 100 nM), for 3 and 6 h, and conversion to DHEA was assessed by HPLC. (B) D4A, D4G and Gal have comparable potencies as 3βHSD inhibitors. LNCaP cells were treated with [3H]-DHEA and the indicated drugs at 0.1, 1.0, and 10 μM for 24 and 48 h. (C) D4G is a more potent inhibitor of SRD5A than Gal and 5α-Gal. LAPC4 cells were treated with [3H]-AD and 1, 5 and 10 μM of the indicated drugs for 8 and 24 h, and flux to 5α-dione was assessed by HPLC. Arrows indicate equal compound concentrations for ease of comparison. Error bars represent s.d. for experiments performed in triplicate. All experiments were performed at least three times.
The effect of galeterone and its metabolites on AR and AR signaling
Gal has been reported to directly bind to and enhance the degradation of AR (Yu et al., 2014). Conversion from Gal to D4G and then to 5αG would provide a 3-keto structure that is shared by both testosterone and DHT, the steroids with the highest affinity for AR (Fig. 1A). To determine the affinity of Gal and its metabolites for AR, we performed a competition assay. The affinity of D4G for mutant AR (expressed in LNCaP, T877A) and wild-type AR (expressed in LAPC4 cells) was greater than that of Gal, comparable to that of D4A, and comparable to or slightly greater than that of 5αG (Fig. 4A–B). With regard to AR stability, at 1 μM, Gal and its metabolites had limited effects on both mutated and wild-type AR stability. However, at a higher concentration (10 μM), Gal and D4G promoted AR degradation in both LNCaP and LAPC4 cells, with D4G being slightly more potent than Gal. 10 μM 5αG enhanced degradation of mutated but not wild-type AR (Fig. 4C). Additional studies of Gal, Abi and their metabolites on the AR variant endogenously expressed in the 22Rv1 model suggest that their effects on the AR variant protein generally parallel effects on the full length AR (Fig. S2).
Figure 4. D4G and 5α-Gal bind AR at least as well as Gal.

(A, B) The affinity of Gal and its metabolites for mutated or wild-type AR. LNCaP (mutated AR) or LAPC4 (wild-type AR) were incubated with [3H]-R1881 with or without the indicated drugs for 30 min. Intracellular radioactivity was normalized to protein concentration. (C, D) The effects of Gal and its metabolites on AR protein. Gal and D4G but not 5α-Gal lead to loss of wild-type AR protein. LNCaP or LAPC4 were treated with indicated drugs for 24h. AR protein was detected by western blot. Experiments were performed at least three times. See Figure S2 for the effects on an AR variant.
To assess the functional consequences of Gal and its metabolites, we investigated their respective effects on expression of androgen-responsive genes. Gal and D4G inhibited DHT-induced AR-target gene expression in LNCaP and LAPC4 cells, comparable to D4A (Fig. 5A–B). To a lesser extent, 5αG also suppressed DHT-induced gene expression. Notably, however, 5αG somewhat increased PSA expression at basal level in the absence of DHT in LNCaP cells. Compared with 3-keto-5α-abiraterone (5α-Abi), a weak AR agonist, 5αG is an even weaker agonist (Fig. 5A–B). Taken together, these data indicate that the conversion from Gal and D4G by SRD5A to 5αG may lead to a diminished effect on AR stability and AR-responsive gene expression.
Figure 5. Direct effects of Gal and its metabolites on AR signaling and xenograft growth.

(A, B) Effects of Gal and its metabolites on AR target gene expression. LNCaP and LAPC4 cells were serum starved for 48 h before treatment with DHT (0.5 or 0.1 nM, respectively) and the indicated drugs (1 μM) for 24 h. Gene expression was assessed by quantitative PCR (qPCR) and normalized to RPLP0 expression and vehicle (Ctrl) treated cells. (C) D4G is as effective as Gal for inhibition of VCaP xenograft progression. NSG mice were castrated and implanted with DHEA pellets before being divided into three groups: vehicle (n=11 mice), Gal (n=11) and D4G (n=11). Tumors were measured three times per week to determine xenograft progression-free survival (PFS; defined as >20% increase in tumor volume). Gal vs vehicle, p=0.011; D4G vs Vehicle, p=0.023; D4G vs Gal, p=0.98. All comparisons were done with log rank test. (D, E) Conversion from Gal and D4G to downstream metabolites occurs in mouse xenografts and serum. Xenograft tissues (VCaP) and sera and xenograft tissues obtained from treated mice were subject to LC-MS/MS analysis for metabolite quantitation.
D4G inhibits tumor progression in a xenograft mouse model
We sought to determine the effect of D4G on tumor growth in a xenograft mouse model, because D4G was comparable to or slightly better than Gal in blocking steroidogenesis, promoting AR degradation, and suppressing DHT-induced gene expression. VCaP xenografts were grown subcutaneously in orchiectomized mice with DHEA pellet implantation to mimic human adrenal androgen physiology. Time from initiation of treatment with D4G, Gal or vehicle to tumor progression (>20% increase in tumor volume) was assessed by generating Kaplan–Meier survival curves and comparing treatment groups with the log-rank test. Gal inhibited xenograft growth (vehicle vs. Gal, p=0.01). Xenograft progression was also significantly delayed in the D4G group compared with the vehicle group (p=0.02) and was no different when compared to the Gal group (D4G vs Gal, p=0.98) (Fig. 5C). LC-MS/MS for Gal and its metabolites on serum and tumors collected at the study end confirmed that conversion to downstream Gal steroidal metabolites is detectable in both xenografts and serum (Fig. 5D–E).
DISCUSSION
CYP17A1 is a clinically validated target for the treatment of CRPC (Alex et al., 2016; Bambury and Rathkopf, 2016; Yoshimoto and Auchus, 2015). Non-steroidal and steroidal CYP17A1 inhibitors are both undergoing pharmacologic and clinical development; however, the consequences of using steroidal vs. non-steroidal drugs are not well understood. Here, we report that Gal, a Δ5, 3β-hydroxyl steroidal drug, is converted to D4G, 5αG and 5 additional 5-(α/β)-reduced metabolites in vitro and in vivo, with important consequences of the downstream metabolites on the androgen axis. We previously showed that abiraterone undergoes steroidogenic metabolism to generate D4A, 5α-Abi and 5 other steroidal metabolites (Li et al., 2016; Li et al., 2015). Therefore, steroidogenic metabolism of drugs with the Δ5, 3β-hydroxyl structure appears to be a class effect, instead of being a property that is unique to a single drug. The activity of these metabolites is a critical issue with broad consequences for drug development across steroid-dependent diseases.
An important property that distinguishes Gal from abiraterone is that Gal has been reported to have direct properties as an AR antagonist and degrader, while CYP17A1 is generally thought to be the main direct target of abiraterone. However, conversion of Gal by 3βHSD and SRD5A to other metabolites that are formed in vitro and in vivo, with varying biochemical activities, clearly has consequences on steroidogenic enzymes and direct effects on AR. Importantly, D4G and 5αG bind AR more potently than Gal, although both of these metabolites downstream of 3βHSD have divergent effects on AR. Interestingly, despite the increased affinity for AR and maintaining AR enhanced degradation activity, the activity of D4G against a xenograft model of CRPC was no better than Gal. It is possible that any increased anti-tumor xenograft activity by D4G was reversed because it is 1 metabolic step closer to 5αG, resulting in higher intratumoral concentrations of the latter (Fig. 5D). This result raises the possibility that SRD5A inhibition to block 5αG synthesis might be beneficial.
Our findings also suggest that the design and use of steroid-based drugs should consider the relative activities of steroidogenic enzymes in prostate cancer. For example, Gal is rapidly converted to D4G and 5αG in cells that have high but not low 3βHSD1 activity (Fig. 1B–C). A published head-to-head comparison of Gal vs. abiraterone in LAPC4 xenografts demonstrated superior activity of Gal (Bruno et al., 2011). However, 3βHSD1 activity is low in LAPC4 cells, and therefore this comparison likely was more selectively focused on Gal and abiraterone. Consequences of downstream Gal metabolites are more likely to be seen in models, tumors and patients that have the 3βHSD1 N367T missense that accumulates and results in high activity(Hearn et al., 2016).
Although we previously confirmed the presence of steroidal abiraterone metabolites in patients (Li et al., 2016; Li et al., 2015), Gal is under study in clinical trials; therefore, we did not have access to clinical samples. However, given the similar metabolic behavior of Gal with abiraterone in prostate cancer models, in vivo and with specific steroidogenic enzymes, it is highly likely that the concentrations of Gal metabolites are significant and similar in patients.
In summary, our results provide for a more complete picture of Gal metabolism and activity. These findings demonstrate that metabolism by steroidogenic enzymes is a class effect of Δ5, 3β-hydroxyl drugs that should be accounted for in preclinical and clinical drug development and distinguishes these agents from non-steroidal inhibitors. The levels of these metabolites and their respective activities are determined by the expression and activity of endogenous steroidogenic enzymes, including 3βHSD and SRD5A. These findings must be considered for the development of better treatment strategies.
STAR * METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Nima Sharifi (Sharifn@ccf.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
LNCaP, 293T and VCaP cells were maintained in RPMI-1640 (LNCaP) or DMEM (293T and VCaP) with 10% FBS. LAPC4 cells were kindly provided by Dr. Charles Sawyers (Memorial Sloan Kettering Cancer Center, New York, NY) and grown in Iscove’s Modified Dulbecco’s Medium with 10% FBS. All experiments with LNCaP and VCaP cells were done in plates coated with poly-DL-ornithine. A 293T cell line stably expressing human CYP17A1 was generated by transfection with plasmid pcDNA3-c17 (a generous gift of Dr. Walter Miller, University of California, San Francisco) and selection with G418 as described (Papari-Zareei et al., 2006). Cell lines were authenticated by DDC Medical (Fairfield, OH) and determined to be mycoplasma-free using primers 5′-ACACCATGGGAGCTGGTAAT-3′ and 5′-GTTCATCGACTTTCAGACCCAAGGCAT3′. All cell lines were grown at 37°C in a humidified 5% CO2 atmosphere.
METHOD DETAILS
High-performance liquid chromatography (HPLC)
Cell Line Metabolism
Cells were seeded and incubated in 12-well plates with 0.2 million cells/well for ~24 h and then incubated with indicated drugs or a mixture of radioactive ([3H]-labeled) and non-radioactive steroids (final concentration, 100 nM; ~1,000,000 cpm/well; PerkinElmer, Waltham, MA) at 37°C. Aliquots of medium were collected at the indicated times. Collected medium was treated with 1,000 units of β-glucuronidase (Helix pomatia; Sigma-Aldrich) at 37°C for 2 h, extracted with 860 μL ethyl acetate:isooctane (1:1), and concentrated under nitrogen gas.
HPLC analysis was performed on a Waters 717 Plus HPLC or an Agilent 1260 HPLC. Dried samples were reconstituted in 100 μL of 50% methanol and injected into the instrument. Steroids and drug metabolites were separated on a Kinetex 100 × 2.1 mm, 2.6 μm particle size C18 reverse-phase column (Phenomenex, Torrance, CA) using a methanol/water gradient at 30°C. The column effluent was analyzed using a dual-wavelength UV-visible detector set at 254 nm or β-RAM model 3 in-line radioactivity detector (IN/US Systems, Inc.) using Liquiscint scintillation cocktail (National Diagnostics, Atlanta, GA). All HPLC studies were conducted in triplicate and repeated at least 3 times in independent experiments. Results are shown as mean ± SD.
Gene expression analysis
Cells were starved with phenol red-free and serum free-medium for at least 48 h and treated with the indicated drugs and/or androgens. RNA was extracted with a GenElute Mammalian Total RNA miniprep kit (Sigma-Aldrich). cDNA was synthesized from 1 μg RNA in a reverse transcription reaction using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Quantitative PCR (qPCR) analysis was conducted in triplicate with primers for PSA, TMPRSS2, and RPLPO (housekeeping gene) in an ABI 7500 Real-Time PCR machine (Applied Biosystems) using iTaq Fast SYBR Green Supermix with ROX (Bio-Rad) in 96-well plates at a final reaction volume of 20 μL. Accurate quantitation of each mRNA was achieved by normalizing the sample values to RPLPO and to vehicle-treated cells.
Western blot analysis
Cells were starved for 48h before treated with indicated drugs for 24h. Whole cell lysate protein was prepared using ice cold RIPA buffer contains proteasome inhibitor cocktail. Western blot was conducted as described previously (Li et al., 2017). Simply, after the measurement of protein concentration with the BCA assay, 30–50 μg total protein were loaded and electrophoresed in 8% SDS-PAGE gel and transferred to the nitrocellulose membrane. After blocking and incubation with the primary antibodies (refer to KEY RESOURCES TABLE), the appropriate secondary antibody was incubated for 1 hour at room temperature. The band with peroxidase activity was detected by chemiluminescent detection system.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-Actin | Sigma-Aldrich | CAT#A2228 |
| AR | Santa Cruz | CAT#sc-815 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | CAT#31430 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | CAT#31460 |
| Biological Samples | ||
| Double charcoal-stripped human serum | Golden West Biological Inc | CAT#SP2050 |
| Mice Serum | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-glucuronidase | Sigma-Aldrich | CAT#G7646-500 |
| Dutasteride | Medkoo Biosciences | CAS#164656-23-9 | |
| Galeterone | Shanghai Forever Synthesis Co., Ltd | NX41765 |
| D4G | Shanghai Forever Synthesis Co., Ltd | NX41765 |
| Acetonitrile | Thermo Fisher Scientific | CAT#A9554 |
| Methanol | Thermo Fisher Scientific | CAT#A454K4 |
| Formic Acid | Thermo Fisher Scientific | CAT#A1170 |
| poly-DL-ornithine | Sigma-Aldrich | CAT#P3655-1G |
| [3H]-DHEA | PerkinElmer | NET 814001MC |
| [3H]-AD | PerkinElmer | NET 926005MC |
| [3H]-Pregnenolone | PerkinElmer | NET 039001MC |
| [3H]-R1881 | PerkinElmer | NET 590250VC |
| NaCl | Thermo Fisher Scientific | CAT#S 271-1 |
| RIPA buffer | Sigma-Aldrich | CAT#R0278 |
| Protease inhibitor cocktail tablet | Roche | 05892791001 |
| DHEA pellets | Innovative research of America | CAT#A-111 |
| Ethyl alcohol | Thermo Fisher Scientific | CAT#AC 615095000 |
| Safflower oil | Sigma-Aldrich | CAT#47120-U |
| Matrigel | Thermo Fisher Scientific | CB40234 |
| Ethyl acetate | Sigma-Aldrich | CAT#34858-4L |
| Iso octane | Thermo Fisher Scientific | CAT#03014 |
| Tert-butyl methyl ether | Thermo Fisher Scientific | CAT#AC 389050025 |
| Liquiscint scintillation cocktail | Thermo Fisher Scientific | CAT#5089990170 |
| See Chemical Synthesis for synthesis for additional compound | This paper | Supplementary methods |
| Critical Commercial Assays | ||
| GenElute Mammalian Total RNA miniprep kit | Sigma-Aldrich | KIT#RTN 350 |
| iScript cDNA Synthesis Kit | Bio-Rad | KIT#1708891 |
| iTaq Fast SYBR Green Supermix with ROX | Bio-Rad | CAT#1725125 |
| LipoD | SignaGen Laboratories | CAT#SL100668 |
| Pierce™ BCA Protein Assay Kit | Thermal Fisher Scientific | CAT#23225 |
| Pierce™ ECL Western Blotting Substrate | Thermal Fisher Scientific | CAT#32209 |
| Experimental Models: Cell Lines | ||
| LNCaP | ATCC | CRL -1740 |
| VCaP | ATCC | CRL -2876 |
| 293T | ATCC | CRL -3216 |
| LAPC4 | Dr. Charles Sawyers | N/A |
| RPMI-1640 | Sigma | CAT#R8758 |
| DMEM | Sigma | CAT#R7509 |
| IMDM | Sigma | CAT#I3390 |
| Fetal bovine serum | Gemini bio products | CAT#900-108 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSG | The Jackson Laboratory | 005557 |
| Software and Algorithms | ||
| Analyst 1.6.2 | ABSciex | https://sciex.com/products/software/analyst-software |
| Breeze 2 | Waters Corporation | http://www.waters.com/waters/en_US/Breeze-2-HPLC/nav.-htm?cid=514256&locale=en_US |
| GraphPad Prism V 5.0 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| 7500 Software V2.0.6 | Thermo | https://www.thermofisher.com/us/en/home/technicalresources/softwaredownloads/applied-biosystems-7500-real-time-pcr-system.html |
| Wallac 1420 V.3 | Perkin elmer | http://www.bionet.nsc.ru/gep/materials/Wallac1420_Manual_Eng_v3_00.pdf |
| Nanodrop 1000 V3.8 | Thermo | http://www.nanodrop.com/Download.aspx?Type=Software&Cat=NanoDrop%201000 |
| SigmaStat V .3.5 | Systat Software Inc. | https://systatsoftware.com/products/sigmastat/ |
Mouse xenograft studies
Male NSG mice, 6 to 8 weeks of age were obtained from the Cleveland Clinic Biological Resources Unit facility. 107 VCaP cells were injected subcutaneously with Matrigel into surgically orchiectomized NSG mice that were implanted with 5 mg 90-day sustained-release DHEA pellets (Innovative Research of American, Sarasota, FL). Once tumors reached 300 mm3 (length × width × width × 0.52), the mice were arbitrarily assigned to vehicle (n=11), galeterone (n=11), or D4G (n=11) treatment groups. Galeterone and D4G (0.15 mmol/Kg/d in 0.10 mL 15% ethyl alcohol in safflower oil solution) were administered via 5 mL/Kg intraperitoneal injection twice daily, 5 days per week for up to 20 days. Control groups were administered 0.1 mL 15% ethyl alcohol in safflower oil solution via intraperitoneal injection every day for 20 days. Once the treatment was started, tumor volume was measured three times per week, and time to increase in tumor volume by 20% was determined. Mice were sacrificed at treatment day 20. The significance of the difference between treatment groups was assessed by Kaplan-Meier survival analysis using a log-rank test in SigmaStat 3.5. Student’s t-test was used to determine significance between different treatments.
AR competition assay
Cells were cultured in serum-free medium for 48 h and then treated with [3H]-R1881 and the indicated concentrations of drugs for 30 min. Cells were washed with 1X PBS 4 times and 0.9% NaCl solution twice before lysis with RIPA buffer. Intracellular radioactivity was measured with a Beckman Coulter LS60001C liquid scintillation counter and normalized to the protein concentration as detected with a Wallac Victor2 1420 Multilabel counter (Perkin Elmer).
Mouse serum and tumor extraction
The metabolites were extracted from 20 μL of mouse serum by adding 280 μL of methanol containing the internal standard (abiraterone). The samples were then vortexed and centrifuged at 12,000 rpm for 10 min, 200 μL from the supernatant were transferred to HPLC vial. In order to extract the metabolites from the tumor, (28.3–63.3 mg) of the tumor were homogenized with 750 μL LC-MS grade water, 75 μL of the internal standard (abiraterone) was added to the mixture, the metabolites were extracted from the homogenate using 2.5 mL of methyl tert butyl ether (Sigma Aldrich, St. Louis, MO), the organic layer was then evaporated and the samples were reconstituted with 300 μL methanol: water (50:50).
Mass spectrometry
Galeterone and its seven steroidal metabolites were determined using the validated LC-MS method for the determination of abiraterone and its seven structurally related metabolites (Alyamani et al., 2016) with slight modifications. The mobile phase consisted of 30% A (0.2% formic acid in water) and 70% B (0.2% formic acid in methanol:acetonitrile, 60:40). Separation of drug metabolites (Fig S1C) was achieved using a Zorbax Eclipse plus 150 × 2.1 mm, 3.5μm C18 column (Agilent, Santa Clara, CA) at a flow rate of 0.2 mL/min. Drug metabolites were ionized using electrospray ionization in positive ion mode.
Chemical Synthesis
The synthesis of 3β-hydroxy, 5α-galeterone (3β-OH, 5αG) started with the key intermediate in 3β-acetoxy-17-chloro-5α-androsta-16-formyl-ene 2, which was obtained by chloroformylation reaction of 3β-acetoxy-5α-androstan-16-one 1 (Obtained from acetylation of 3β-hydroxy-5α-androstan-16-one) using phosphorus oxychloride in chloroform (Fig S1A). Treatment of 2 with benzimidazole in the presence of K2CO3 in DMF at 25°C gave the desired product 3 in nearly quantitative yield. Compound 3 undergo deformylation with 10% palladium on activated charcoal in refluxing benzonitrile to give compound 4 in 84% yield, from which hydrolysis gave the required compound 3β-hydroxy, 5α-galeterone 5. Compound 5 was oxidized by Jone’s reagent (chromium trioxide) to get 3-keto, 5α-galeterone 6. 3α-hydroxy-5α-galeterone was prepared from 3β-hydroxy-5α-galeterone by inverting the stereochemistry at C-3 via Mitsunobu Reaction [PPh3, DIAD (diisopropylazodicarboxylate), and benzoic acid] followed by hydrolysis of benzoate ester by 10% KOH in methanol at 80°C.
The corresponding 5β-galeterone analogs were synthesized using same synthetic strategy (Fig. S1B) starting from etiocholenone (3α-hydroxy-5-androstan-16-one) which was obtained from dehydroxyepiandrosterone (DHEA) in four steps using CuCl-mediated sodium borohydride stereo-selective reduction of 3-keto-Δ4-intermediate as key step. (C. Wang et al. Steroids 78 (2013) 1339–1346). Reaction of compound 10 with benzimidazole in presence of K2CO3 in DMF was done at 80°C instead of 25°C for the 5α-reduced series.
Synthesis of 3β-Acetoxy-17-chloro-5α-androsta-16-formyl-ene (2)
A solution of acetate 1 (12.0 g, 9.02 mmol) in anhydrous chloroform (200 mL) was added dropwise to a cold (0°C) and stirred solution of phosphorous oxychloride (69.0 mL) and dimethylformamide (69.0 mL) under nitrogen. The mixture was allowed to warm to 25°C, then heated to reflux for 5 h, and then stirred at 50°C overnight. The resultant mixture was concentrated under reduced pressure, poured onto ice, and extracted with ethyl acetate. The combined extracts were washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to give a white solid. Purification by flash chromatography using 10% EtOAc/hexanes gave compound 2 (10.0 g, 81%). 1H-NMR (400 MHz, CDC13) δ 0.85 (s, 3H),0.95 (s, 3H), 0.96-1.55 (m, 11H), 1.60–1.73(m, 5H), 1.80–1.84 (m, 2H), 1.97–2.06 (m, 1H), 2.0 (s, 3H), 2.52 (m, 1H), 4.66 (m, 1H), 9.96 (s, 1H).
Synthesis of 3β-Acetoxy- 17-(1H-benzimidazol-1-yl)-5α-androsta-16-formyl-ene (3)
A mixture of compound 2 (10.0 gm, mmol), benzimidazole (9.0 g, mmol), and potassium carbonate (13.0 g, mmol) in dry DMF (60 mL) was stirred at 25 °C under N2 for 1.5 h. The mixture was cooled to 25 °C and added to water, and the solid obtained was extracted with EtOAc. The combined extracts were washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to give a brown solid. Purification by flash chromatography using 1–3% CH3OH/CH2Cl2 gave compound 3 as a pale yellow solid (10.5 g, quant.) 1H-NMR (400 MHz, CDC13) δ 0.86 (s, 6H), 0.89-1.6 (m, 10H), 1.61–1.80 (m, 8H), 2.01(s, 3H), 2.24–2.33 (m, 1H), 2.75 (dd, J = 15.1, 6.06 Hz, 1H), 4.66 (m, 1H), 7.31 (m, 3H), 7.82 (m, 1H), 7.86 (s, 1H), 9.54 (s, 1H).
Synthesis of 3β-Acetoxy- 17-(1H-benzimidazol-1-yl)-5α-androsta-16-ene (4)
A solution of compound 3 (10.5 g, 22.7 mmol) in dry benzonitrile (80 mL) was refluxed in the presence of Pd/C (6 g) for 6 h. After cooling to 25 °C, the catalyst was removed by filtration through a pad of Celite. The filtrate was evaporated, and the residue was purified by flash chromatography using 1% CH3OH/CH2Cl2 to give compound 4 as a pale off-white solid (8.34 gm, 84%). 1H-NMR (400 MHz, CDCl3) δ 0.86 (s, 3H), 0.96 (s, 3H), 0.78-0.9 (m, 1H), 1.0–1.51 (m, 2H), 1.27–1.83 (m, 15H), 2.03 (s, 3H), 2.10–2.18 (m, 1H), 2.34–2.42 (m, 1H), 4.68 (m, 1H), 5.96 (s, 1H), 7.27 (m, 2H), 7.45 (m, 1H), 7.80 (m, 1H), 7.95 (s, 1H).
Synthesis of 17-(1H-benzimidazol-1-yl)-5α-androsta-16-ene-3β-ol (5)
To a solution of acetate 4 (8.32g, 19.2 mmol) in methanol (100.0 mL) and methylene chloride (10 mL) at 0°C was added a solution of KOH in methanol (10%, 50 mL) dropwise. The mixture was allowed to warm to 25°C and was stirred 3 h. The solvent was evaporated under reduced pressure and to the residue was added water, and the mixture was extracted with ethyl acetate. The organic phase was washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to obtain crude material, which was purified by flash chromatography with 2% CH3OH/CH2Cl2 to isolate 5 as white solid (6.52 gm, 87%). 1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 3.62 (m, 1H), 5.95 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H). HPLC = 98 %.
Synthesis of 17-(1H-benzimidazol-1-yl)-5α-androsta-16-ene-3-one (6)
To a solution of compound 4 (800 mg, 1.85 mmol) in acetone (25 mL) and methylene chloride (5 mL), chromic acid 10% (w/v) (8.0 mL) was drop-wise added at 0°C. The mixture was stirred at room temperature for 3 h, and a solution of sodium bicarbonate was then added to a pH = 7. The reaction mixture was extracted with ethyl acetate, and the organic phase was washed with water and dried over Na2SO4. Product was purified by flash chromatography using 1–2% CH3OH/CH2Cl2 as eluent to isolate 6 as an white solid (640 mg, 80%). 1H-NMR (400 MHz, CDC13) δ 0.99 (s, 3H), 1.05 (s, 3H), 1.07–1.15 (m, 2H), 1.30–1.52 (m, 4H), 1.62–1.85 (m, 6H), 1.96–2.43 (m, 8H), 5.96 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.93 (s, 1H). HPLC = 97.4 %.
Synthesis of 17-(1H-benzimidazol-1-yl)-5β-androsta-16-ene-3β-ol (8)
To a solution of 5 (1.45g, 3.71mmol) in THF (25mL) was added triphenylphosphine (1.071g, 4.08mmol) and benzoic acid (453 mg, 3.71mmol). Cooled the reaction to 0°C for 15 min and DIAD (0.85mL, 4.08mmols) was added dropwise. The reaction was slowly warmed to room temperature and stirred for 4 h. On completion of reaction quenched with water and extracted with Ethyl Acetate. Crude product was purified by flash column chromatography using 20% EtOAc/Hexane to get 7 as white solid. 1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 5.25 (bs, 1H), 5.95 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.42 (m,3H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H), 8.04 (dd, 2H). To a solution of benzoate 7 (2.0 g, 4.01mmol) in methanol (20 mL) at 0°C was added a solution of KOH in methanol (10%, 20 mL) drop-wise. The mixture was allowed to warm to 80°C and was stirred 2 h. The solvent was evaporated under reduced pressure and to the residue was added water, and the mixture was extracted with ethyl acetate. The organic phase was washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to obtain crude material, which was purified by flash chromatography with 2% CH3OH/CH2Cl2 to isolate to 8 as white solid (1.5 g, 96%). 1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 3.62 (m, 1H), 5.95 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H). HPLC = 96.7 %.
Synthesis of 3α-Acetoxy-17-chloro-5β-androsta-16-formyl-ene (10)
A solution of acetate 9 (3.0 g, 9.0 mmol) in anhydrous chloroform (60 mL) was added dropwise to a cold (0°C) and stirred solution of phosphorous oxychloride (15 mL) and dimethylformamide (15 mL) under nitrogen. The mixture was allowed to warm to 25°C, then heated to reflux for 5 h, and then stirred at 50°C overnight. The resultant mixture was concentrated under reduced pressure, poured onto ice, and extracted with ethyl acetate. The combined extracts were washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to give a white solid. Purification by flash chromatography using 10% EtOAc/hexanes gave compound 10 (2.59 g, 81%). 1H-NMR (400 MHz, CDC13) δ 0.85 (s, 3H), 0.95 (s, 3H), 0.96–1.55 (m, 11H), 1.60–1.73 (m, 5H), 1.80–1.84 (m, 2H), 1.97–2.06 (m, 1H), 2.0 (s, 3H), 2.51 (m, 1H), 4.71 (m, 1H), 9.97 (s, 1H).
Synthesis of 3α-Acetoxy- 17-(1H-benzimidazol-1-yl)-5β-androsta-16-formyl-ene (11)
A mixture of compound 10 (2.0 g, 5.27 mmol), benzimidazole (1.8 g, 15.2 mmol), and potassium carbonate (2.6 g, 18.8 mmol) in dry DMF (12 mL) was heated at 80°C under nitrogen for 1.5 h. The mixture was cooled to 25 °C and added to water, and the solid obtained was extracted with EtOAc. The combined extracts were washed with brine and dried (Na2SO4). The solvent was removed under vacuum to give a brown solid. Purification by flash chromatography using 1–3% CH3OH/CH2Cl2 gave compound 11 as a pale yellow solid (2.14gm, quant.) 1H-NMR (400 MHz, CDC13) δ 0.86 (s, 6H), 0.89–1.6 (m, 10H), 1.61–1.80 (m, 8H), 2.01(s, 3H), 2.24–2.33 (m, 1H), 2.75 (dd, J = 15.1, 6.06 Hz, 1H), 4.74 (m, 1H), 7.33 (m, 3H), 7.84 (m, 1H), 7.86 (s, 1H), 9.56 (s, 1H).
Synthesis of 3α-Acetoxy- 17-(1H-benzimidazol-1-yl)-5β-androsta-16-ene (12)
A solution of compound 11 (2.14 g, 4.64 mmol) in dry benzonitrile (20 mL) was refluxed in the presence of Pd/C (1.30 g) for 16 h. After cooling to 25 °C, the catalyst was removed by filtration through a pad of Celite. The filtrate was evaporated, and the residue was purified by flash chromatography using 1% CH3OH/CH2Cl2 to give compound 12 as a pale yellow solid (1.5 gm, 75%). 1H-NMR (400 MHz, CDCl3) δ 0.86 (s, 3H), 0.96 (s, 3H), 0.78–0.9 (m, 1H), 1.0–1.51 (m, 2H), 1.27–1.83 (m, 15H), 2.03 (s, 3H), 2.10–2.18 (m, 1H), 2.34–2.42 (m, 1H), 4.74(m, 1H), 5.96 (s, 1H), 7.27 (m, 2H), 7.45 (m, 1H), 7.80 (m, 1H), 7.95 (s, 1H).
Synthesis of 17-(1H-benzimidazol-1-yl)-5β-androsta-16-ene-3α-ol (13)
To a solution of acetate 12 (1.5 g, 3.4 mmol) in methanol (20 mL) and methylene chloride (5 mL) at 0°C was added a solution of KOH in methanol (10%, 15 mL) dropwise. The mixture was allowed to warm to 25°C and was stirred 3 h. The solvent was evaporated under reduced pressure and water was added to the residue, and the mixture was extracted with ethyl acetate. The organic phase was washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to obtain crude material, which was purified by flash chromatography with 1–2% CH3OH/CH2Cl2 to isolate to 13 as yellow solid (410 mg, 92%).1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 3.65 (m, 1H), 5.97 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H). HPLC = 98 %.
Synthesis of 17-(1H-benzimidazol-1-yl)-5β-androsta-16-ene-3-one (14)
To a solution of compound 13 (270 mg, 0.14 mmol) in acetone (25 mL), chromic acid 10% (w/v) (4.5 mL) was drop-wise added at 0°C. The mixture was stirred at room temperature for 3 h, and a solution of sodium bicarbonate was then added to a pH = 7. The reaction mixture was extracted with ethyl acetate, and the organic phase was washed with water and dried over Na2SO4. The crude product was purified by flash chromatography using 1–2% CH3OH/CH2Cl2 as eluent to isolate 14 as an offwhite solid (240 mg, 88%). 1H-NMR (400 MHz, CDC13) δ 0.99 (s, 3H), 1.05 (s, 3H), 1.07–1.15 (m, 2H), 1.30–1.52 (m, 4H), 1.62–1.85 (m, 6H), 1.96–2.43 (m, 8H), 5.96 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.93 (s, 1H). HPLC = 98 %.
Synthesis of 17-(1H-benzimidazol-1-yl)-5β-androsta-16-ene-3β-ol (16)
To a solution of 13 (500 mg, 1.28mmol) in THF (25mL) was added triphenylphosphine (390 mg, 1.48mmol) and benzoic acid (151 mg, 1.23 mmol). After cooling the reaction to 0°C for 15 min DIAD (diisopropylazodicarboxylate) (0.85mL, 1.48mmols) was added dropwise. The reaction was slowly warmed to room temperature and stirred for 2 h. On completion the reaction was quenched with water and extracted with ethyl acetate. The crude product was purified by flash column chromatography using 20% EtOAc/hexanes to get 15 as white solid. 1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 5.27 (bs, 1H), 5.95 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.42 (m,3H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H), 8.04 (dd, 2H). To a solution of benzoate 15 (260 mg, 0.52 mmol) in methanol (5 mL) at 0°C was added a solution of KOH in methanol (10%, 2.5 mL) drop-wise. The mixture was allowed to warm to 80°C and was stirred 8 h. The solvent was evaporated under reduced pressure water was added to the residue, and the mixture was extracted with ethyl acetate. The organic phase was washed with water, brine, and dried (Na2SO4). The solvent was removed under reduced pressure to obtain crude material, which was purified by flash chromatography with 2% CH3OH/CH2Cl2 to isolate to 16 as white solid (205 mg, 97%). 1H-NMR (400 MHz, CDCl3) δ 0.85 (s, 3H), 0.97 (s, 3H), 0.78–0.81 (m, 1H), 0.99–1.50 (m, 10H), 1.60–1.90 (m, 8H), 2.11–2.20 (m, 1H), 2.34–2.41 (m, 1H), 3.62 (m, 1H), 5.95 (dd, J = 1.6, 3.3 Hz, 1H), 7.28 (m, 2H), 7.47 (m, 1H), 7.80 (m, 1H), 7.94 (s, 1H). HPLC = 95.3 %.
QUANTIFICATION AND DATA ANALYSIS
All data fitting and statistical analysis performed using GraphPad version 5.0 for Windows, GraphPad Software Inc, www.graphpad.com. Survival plots were made using SigmaStat Version 3.5. Systat Software Inc, www.Systatsoftware.com. Statistical values including the exact n and statistical significance are also reported in the Figure legends.
Supplementary Material
SIGNIFICANCE.
Androgen synthesis from extragonadal precursor steroids is a central mechanism that drives prostate cancer resistance to medical or surgical castration and the development of castration-resistant prostate cancer (CRPC). A major part of the therapeutic armamentarium for treatment of CRPC is pharmacologic inhibition of CYP17A1, an enzyme required for androgen and estrogen synthesis, which has also been investigated as a target for ER+ breast cancer. Both steroidal and non-steroidal CYP17A1 inhibitors continue to undergo preclinical and clinical development. The major significance of this work is that it demonstrates a class effect of steroidal CYP17A1 inhibitors – namely that they are metabolized by steroidogenic enzymes that normally act upon endogenous steroids – thus drawing a major distinction between steroidal and non-steroidal CYP17A1 inhibitors. These findings are critical for the development of the preclinical and clinical roadmap for CYP17A1 inhibitors.
Highlights.
Galeterone is metabolized by 3βHSD to Δ4-galeterone, also inhibiting the AR axis
Δ4-galeterone (D4G) inhibits CRPC growth comparably to galeterone
D4G is metabolized by 5α-reductase to 5α-reduced galeterone metabolites
5α-reduced metabolites lose AR axis inhibition activity
Acknowledgments
We thank Trevor Penning (University of Pennsylvania) for the AKR1C2 construct and David Russell (University of Texas Southwestern Medical Center) for LY191704. We also thank Dave Schumick for the graphical abstract illustration. This work has been supported in part by funding from a Howard Hughes Medical Institute Physician-Scientist Early Career Award (to N.S.), a Prostate Cancer Foundation Challenge Award (to N.S.), an American Cancer Society Research Scholar Award (12-038-01-CCE; to N.S.), grants from the National Cancer Institute (R01CA168899, R01CA172382, and R01CA190289; to N.S.) and a grant from the US Army Medical Research and Materiel Command (PC121382 to Z.L.), a Prostate Cancer Foundation Young Investigator Award (to Z.L.).
Footnotes
Author Contributions
M.A. generated the hypothesis and performed the mass spectrometry metabolism studies. Z.L. performed gene expression and metabolism studies. J.L. and T.J. performed immunoblot analyses. M.A. and M.B. performed the mouse experiments. S.U. performed chemical syntheses. Z.L., M.A., R.J.A. and N.S. designed the studies and wrote the manuscript. All authors analyzed and discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alex AB, Pal SK, Agarwal N. CYP17 inhibitors in prostate cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2016;8:267–275. doi: 10.1177/1758834016642370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyamani M, Li Z, Upadhyay SK, Anderson DJ, Auchus RJ, Sharifi N. Development and validation of a novel LC-MS/MS method for simultaneous determination of abiraterone and its seven steroidal metabolites in human serum: Innovation in separation of diastereoisomers without use of a chiral column. The Journal of steroid biochemistry and molecular biology. 2016 doi: 10.1016/j.jsbmb.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, de Bono JS. Prostate cancer. Lancet. 2015 doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- Attard G, Reid AH, Auchus RJ, Hughes BA, Cassidy AM, Thompson E, Oommen NB, Folkerd E, Dowsett M, Arlt W, et al. Clinical and Biochemical Consequences of CYP17A1 Inhibition with Abiraterone Given with and without Exogenous Glucocorticoids in Castrate Men with Advanced Prostate Cancer. The Journal of clinical endocrinology and metabolism. 2012;97:507–516. doi: 10.1210/jc.2011-2189. [DOI] [PubMed] [Google Scholar]
- Bambury RM, Rathkopf DE. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol Oncol. 2016;34:348–355. doi: 10.1016/j.urolonc.2015.05.025. [DOI] [PubMed] [Google Scholar]
- Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. The New England journal of medicine. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchovsky N, Wilson JD. The conversion of testosterone to 5-alpha-androstan-17-beta-ol-3-one by rat prostate in vivo and in vitro. The Journal of biological chemistry. 1968;243:2012–2021. [PubMed] [Google Scholar]
- Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AM, Njar VC. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76:1268–1279. doi: 10.1016/j.steroids.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, et al. Tumor clone dynamics in lethal prostate cancer. Science translational medicine. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Heemers H, Sharifi N. Prostate Cancer. Cold Spring Harbor Laboratory; Androgen signaling in prostate cancer. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn JW, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang KH, Carlson R, Rangel L, Reagan K, Davis BJ, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. The Lancet Oncology. 2016;17:1435–1444. doi: 10.1016/S1470-2045(16)30227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6:27440–27460. doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Alyamani M, Zhang A, Chang KH, Berk M, Li Z, Zhu Z, Petro M. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. 2017;6 doi: 10.7554/eLife.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–551. doi: 10.1038/nature17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, Stephenson JJ, Vogelzang NJ, Koletsky AJ, Nordquist LT, Edenfield WJ, et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1356–1363. doi: 10.1158/1078-0432.CCR-15-1432. [DOI] [PubMed] [Google Scholar]
- Mostaghel EA, Geng L, Holcomb I, Coleman IM, Lucas J, True LD, Nelson PS. Variability in the androgen response of prostate epithelium to 5alpha-reductase inhibition: implications for prostate cancer chemoprevention. Cancer research. 2010;70:1286–1295. doi: 10.1158/0008-5472.CAN-09-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem. 2015;58:2077–2087. doi: 10.1021/jm501239f. [DOI] [PubMed] [Google Scholar]
- Papari-Zareei M, Brandmaier A, Auchus RJ. Arginine 276 controls the directional preference of AKR1C9 (rat liver 3alpha-hydroxysteroid dehydrogenase) in human embryonic kidney 293 cells. Endocrinology. 2006;147:1591–1597. doi: 10.1210/en.2005-1141. [DOI] [PubMed] [Google Scholar]
- Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, Ramalingam S, Ates-Alagoz Z, Njar VC. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. Journal of medicinal chemistry. 2013;56:4880–4898. doi: 10.1021/jm400048v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, et al. Abiraterone in Metastatic Prostate Cancer without Previous Chemotherapy. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Hu Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol. 2014;11:32–42. doi: 10.1038/nrurol.2013.274. [DOI] [PubMed] [Google Scholar]
- Yoshimoto FK, Auchus RJ. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1) The Journal of steroid biochemistry and molecular biology. 2015;151:52–65. doi: 10.1016/j.jsbmb.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4075–4085. doi: 10.1158/1078-0432.CCR-14-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.