

SUMMARY
Poor response to cancer therapy due to resistance remains a clinical challenge. The present study establishes a widely prevalent mechanism of resistance to gemcitabine in pancreatic cancer, whereby increased glycolytic flux leads to glucose addiction in cancer cells and a corresponding increase in pyrimidine biosynthesis to enhance the intrinsic levels of deoxycytidine triphosphate (dCTP). Increased levels of dCTP diminish the effective levels of gemcitabine through molecular competition. We also demonstrate that MUC1-regulated stabilization of HIF-1α mediates such metabolic reprogramming. Targeting HIF-1a or de novo pyrimidine biosynthesis, in combination with gemcitabine, strongly diminishes tumor burden. Finally, reduced expression of TKT and CTPS, which regulate flux into pyrimidine biosynthesis, correlates with better prognosis in pancreatic cancer patients on fluoropyrimidine analogs.
Keywords: Pancreatic Cancer, Chemotherapy Resistance, Gemcitabine, Pyrimidine Biosynthesis, Nucleotide Synthesis, Non-oxidative Pentose Phosphate Pathway, Cancer Metabolism, Metabolomics, HIF-1α, MUC1, Mucin
In brief/eTOC blurb
Shukla et al. identify that HIF-1α mediates increased glycolytic flux and de novo pyrimidine biosynthesis, leading to gemcitabine resistance in pancreatic cancer cells Targeting HIF-1α or de novo pyrimidine biosynthesis increases the efficacy of gemcitabine.
INTRODUCTION
Poor response to therapies due to development of resistance in tumors remains a significant clinical challenge and contributes to overall poor patient prognosis. Gemcitabine, a deoxycytidine analog that inhibits DNA replication and thereby arrests tumor growth, is a widely used single-agent chemotherapy for pancreatic cancer (de Sousa Cavalcante and Monteiro, 2014). While still being utilized for the treatment of locally advanced or metastatic pancreatic cancer, the effectiveness of gemcitabine has been constrained by the frequent development of resistance to this drug in most of the treated patients (Heinemann et al., 2000). Recently, FOLFIRINOX (fluorouracil, leucovorin, irinotecan, and oxaliplatin) has been approved as a treatment for patients with advanced pancreatic ductal adenocarcinoma, showing significantly improved overall and median progression-free survival as compared to gemcitabine, albeit, with a less favorable toxicity profile (Conroy et al., 2011). Furthermore, not all patients are equally responsive to FOLFIRINOX. Abraxane or albumin-bound paclitaxel along with gemcitabine is another combination therapy recently approved by the FDA (Von Hoff et al., 2013). Gemcitabine, however, continues to be a part of this therapy. The efficacy of several anti-cancer therapies is linked to tumor cell survival and therefore to their effect on metabolic alterations in tumor cells (Fanciulli et al., 2000). Many previous studies have addressed only changes in the rate of influx or efflux of drug as a way of regulating the concentration of drug in the tumor cells for acquiring chemotherapy resistance. Hence, there is an urgent need to determine the metabolic mechanisms that hamper the efficiency of chemotherapy and to identify combinations that may significantly improve the efficacy of gemcitabine and other fluoropyrimidine analogs.
Large portions of solid tumors are hypoxic (Koong et al., 2000). Furthermore, most solid tumors, particularly pancreatic tumors, also demonstrate an increased accumulation of stromal tissue i.e. desmoplasia. Increased desmoplasia and hypoxia have been implicated in the development of resistance to chemotherapy and radiotherapy (Yokoi and Fidler, 2004). Both desmoplasia and hypoxia also lead to the stabilization of HIF-1α, the master regulator of glucose metabolism (Semenza, 2010). Previous studies from our lab have demonstrated higher expression of HIF-1α and the corresponding downstream glucose metabolism genes in pancreatic tumor cells as compared to the surrounding stroma (Chaika et al., 2012a; Chaika et al., 2012b). Pancreatic cancers also metabolize glucose at higher rates (Dang, 2010; DeBerardinis et al., 2008; Vander Heiden et al., 2009). However, the precise fate of glucose and downstream metabolites and their role in therapy responsiveness is yet to be fully explored. Here, we delineate the metabolic alterations that facilitate gemcitabine resistance, utilizing pancreatic cancer as a model.
RESULTS
Increased glucose metabolism fuels gemcitabine resistance in Gem-R pancreatic cancer cells
To investigate the metabolic basis of gemcitabine resistance, we generated pancreatic cancer cell line (Capan-1, T3M4 and MIA PaCa-2) models with acquired gemcitabine resistance. For this, wild-type (WT) pancreatic cancer cells were cultured with increasing concentrations of gemcitabine over a period of approximately 6 months. The resistance status at each dose was determined by calculating IC50 of gemcitabine using MTT cytotoxicity assays (Figure 1A). At the end of the 6-month treatment period, the cell lines generated (Gem-R) were approximately 500–1000-fold more resistant as compared to WT cells (Figure 1B). We observed an increase in clonogenic ability of the Capan-1 and T3M4 Gem-R cells under control and gemcitabine treatment and increased colony number and size in soft agar assays (Figures S1A and S1B). To quantify the relative physiological glucose uptake by Gem-R cells as compared to the WT cells, we performed [3H]-2-deoxyglucose (2DG) uptake assays after culturing the cells under normoxia (20% oxygen) or hypoxia (1% oxygen) for 12 hr. Capan-1 Gem-R and T3M4 Gem-R cells displayed significantly increased glucose uptake in comparison to the controls, under normoxic as well as hypoxic conditions (Figure 1C). Similar alterations were observed for MIA PaCa-2 Gem-R cells (Figures S1C and S1D). Gem-R cells also demonstrated increased lactate release (Figures 1D and S1E). Expression of genes involved in glucose metabolism was also increased in Gem-R cells (Figure 1E).
Figure 1. Increased glucose metabolism fuels gemcitabine resistance in Gem-R pancreatic cells.
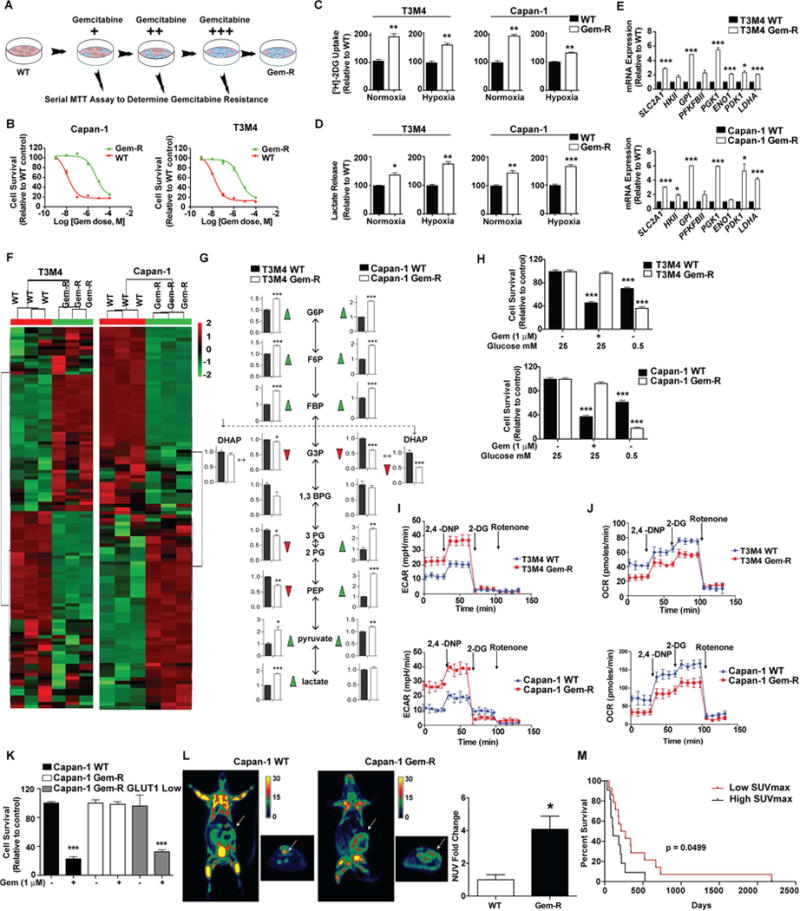
(A) Acquired gemcitabine resistant (Gem-R) Capan-1, and T3M4 pancreatic cancer cells were generated by exposing the corresponding wild-types (WT) to an increasing concentration of gemcitabine over 6 months. The resistance status was confirmed by performing MTT assays at each step. Red and blue colors of cells in the illustration denote sensitivity (or cell death) and resistance (or cell survival), respectively, based of response to gemcitabine treatment for 72 hr. (B) Relative sensitivity of WT as compared to the Gem-R pancreatic cancer cells as determined by MTT cytotoxicity assays. Cells were treated with increasing concentration of gemcitabine and MTT assays were performed 72 hr after treatment. (C) Relative glucose uptake in Gem-R and WT cells cultured under normoxia or hypoxia (1% oxygen). Counts for each group were normalized to the cell count in the respective group and plotted as a percent of WT control. (D) Relative lactate release from Gem-R vs. WT cells as determined by colorimetric analysis. Raw values were normalized to cell counts and plotted as percent of WT control. (E) qPCR analysis for glycolytic genes in Gem-R cells relative to WT cells. (F) Unsupervised hierarchical clustering of significantly deregulated metabolites between cells lines, in Capan-1 WT and Gem-R cells. (G) Major metabolites altered in the glycolytic pathway in Gem-R cells as compared to the WT cells. (H) Effect of glucose deprivation on the growth of WT and Gem-R cells. Cells were cultured in normal and low glucose (0.5 mM) conditions for 36 hr with or without gemcitabine followed by MTT assays. Relative survival is plotted as percent of WT or Gem-R cells cultured in normal glucose. (I–J) WT and Gem-R cells were seeded in 24-well plates and exposed to 2,4-DNP, 2-DG and rotenone to measure ECAR (I) and OCR (J). (K) Relative gemcitabine sensitivity of Capan-1 Gem-R cells FACS-sorted for low GLUT1 expression, as compared to unsorted WT and Gem-R controls as determined by MTT assay values. Cell were treated with gemcitabine for 72 hr. (L) Representative coronal (left) and axial (right) images of [18F]FDG uptake by PET imaging in WT and Gem-R orthotopic implantation models (n=6 mice per group). Normalized uptake values (NUV) fold change for xenografts examined by FDG-PET are presented in the graph on the right. Uptake values were normalized with tumor volume. (M) Kaplan-Meier progression-free survival analysis of PDAC patients on gemcitabine/5-FU chemotherapy with high (SUVmax ≥ 6; n=14) or low (SUVmax < 6; n=11) [18F]FDG-PET signal. For all in vitro studies n=3. Data are represented as mean ± SEM. The bar charts in panels C, D, E, G, and L were compared by Student’s t-test. Data in panels H and K were compared by one-way ANOVA followed by Tukey’s post hoc test. Survival in panel M was compared by Log-rank (Mantel-Cox) test. *p < 0.05, **p < 0.01, and ***p < 0.001 as compared to WT. See also Figure S1 and Table S1.
To investigate the metabolic alterations underlying gemcitabine resistance, we performed Liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS)-based analysis (Gunda et al., 2016) followed by unsupervised hierarchical clustering. LC-MS/MS analysis indicated differential metabolite pools in Gem-R cells compared to WT controls (Figure 1F). Further analyses revealed an increase in the intermediate metabolites of glycolysis. Specifically, we observed an increase in metabolites upstream of dihydroxyacetone phosphate and glyceraldehyde-3-phosphate and increased intracellular pyruvate/lactate levels in Gem-R cells (Figure 1G).
To determine the dependence of the gemcitabine resistance phenotype on glucose metabolism, we cultured the Gem-R cells and the corresponding WT cells under low glucose conditions (0.5 mM glucose) for a short period (36 hr). Gem-R cells failed to respond to gemcitabine treatment but demonstrated significantly diminished survival under low glucose conditions (Figure 1H). Also, the intrinsically gemcitabine resistant AsPC-1 and moderately resistant MIA PaCa-2 cells demonstrated significantly diminished cell survival under glucose-deprived conditions or under treatment with 2-DG (Figures S1F–S1I). Furthermore, in comparison to the WT cells, Gem-R cells demonstrated increased extracellular acidification rate (ECAR) and decreased oxygen consumption rate (OCR) (Figures 1I–1J, S1J). The partially gemcitabine-resistant cells (PR) demonstrated an intermediate increase in ECAR and decrease in OCR compared to the WT cells (Figures S1J–S1L). We sorted Capan-1 Gem-R cells for high and low Glucose transporter 1 (GLUT1) expression. We observed an increased sensitivity to gemcitabine in Gem-R cells sorted for low surface GLUT1 expression (Figures 1K and S1M). However, we did not observe any significant difference in the sensitivity of WT and Gem-R cells against mitochondrial electron transport inhibitors such as metformin or oligomycin (Figure S1N). We also observed increased glucose uptake in tumors derived from orthotopically implanted Gem-R cells in athymic nude mice, compared to that of the WT cells, by performing 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) imaging (Figure 1L). Furthermore, pancreatic cancer patients subjected to fluoropyrimidine analog-based therapies (gemcitabine or 5-FU) demonstrated a poor progression-free survival when high PET signal (SUVmax ≥ 6) was observed in primary tumors (Figure 1M, Table S1-cohort 1). Together these findings establish that gemcitabine resistant cells have an increased dependence on glucose metabolism.
Increased glucose carbon flux through the non-oxidative pentose phosphate pathway (PPP) in Gem-R cells
LC-MS/MS-based metabolomics profiling of Gem-R cells revealed an increase in the steady-state levels of glucose metabolites through the non-oxidative arm of the PPP (Figures 2A and 2B). Moreover, we observed a significant reduction in the levels of oxidative PPP metabolites. Corroborating the metabolite levels, our qPCR findings demonstrated a significant increase in the expression of genes involved in the non-oxidative PPP in Gem-R cells and Gem-R cell-originated tumors (Figures 2C and S2A). Sedoheptulose-1,7-bisphosphate (SBP) was among the PPP metabolites increased in the Gem-R cells (Figure 2B), and it has been proposed that an increase in SBP indicates increased flux in the non-oxidative PPP (Ying et al., 2012). Gemcitabine IC50 did not correlate with the expression levels of glucose-6-phosphate dehydrogenase (G6PD), a key gateway enzyme to the oxidative PPP, in 17 pancreatic cancer cell lines (Figure S2B). Likewise, we found no alteration in the activity of G6PD in Gem-R as compared to WT cells (Figure S2C).
Figure 2. Increased flux of glucose carbon through the non-oxidative pentose phosphate pathway in Gem-R pancreatic cells.
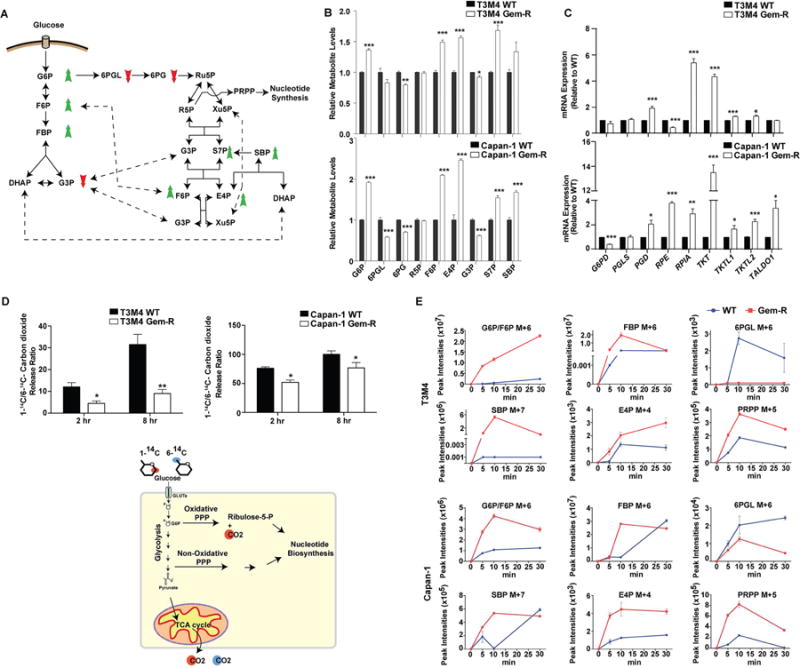
(A) Summary of altered metabolites in Gem-R vs. WT cells. An increase in metabolite levels is indicated with a green upward arrow and a reduction is indicated with a red downward arrow. (B) The PPP metabolite levels in Gem-R relative to WT cells based on targeted LC-MS/MS metabolomics. (C) Relative expression of the PPP genes in Gem-R vs. WT cells determined by qPCR analysis. (D) Radiolabeled CO2 released from 1-14C or 6-14C glucose labeling in Gem-R vs. WT cells. Raw scintillation counts were plotted as a ratio of 1-14C/6-14C-labeled carbon dioxide released at the indicated time points. Below is a schematic illustration of labeled carbon dioxide release generated from 14C-labeled glucose at C1 or C6 positions. (E) Kinetics of incorporation of 13C label from U-13C glucose into PPP metabolites in WT and Gem-R cells, as identified by LC-MS/MS analysis. M+X represents the number of 13C labeled carbon atoms in each metabolite, presented in arbitrary peak intensity units. For all in vitro studies n=3 per sample. Data are represented as mean ± SEM. The bar charts in panels B and C were compared by Student’s t-test. *p < 0.05, **p < 0.01, and ***p < 0.001. G6P: Glucose 6-phosphate; F6P: Fructose 6-phosphate; FBP: Fructose 1,6-bisphosphate; DHAP: dihydroxyacetone phosphate; G3P: glyceraldehyde 3-phosphate; 6PGL: 6-Phosphogluconolactone; 6PG: 6-Phosphogluconate; Ru5P: Ribulose 5-phosphate; R5P: Ribose 5-phosphate; Xu5P: Xylulose 5-phosphate; PRPP: Phosphoribosyl pyrophosphate; S7P: Sedoheptulose 7-phosphate; SBP: Sedoheptulose 1,7-bisphosphate; E4P: Erythrose 4-phosphate. See also Figure S2.
Steady state levels are not always indicative of the flux of metabolites in a pathway, and hence we examined the relative contribution of non-oxidative vs. oxidative PPP by performing 1-14C- and 6-14C-glucose labeling followed by measurement of 14CO2 release. 14CO2 from 1-14C-glucose is released by both the oxidative PPP and the TCA cycle, whereas 14CO2 from 6-14C-glucose can only be released via the TCA cycle. The release ratio of 1-14C/6-14C CO2 was reduced in Gem-R cells as compared to WT cells, indicating decreased flux of glucose through the oxidative PPP in Gem-R cells (Figure 2D). Furthermore, we examined the kinetics of U-13C-glucose labeling of PPP metabolites in WT and Gem-R cells. We observed faster glucose flux into glycolytic metabolites glucose-6-phosphate/fructose-6-phosphate and fructose-1,6-bisphosphate in Gem-R cells, while slower kinetics were observed for oxidative PPP metabolite 6-phosphogluconolactone (Figure 2E). We also observed faster and increased 13C label incorporation in SBP, erythrose 4-phosphate (E4P), and phosphoribosyl pyrophosphate (PRPP) in Gem-R cells compared to WT cells. These results suggest increased glucose carbon flux through non-oxidative PPP in Gem-R cells.
Increased pyrimidine synthesis contributes to gemcitabine resistance in Gem-R cells
LC-MS/MS-based metabolomics followed by metabolic pathway and functional enrichment analysis demonstrated that the pyrimidine synthesis pathway is among the most significantly altered pathways in Gem-R cells as compared to controls (Figure 3A). We performed oligonucleotide array analysis to determine the genes altered in Capan-1 Gem-R vs. WT cells. We observed that Capan-1 Gem-R cells, compared to the WT cells, have increased expression of several genes involved in PPP and nucleotide biosynthesis pathways, including TKT (transketolase), CTPS1 (cytidine triphosphate synthase), TYMS (thymidylate synthetase), NME4 (NME/NM23 nucleoside diphosphate kinase 4), and PRPSAP1 (phosphoribosyl pyrophosphate synthetase-associated protein 1) (Table S2). Correspondingly, we observed significant increases in N-carbamoyl-L-aspartate, dihydroorotate (DHOA), and uridine and cytidine metabolites (Figure 3B), all of which are metabolic intermediates in the pyrimidine synthesis pathway. We also performed qPCR to confirm the levels of genes involved in PPP and purine/pyrimidine nucleotide biosynthesis pathways in WT and Gem-R cells and tumors (Figures 3C and S3A). Combining the microarray data and the qPCR data in both the cell lines, we observed a significant induction of TKT and CTPS (Figures 3C and Table S2), which would increase the flux of glucose carbon into the pyrimidine biosynthesis pathway. Previous studies have indicated that increased cytidine deaminase (CDA) levels may also contribute to gemcitabine resistance (Weizman et al., 2014). However, we did not observe any significant difference in CDA mRNA levels in Gem-R cells compared to WT cells (Figure S3B).
Figure 3. Gem-R cells have higher de novo pyrimidine biosynthesis in vitro and in vivo.
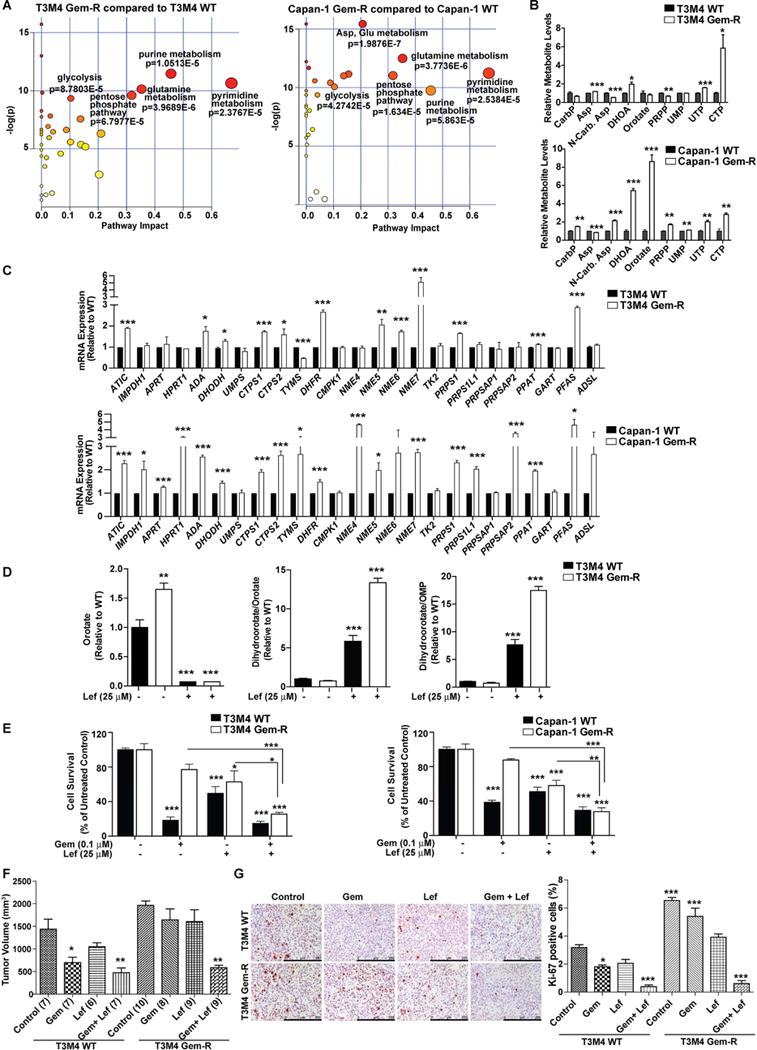
(A) Metabolic pathway impact analysis of significantly upregulated metabolites by Metaboanalyst 3.0 in Gem-R as compared to WT cells. (B) Levels of metabolites of de novo pyrimidine synthesis pathway in Gem-R cells relative to WT cells as determined by LC-MS/MS-based metabolomics. (C) Relative mRNA expression levels of genes in the pyrimidine and the purine synthesis pathways analyzed by qPCR. Data analyzed by Student’s t-test and plotted relative to expression levels in WT cells. (D) Levels of orotate and the ratios of dihydroorotate/orotate and dihydroorotate/Orotidine 5′-monophosphate (OMP) in WT and Gem-R cells in the presence or absence of leflunomide relative to untreated WT cells. Data were analyzed by one-way ANOVA, followed by Bonferroni’s post hoc test. (E) Relative survival of Gem-R and WT cells by MTT assays, under treatment with gemcitabine, leflunomide, or gemcitabine with leflunomide. Data is presented relative to respective untreated shScr controls for WT and Gem-R cells. Comparisons made to the respective controls or indicated groups by two-way ANOVA, followed by Bonferroni’s post hoc test. (F) Tumor volumes upon necropsy, after three weeks of treatment, in orthotopically implanted mice subjected to treatments with control, gemcitabine (Gem), leflunomide (Lef) or gemcitabine with leflunomide (Gem + Lef). Numbers in parentheses indicate the number of mice in each cohort. All the groups were compared to the control WT cohort by one-way ANOVA and Dunnett’s post hoc test. (G) IHC staining for Ki-67 and quantification of percent positive cells in the formalin-fixed tumor sections from the indicated treatment groups. Scale bars: 250 μm. Ki-67 positive and negative cells were counted manually in ten fields of 5 tumors of each group. All the groups were compared to the control WT cohort by one-way ANOVA and Tukey’s post hoc test. For all in vitro studies n=3 per sample. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. CarP: Carbamoyl phosphate; Asp: L-Aspartate; N-Carb. Asp: N-carbamoyl-L-aspartate; DHOA: 4,5-Dihydrooratate; PRPP: Phosphoribosyl pyrophosphate; UMP: Uridine 5′-monophosphate; UTP: Uridine 5′-triphosphate; CTP: Cytidine 5′-triphosphate. See also Figure S3 and Table S2.
Generation of orotate via dihydroorotate dehydrogenase (DHODH) is a crucial step in de novo pyrimidine biosynthesis. Therefore, in order to evaluate if the inhibition of DHODH itself could overcome gemcitabine resistance in pancreatic cancer, we treated the Gem-R and WT pancreatic cancer cell lines with 25 μM leflunomide, an inhibitor of DHODH (Ruckemann et al., 1998). Treatment with leflunomide significantly diminished orotate and downstream orotidine 5′-monophosphate (OMP) levels while causing an increase in the DHOA levels (Figure 3D), suggesting a blockade of DHODH activity. Leflunomide increased the efficacy of gemcitabine and inhibited cell survival in Capan-1 and T3M4 Gem-R cells (Figure 3E). Additionally, there was a significant inhibition of cell survival in other pancreatic cancer cell lines when treated with both gemcitabine and leflunomide (Figure S3C). Furthermore, Gem-R cells showed significant response to gemcitabine (50 mg/kg/day) in the presence of leflunomide (10 mg/kg/day) in orthotopic implantation models of pancreatic cancer, as observed by tumor volume, survival, and proliferation based on Ki-67 staining (Figures 3F–3G, S3D). However, we observed no noticeable body weight changes between different treatment groups (Figure S3E). These results indicate that Gem-R cells have increased pyrimidine biosynthesis and that their poor responsiveness to gemcitabine can be reversed at least in part by targeting pyrimidine biosynthesis.
Increased deoxycytidine reduces the effectiveness of gemcitabine in Gem-R cells
Gemcitabine is a nucleoside analog, and hence both nucleoside synthesis and uptake direct its efficacy. Increased glycolysis in the Gem-R cells increases the flux of glycolytic intermediates through the non-oxidative PPP, causing an increase in the pyrimidine biosynthesis. 1H-13C HSQC NMR analysis indicated an increase in all the nucleoside pools in Gem-R vs. WT cells (Figures 4A and S4A). Increased synthesis of CTP might mitigate gemcitabine efficacy, so we investigated if increasing the cellular deoxycytidine levels could directly increase gemcitabine resistance in pancreatic cancer cells. We treated Capan-1 and T3M4 with 25 μM or 100 μM of deoxycytidine, and S2-013, SUIT2, FG/COLO357 and MIA PaCa-2 cells with 100 μM of each of deoxycytidine, thymidine, deoxyguanosine or deoxyadenosine, individually. Treatment with deoxycytidine, but not other nucleosides, increased resistance to gemcitabine in all of these cells (Figures 4B, 4C, S4B, and S4C). Resistance to gemcitabine might also occur due to reduced influx of gemcitabine into cancer cells. However, our data demonstrates that even under untreated conditions Gem-R cells have an increased expression of SLC28A3 and SLC29A1, the principal nucleoside transporters involved in gemcitabine and deoxycytidine uptake (Figure S4D). Furthermore, we observed that compared to the WT cells, Gem-R cells maintained a higher deoxycytidine pool and a lower gemcitabine/deoxycytidine ratio in the presence or absence of exogenous deoxycytidine (Figure 4D). We also observed that deoxycytidine nucleotide levels positively correlated with gemcitabine IC50 values in a panel of 15 pancreatic cancer cell lines (Figure 4E). Additionally, we analyzed the deoxycytidine nucleotide pools in the flash-frozen tissue specimens from the Rapid Autopsy Program at UNMC (Table S1-cohort 2). We observed significantly decreased progression-free survival with higher deoxycytidine or deoxyuridine, but not deoxyguanosine or deoxyadenosine, nucleotide pools in the primary tumors in patients that were subjected to gemcitabine or 5-FU (Figures 4F and S4E).
Figure 4. Increased deoxycytidine reduces the efficacy of gemcitabine.
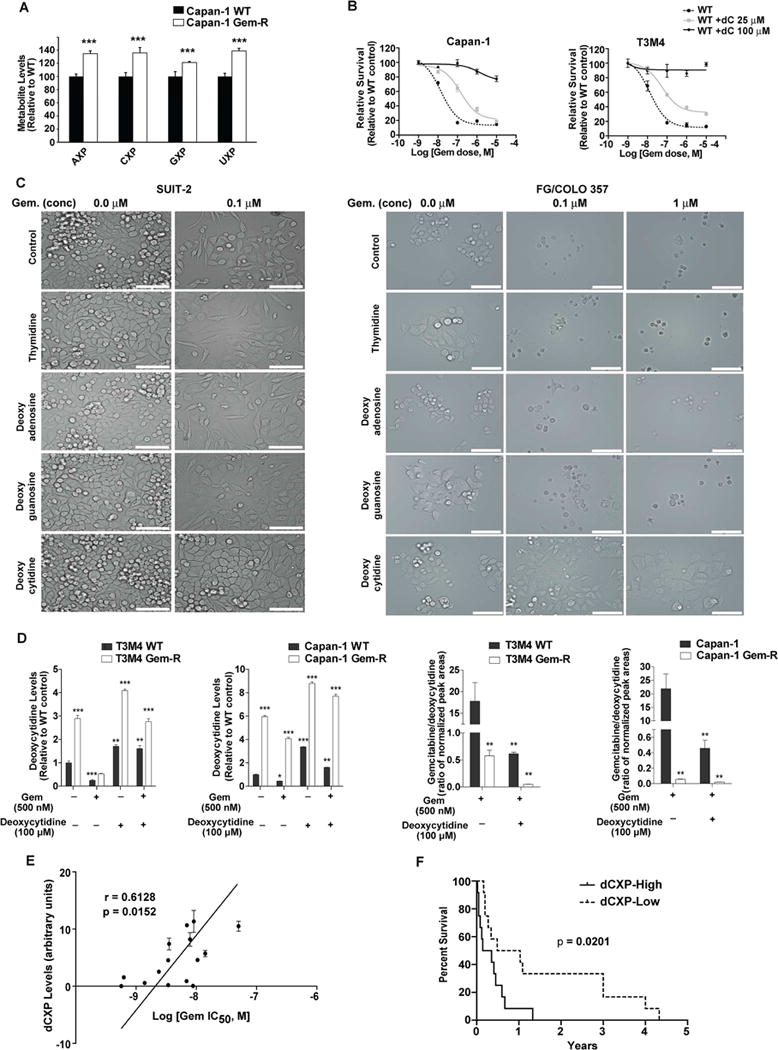
(A) NMR-based metabolite detection for nucleosides in Capan-1 Gem-R vs. WT cells. X indicates the number of phosphate groups and can be mono, di or triphosphates. Levels in Gem-R cells are presented relative to the WT control and analyzed by Student’s t-test. (B) Effect of deoxycytidine (dC) on gemcitabine sensitivity in WT cells by MTT assays at 72 hr post-treatment. (C) Bright-field images of SUIT-2 and FG/Colo357 cells treated with gemcitabine and different nucleosides (100 μ M) for 72 hr. Scale bars: 100 μm (D) Relative deoxycytidine levels and gemcitabine/deoxycytidine ratios as determined by LC-MS/MS in WT and Gem-R cells under treatment with gemcitabine and/or deoxycytidine. compared to untreated WT cells. Data was analyzed by one-way ANOVA and Bonferroni’s post hoc test. (E) Correlation of dCXP levels versus the IC50 of gemcitabine in 15 pancreatic cancer cell lines. ‘r’ depicts Pearson correlation value and p values denote significance of correlation. (F) Kaplan-Meier progression-free survival analysis of PDAC patients on gemcitabine/5-FU chemotherapy with high (above median; n=12) or low (below median; n=12) dCXP levels, as determined by LC-MS/MS in human pancreatic tumors. Data was compared with log-rank (Mantel-Cox) test. For all in vitro studies n=3 per sample. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 as compared to WT controls. See also Figure S4 and Table S1.
HIF-1α is the metabolic master regulator of enhanced glucose metabolism and pyrimidine biosynthesis in gemcitabine resistant pancreatic cancer cells
HIF-1α is a key transcriptional regulator of glycolysis (Zhang et al., 2008). We observed that Gem-R cells express significantly higher levels of HIF-1α, even under normoxic conditions (Figures 5A and S4F). The Gem-R cells also demonstrate higher expression of HIF-1α-dependent enzymes, including GLUT1 and LDHA, which regulate glycolytic flux. The differences are further increased under hypoxic conditions. To determine if the increased glucose dependence of Gem-R cells is HIF-dependent, we stably knocked down HIF-1α and HIF-2α in Capan-1 and T3M4 WT and Gem-R cells (Figure 5B). Knocking down HIF-1α inhibited glucose uptake in both WT and Gem-R pancreatic cancer cells (Figure 5C). Knocking down HIF-2α also decreased glucose uptake, but the effect was not as robust as for HIF-1α. Furthermore, knocking down HIF-1α diminished Gem-R cell survival under gemcitabine-treated conditions (Figure 5D). These findings indicate that, like WT cells, Gem-R cells depend on HIF-1α for enhanced uptake of glucose and that HIF-1α is required for gemcitabine resistance in these models.
Figure 5. HIF-1α regulates the metabolic phenotype and gemcitabine resistance in pancreatic cancer.
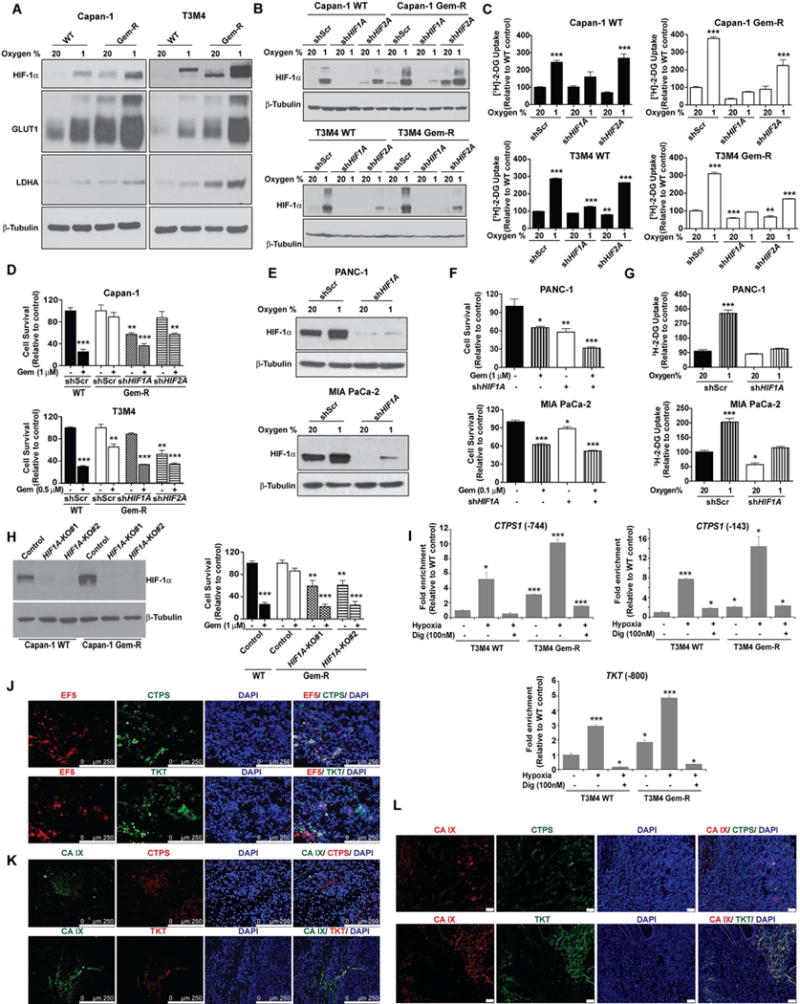
(A) Expression of HIF-1α and HIF-1α-dependent gene products in WT vs. Gem-R cells. Cells were cultured under normoxia (20% oxygen) or hypoxia (1% oxygen) for 12 hr and the lysates were utilized for immunoblotting to determine the levels of HIF-1α, GLUT1 and LDHA. β-tubulin was used as a loading control. (B) WT and Gem-R cells were stably knocked down for HIF1A or HIF2A using shRNA. A scrambled shRNA (shScr) was used as a control. Knockdown status of HIF-1α was confirmed by immunoblotting lysates from cells cultured under normoxia and hypoxia for 6 hr, using β-tubulin as a loading control. (C) Glucose uptake in WT and Gem-R cells as measured by [3H]-2DG labeling. Scrambled control (shScr), shHIF1A or shHIF2A cells in each group were cultured under normoxia or hypoxia (1% oxygen) for 12 hr. Raw scintillation values were normalized to cell counts and depicted as percent of shScr WT/Gem-R controls. (D) Gemcitabine responsiveness in shScr, shHIF1A or shHIF2A Gem-R cells as compared to the shScr WT cells. Cells were treated with gemcitabine for 72 hr, followed by MTT assays. Data is presented relative to respective untreated shScr controls for WT and Gem-R cells. (E) PANC-1 and MIA PaCa-2 pancreatic cancer cells were assessed for knockdown of HIF1A by immunoblotting lysates from cells under normoxic and hypoxic conditions. (F) Effect of HIF1A-knockdown on cell survival under gemcitabine-treated or untreated conditions for 72 hr as determined by MTT assays. (G) Glucose uptake in shScr and shHIF1A cells as measured by [3H]-2DG labeling. Cells in each group were cultured under normoxia or hypoxia (1% oxygen) for 12 hr. Raw scintillation values were normalized to cell counts and are plotted as percent of normoxic shScr cells. (H) Evaluation of HIF-1α expression upon CRISPR/Cas9-mediated knockout of HIF1A (with two independent target regions of HIF1A i.e. KO#1 and KO#2) in Capan-1 WT and Gem-R cells by western blotting (left). β-tubulin was used as a loading control. Evaluation of the effect of HIF1A knockout on gemcitabine responsiveness by MTT assays in Capan-1 WT and Gem-R cells after 72 hr treatment (right). Data is presented relative to respective untreated controls for WT and Gem-R cells. (I) Occupancy of CTPS1 and TKT promoters by HIF-1α was assessed by ChIP using anti-HIF-1α Ab or IgG control, followed by qPCR analysis. Occupancy of HIF-1α at proximal (−143) and distal (−744) CTPS1 promoter regions and TKT promoter region from T3M4 WT and T3M4 Gem-R cells under normoxic and hypoxic conditions (6 hr) is presented as relative to that in T3M4 WT cells under normoxic conditions. (J) Colocalization of CTPS and TKT with 2-nitroimidazole (EF5; a hypoxia probe) in tumor sections from orthotopically implanted Capan-1 cells by immunofluorescence microscopy. Tumors were collected after four weeks of implantation. Scale bars: 250 μm. (K) Colocalization of CTPS and TKT with CA IX in tumor sections from orthotopically implanted Capan-1 cells by immunofluorescence microscopy. Tumors were collected after four weeks of implantation. Scale bars: 250 μm. (L) Colocalization of CTPS and TKT with CA IX in primary tumor sections from human pancreatic cancer patients. Scale bars: 50 μm. For all in vitro studies n=3 per sample. Data in bar charts were compared to the controls by one-way ANOVA with Tukey’s post hoc analysis and are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figure S4.
Next, to determine if the role of HIF-1α in mediating gemcitabine resistance was exclusive to acquired resistant cells, we knocked down HIF-1α in intrinsically gemcitabine resistant cells. As observed in Gem-R cells, knocking down HIF-1α significantly diminished cell survival and glucose uptake in PANC1 and, to a somewhat lesser extent, MIA PaCa-2 pancreatic cancer cell lines (Figures 5E–5G). We also observed a positive correlation between HIF-1α expression and gemcitabine IC50 in 15 pancreatic cancer cell lines (Figures S4G and S4H). Additionally, we performed knockout of HIF1A by CRISPR/Cas9 in Gem-R cells and observed a similar increase in their responsiveness to gemcitabine (Figure 5H).
Next, we determined if the increased expression of TKT and CTPS1 in Gem-R cells was regulated by HIF-1α. While regulation of CTPS1 by HIF-1α is not known, TKT is a known HIF-1α target (Semenza, 2013; Zhao et al., 2010). We performed chromatin immunoprecipitation (ChIP) assays to determine HIF-1α occupancy on TKT and CTPS1 gene promoters. We observed significantly increased occupancy of proximal and distal HIF-1α response elements (HRE) in the CTPS1 promoter by HIF-1α in Gem-R cells compared to WT cells (Figure 5I). HIF-1α binding was further induced under hypoxic conditions and diminished by digoxin, a translational inhibitor of HIF-1α (Zhang et al., 2008). Similar trends were observed for HRE in the TKT promoter. Additionally, we observed co-localization of TKT and CTPS with EF5, a hypoxia marker, and with CA IX, a marker of HIF-1α activity in orthotopically implanted Capan-1 cell-derived tumors (Figures 5J and 5K). We also observed a co-localization of TKT and CTPS with CA IX in human PDAC tissue sections (Figure 5L). These data indicate a role of HIF-1α in mediating resistance to gemcitabine by modulating the expression of CTPS1, TKT, and other glycolytic genes in pancreatic cancer cells.
Pharmacological inhibition of HIF-1α improves gemcitabine sensitivity
We next examined if HIF-1α inhibitor digoxin could abrogate gemcitabine resistance. Treatment with digoxin diminished HIF-1α levels in Capan-1 and T3M4 Gem-R cells (Figure 6A). Furthermore, digoxin reduced glucose uptake in Gem-R and WT cell lines (Figures S5A–S5C). Gem-R cells also demonstrated a significant reduction in the expression of genes involved in glucose metabolism under digoxin treatment (Figure S5D). Digoxin reduced deoxycytidine nucleotide levels and survival in response to gemcitabine in Gem-R cells (Figures 6B–6D). Altogether, these results provide evidence that HIF-1α promotes increased glucose dependence and gemcitabine resistance in pancreatic cancer cells.
Figure 6. Pharmacological inhibition of HIF-1α improves gemcitabine sensitivity.
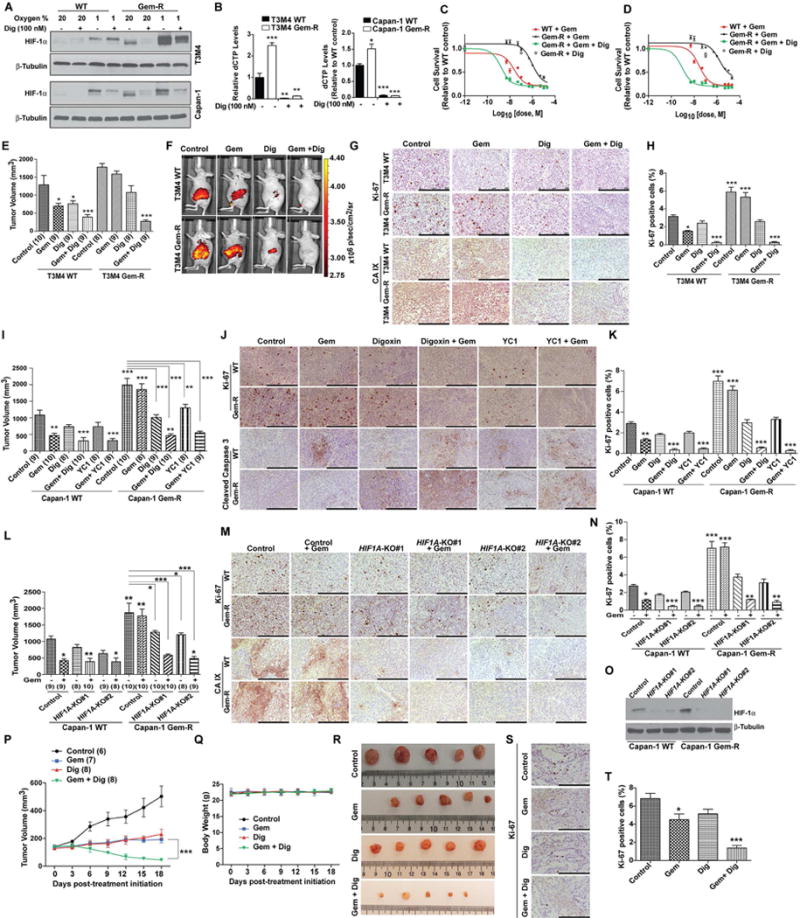
(A) Immunoblotting for HIF-1α in T3M4 and Capan-1 WT and Gem-R cells cultured under normoxia or hypoxia (for 12 hr), treated with and without digoxin (Dig) for 12 hr. (B) The dCTP levels relative to control WT cells, under treatment with solvent control or digoxin, as determined by LC-MS/MS. Values are presented relative to solvent control treated WT cells. (C–D) Effect of digoxin treatment (100 nM) on gemcitabine responsiveness of T3M4 (C) and Capan-1 (D) Gem-R cells as determined by MTT assays, 72 hr after treatment with digoxin, a range of doses of gemcitabine, or both. Effect of digoxin alone on Gem-R cell survival, relative to DMSO-treated controls, is indicated by a single open diamond symbol. (E–K) Effect of digoxin and YC1 on gemcitabine responsiveness in orthotopically implanted T3M4 (E–H) and Capan-1 (I–K) WT and Gem-R tumor models. Tumor volumes upon necropsy (after three weeks of treatment) in orthotopically implanted mice subjected to treatments with control, gemcitabine alone (50 mg/kg, biweekly), digoxin (2 mg/kg, daily) or YC1 (15 mg/kg, daily) alone or gemcitabine with digoxin or YC1 (E and I). In vivo glucose uptake in tumor-bearing mice was determined three weeks after implantation; the mice were injected intraperitoneally with 100 μl of 10 nmoles RediJect 2DG 750 probe and imaged 3 hr later (F). IHC staining for Ki-67 and CA IX or cleaved caspase 3 (G and J) and Ki-67 staining quantitation (H and K) in the formalin-fixed tumor sections from the indicated treatment groups. (L-O) Effect of CRISPR/Cas9-mediated HIF1A knockout (with two independent target regions of HIF1A i.e. KO#1 and KO#2) on gemcitabine responsiveness in orthotopically implanted Capan-1 WT and Gem-R tumor models. Tumor volumes upon necropsy, after three weeks of treatment, in orthotopically implanted mice subjected to treatments with control or gemcitabine (L). IHC staining for Ki-67 and CA IX (M) and Ki-67 staining quantitation (N) in the formalin-fixed tumor sections from the indicated treatment groups. Evaluation of HIF1A knockout status by western blotting in tumor extracts (O). (P–T) Effect of digoxin on gemcitabine responsiveness in a patient-derived xenograft model. Tumor volumes measured by calipers at indicated time points in tumor-implanted mice subjected to treatments with control, gemcitabine alone (50 mg/kg, biweekly), digoxin (2 mg/kg, daily) or gemcitabine with digoxin. Tumor volumes were quantified by caliper measurements and statistically compared by two-way ANOVA analysis with Bonferroni’s post hoc test; p value presented indicates comparison of tumor volumes in gemcitabine and digoxin treated mice at day 18 post-implantation with that of mice treated with gemcitabine alone (P). Body weights of mice with the indicated treatments (Q). Representative tumor images upon necropsy (R). IHC staining for Ki-67 (S) and Ki-67 staining quantitation (T). Ki-67 positive and negative cells were counted manually in ten fields of 5 tumors of each group. Numbers in parentheses indicate the number of mice in each cohort. For all in vitro studies n=3 per sample. Data are represented as mean ± SEM. All cohorts in bar charts were compared to the respective controls by one-way ANOVA with Tukey’s post hoc analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. Scale bars: 250 μm. See also Figure S5.
Digoxin treatment reduces cell proliferation in breast and prostate cancer cell lines (Lin et al., 2009; Zhang et al., 2008; Zhang et al., 2012), and digoxin is currently in clinical trials for prostate cancer (Lin et al., 2014). To determine the efficacy of digoxin in gemcitabine resistant pancreatic tumors, we orthotopically implanted WT or Gem-R cells in the pancreas of athymic nude mice. Starting day 7 post-implantation, the mice were treated with digoxin, gemcitabine or saline. Treatment with gemcitabine reduced tumor volume, glucose uptake and proliferation in WT mice (Figures 6E–6K). However, we observed no significant reduction in tumor volume in the Gem-R tumor-bearing group due to gemcitabine treatment (Figures 6E and 6I). Digoxin treatment diminished tumoral glucose uptake and CA IX expression in mice in both groups (Figures 6F and 6G). A combination of gemcitabine and digoxin significantly diminished tumor volume and proliferation and increased survival in both WT and Gem-R groups (Figures 6E–6K and S5E). Treatment with YC1, another HIF-1α inhibitor, showed similar results (Figures 6I–6K). Treatment with combinations of digoxin or YC1 with gemcitabine also induced apoptosis (Figure 6J). Digoxin or YC1 did not cause any significant decrease in overall mouse body weight (Figures S5F and S5G). We also evaluated if genetic knockout of HIF1A would also sensitize tumor cells to gemcitabine. We observed a significant decrease in tumor burden, proliferative index and CA IX staining upon treatment of HIF1A knockout WT or Gem-R cell-derived tumors with gemcitabine (Figures 6L–6O). We also evaluated the effect of gemcitabine and digoxin on patient-derived xenografts (PDX) in athymic nude mice. Without affecting body weight, the combination of gemcitabine and digoxin significantly reduced growth, size, and proliferative index of tumors in mice (Figures 6P–6T and S5H). Taken together, these data indicate that targeting HIF-1α in combination with gemcitabine in Gem-R-tumor-bearing mice and PDX-implanted mice can abrogate tumor growth.
MUC1 enhances HIF-1α levels and diminishes gemcitabine sensitivity in Gem-R cells
We have previously demonstrated that MUC1, a large type 1 transmembrane protein, stabilizes and activates HIF-1α and increases glucose uptake and metabolism (Chaika et al., 2012a; Mehla and Singh, 2014). Our studies indicate that Gem-R cells have increased glucose and HIF-1α dependence. Interestingly, Gem-R cells also have increased MUC1 expression compared to the WT cells (Figure 7A). Hence, we further investigated if MUC1 expression is at least in part responsible for HIF-1α stabilization and the metabolic phenotype in our acquired gemcitabine resistance models. To achieve this, we generated Gem-R cells with stable knockdown of MUC1 (Figure 7B). MUC1 knockdown Gem-R cells presented decreased HIF-1α protein expression (Figure 7B) and reduced glucose uptake and lactate release (Figures 7C and 7D). Furthermore, knocking down MUC1 significantly diminished gemcitabine resistance in Gem-R cells (Figure 7E).
Figure 7. Knocking down MUC1 abrogates HIF-1α levels and increases gemcitabine sensitivity in Gem-R cells.
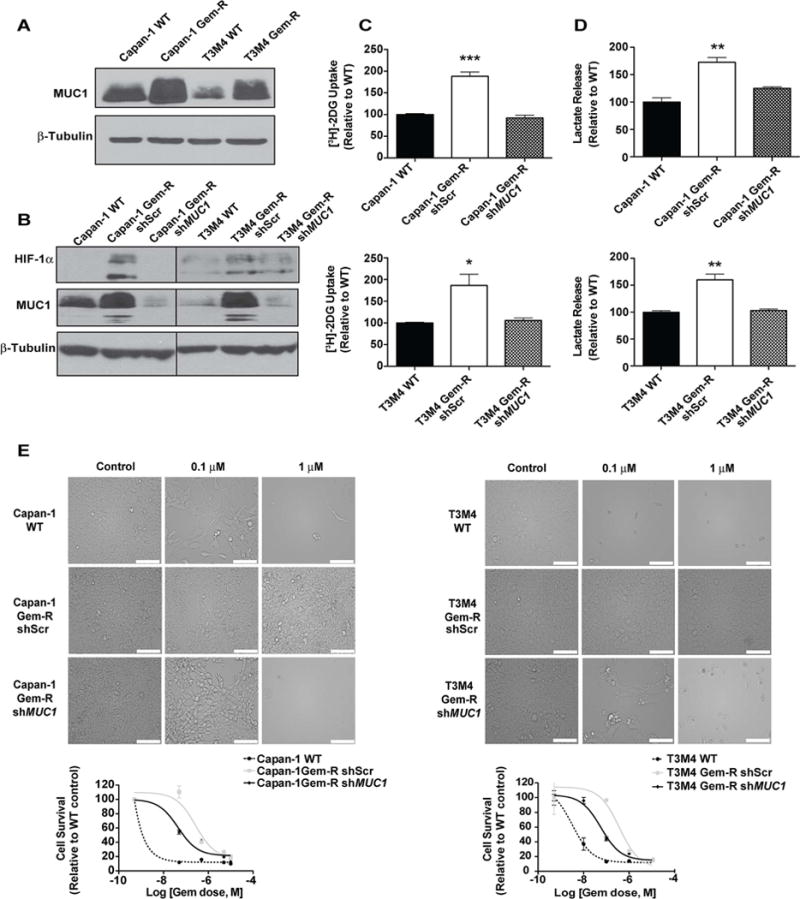
(A) Relative MUC1 expression in Gem-R vs. WT cells as determined by immunoblotting. (B) MUC1 and HIF-1α protein levels in WT, Gem-R and MUC1 knockdown Gem-R cells, as determined by immunoblotting. β-tubulin was used as a loading control. (C) Relative glucose uptake determined by [3H]-2DG uptake assays in MUC1 knockdown Gem-R as compared to scrambled controls of Gem-R and WT cells. Raw scintillation values were normalized to cell counts and depicted as percent of shScr WT controls. (D) Relative lactate release from shScr WT, shScr Gem-R and MUC1 knockdown Gem-R cells. Raw values were normalized to cell counts and plotted as percent of shScr WT controls. (E) Effect of MUC1 knockdown on gemcitabine sensitivity in Gem-R vs. WT cells as denoted by pictomicrographs and survival curves. Scale bars: 100 μm. Cell survival was measured by MTT assay after 72 hr gemcitabine treatment. Data in bar charts were compared by one-way ANOVA with Tukey’s post hoc analysis. For all in vitro studies, n=3 per sample. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Reduced CTPS and TKT levels in human pancreatic cancer patients correlate with increased gemcitabine sensitivity
We investigated the levels of most genes involved in the PPP, pyrimidine and purine synthesis pathways in 17 pancreatic cancer cell lines and correlated them with the IC50 of gemcitabine. We observed that gemcitabine resistance has a direct correlation with the expression of TKT and CTPS (Figure 8A). We also performed IHC studies on flash-frozen tissue sections from tumors from pancreatic cancer patients obtained through the Rapid Autopsy Program at UNMC (Table S1-cohort 3). Our data revealed a significant increase in TKT and CTPS expression in primary and metastatic pancreatic cancer tissues compared to the normal pancreas (Figure 8B). Utilizing these patient data, we investigated if TKT/CTPS expression in primary tumors correlated with survival of patients treated with pyrimidine analogue-based chemotherapy. Patients with higher CTPS or TKT expression in primary tumors had a significantly decreased progression-free survival (Figure 8C).
Figure 8. Reduced CTPS and TKT levels correlate with increased survival in human pancreatic cancer patients.
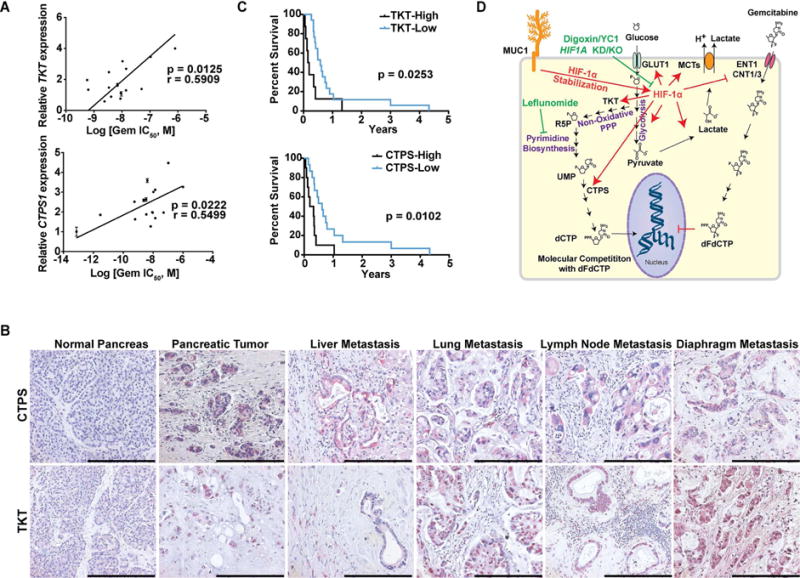
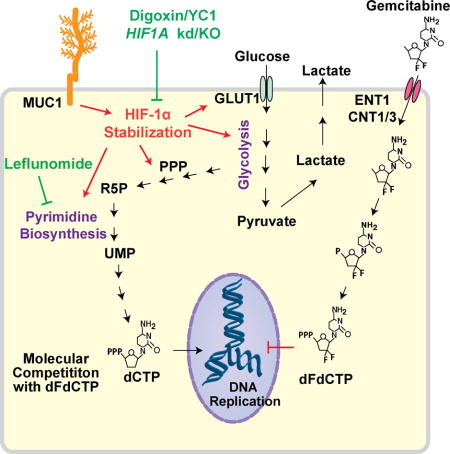
(A) Correlation of TKT and CTPS1 expression levels versus the IC50 of gemcitabine in 17 pancreatic cancer cell lines. ‘r’ depicts Pearson correlation value, and p values denote significance of correlation. (B) IHC staining of TKT and CTPS in formalin-fixed paraffin-embedded sections obtained from normal human pancreas, PDAC and metastatic lesions. Scale bars: 250 μm (C) Kaplan-Meier progression-free survival analysis of PDAC patients on gemcitabine/5-FU chemotherapy with high (above a composite score of 9; n=8 for TKT, n=10 for CTPS) or low (below a composite score of 9; n=17 for TKT, n= 15 for CTPS) TKT or CTPS expression levels based on IHC in pancreatic tumors. Comparisons were made by log-rank (Mantel-Cox) test and p values denote significance of alterations in survival in TKT or CTPS high- vs. low-expressing population. (D) Graphical summary for the metabolic basis of gemcitabine resistance in pancreatic cancer. Gemcitabine resistant cells demonstrate increased HIF–1α-mediated glucose uptake and, resultantly, increased flux of glucose through the PPP and pyrimidine biosynthesis to generate dCTP. Inhibition of HIF-1α or pyrimidine biosynthesis increases gemcitabine sensitivity in pancreatic cancer cells. R5P: Ribose-5-Phosphate; UMP: Uridine 5′-monophosphate; dCTP: Deoxycytidine 5′-triphosphate; dFdCTP: 2′,2′-Difluorodeoxycytidine 5′-triphosphate or Gemcitabine 5′-triphosphate. See also Table S1.
To summarize, gemcitabine resistant tumors demonstrate increased HIF-1α-mediated glucose uptake. Glucose carbon is fed through both glycolysis and the non-oxidative PPP. Differentially increased carbon flux through the non-oxidative PPP and pyrimidine biosynthesis pathways leads to an increase in the cytoplasmic pools of dCTP, which can outcompete gemcitabine from incorporating into the replicating DNA (Figure 8D). Inhibition of glycolysis or pyrimidine biosynthesis leads to increased gemcitabine sensitivity.
DISCUSSION
Drug resistance is the major cause of the failure of clinical effectiveness of chemotherapy. Pyrimidine analogs gemcitabine or 5-fluorouracil, alone or in combination with other drugs, are the current standard of care for advanced metastatic pancreatic cancer. However, the response to gemcitabine in patients is very poor with no drastic reduction in metastasis or increase in patient survival (Heinemann et al., 2000). Multiple molecular mechanisms of gemcitabine resistance encompassing different pathways have been suggested. However, most previous studies have mainly addressed acquired resistance due to changes in the rate of drug influx or efflux. Our current study presents strong evidence for the utility of agents targeting tumor cell metabolism in abrogating gemcitabine resistance in pancreatic cancer. Here, we present a mechanism of chemoresistance by which cancer cells increase intracellular cytidine pools that can in turn render gemcitabine ineffective by molecular competition.
Altered metabolism is one of the important hallmarks of cancer (Hanahan and Weinberg, 2011). The hypoxic microenvironment of pancreatic tumors stabilizes HIF-1α, which is the master regulator of glucose metabolism and induces glucose dependence in cancer cells (Semenza, 2003; Semenza, 2009). The increased glucose uptake under hypoxia not only feeds into the glycolysis pathway, but also into intermediate pathways to generate biomass. Hypoxia has been linked with resistance in various cancers (Song et al., 2006), including pancreatic cancer (Yokoi and Fidler, 2004). A recent study by Lin et al. demonstrates that HIF-1α makes cells resistant to cisplatin, oxaliplatin and paclitaxel (Lin et al., 2011). However, none of these studies directly determine the effect of HIF-1α or the underlying metabolic phenotype on gemcitabine sensitivity. Our data demonstrates that gemcitabine resistant pancreatic cancer cells have increased expression of HIF-1α, accompanied by increased glycolytic phenotype and dependence on glucose. Previous studies from our lab indicate that the cytoplasmic tail of MUC1, a type I transmembrane protein, physically interacts with and prevents the proteasomal degradation of HIF-1α in pancreatic cancer cell lines and animal models (Chaika et al., 2012a), and here, we observed increased expression of MUC1 in gemcitabine resistant cells. Hence, we demonstrate that Gem-R cells stabilize HIF-1α by upregulating MUC1 expression. We also observed increased gemcitabine sensitization upon inhibition of HIF-1α in culture conditions, orthotopic implantation models and PDX models.
Increased glycolysis serves to rapidly generate the biosynthetic intermediates to supply the ingredients for cell growth and proliferation. Our results indicate that increased glucose uptake in Gem-R cells is fed through the non-oxidative arm of PPP for de novo synthesis of pyrimidine nucleotides. TKT and CTPS are upregulated in Gem-R cells. Together, TKT and CTPS could serve to increase carbon flux into the pyrimidine biosynthetic pathway. Likewise, inhibition of DHODH, a key enzyme in the pyrimidine biosynthesis pathway, with leflunomide increased gemcitabine sensitivity in the Gem-R cells. Previous studies show a TKT-mediated increase in the non-oxidative PPP in BCR-ABL upregulated imatinib-resistant tumors (Zhao et al., 2010). It is possible that HIF-1α-mediated upregulation of TKT might be a common mechanism of survival in chemotherapy resistant cancers. However, it is unlikely that pyrimidine synthesis would be essential for imatinib-resistance in BCR-ABL tumors. Increased nucleotide synthesis in Gem-R cells leads to accumulation of dCTP and causes competitive inhibition of gemcitabine activity, thereby promoting gemcitabine resistance. While the Gem-R cells are able to take deoxycytidine and gemcitabine from extracellular milieu, they are able to maintain a higher deoxycytidine/gemcitabine ratio and hence sustain gemcitabine resistance. In addition, cell-extrinsic mechanisms of resistance have been identified, including drug scavenging by fibroblasts (Hessmann et al., 2017). The cell-intrinsic mechanisms of resistance identified in the current study are expected to operate in concert with the cell-extrinsic mechanisms of resistance.
For the last three decades gemcitabine has been the only single-agent treatment option for advanced metastatic pancreatic cancer patients (Burris and Storniolo, 1997). However, frequent development of resistance to this therapy contributes to poor prognosis of patients. Multiple molecular targets have been proposed to increase the efficacy of gemcitabine in pancreatic cancer; however, no significant advance has resulted so far from such efforts. Our study delineates the core metabolic alterations that mediate gemcitabine resistance in acquired as well as intrinsically resistant pancreatic cancer cells, thereby elucidating a widely prevalent metabolic mechanism of gemcitabine resistance. Targeting these core metabolic pathways could overcome gemcitabine resistance in pancreatic cancer.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to the lead contact: Pankaj K. Singh (pankaj.singh@unmc.edu).
EXPERIMENTAL MODEL ANND SUBJECT DETAILS
Cell Lines
Capan-1 (source: male), Capan-2 (source: male), PANC1 (source: male), CFPAC (source: male), BxPC3 (source: female), HPAFII (source: male), AsPC-1 (source: female), SUIT-2 (source: male), FG/Colo 357 (source: female) and MIA PaCa-2 (source: male) pancreatic cancer cells were obtained from the American Type Culture Collection (Rockville, MD). S2-013 and S2-007 are cloned sub-lines of a human pancreatic tumor cell line (SUIT-2; source: male) derived from a liver metastasis. S2-013, S2-007, SUIT-2, Panc03.27 (source: female), HuP-T3 (source: male), QGP-1 (source: male), PaTu 8902 (source: male), and T3M4 (source: male) cell lines were a generous gift from Dr. Michael Hollingsworth (Eppley Institute, UNMC, Omaha, NE). All the cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 mg/ml) and streptomycin (100 mg/ml), and incubated at 37°C in a humidified chamber with 5% CO2. All cells were passaged with 0.25% trypsin/2.21 mM EDTA in PBS when they reached a confluency of 75–80%. The cell lines were validated by STR profiling at University of Arizona Genetics Core (UAGC).
In vivo mouse studies
Female athymic nude mice (Foxn1nu/Foxn1nu) age 4–6 weeks were purchased from Charles River Laboratories and housed in the Animal Facility at the University of Nebraska Medical Centre, Omaha, USA. All procedures were approved by the University of Nebraska Medical Centre Institutional Animal Care and Use Committee and in accordance to NIH guidelines. For all the treatment studies mice were randomly assigned to different treatment group. For xenograft studies, after establishing tumor mice were randomly distributed in different group.
Human Studies
The age, tumor grade, and sex of the subjects is provided in Table S1. Sample size information is indicated in the figure legends. All the studies with human subjects were approved by the UNMC IRB committee the committee. Also, informed consent waiver was approved by the UNMC IRB committee for all subjects.
METHOD DETAILS
Glucose uptake assay
WT and Gem-R cells were treated with the indicated agents for 12 hr and glucose uptake was performed by utilizing [3H]-2-DG as previously described (Shukla et al., 2014).Briefly, 5×104 cells were seeded per well in 24 well plate and incubated over-night at 37°C with 5% CO2. Next day, cell were starved for glucose for 2 hr and then treated with 1 μCi [3H]-2-deoxyglucose (DG) for 20 min. After incubation, cells were washed twice with PBS and lysed in 1% SDS. Lysates were transferred to scintillation vials containing scintillation fluid. The vials were subjected to [3H] counting by utilizing automated scintillation counter. The values were normalized with respective cell counts.
Mass spectrometric metabolomics analysis
Cells were seeded in 6 cm plates and two hours before the collection of metabolites the culture medium was replaced with fresh medium. Polar metabolites were extracted and then analyzed with LC-MS/MS using the selected reaction monitoring (SRM) method with positive/negative ion polarity switching on a Xevo TQ-S mass spectrometer (Gunda et al., 2016; Yuan et al., 2012). Peak areas integrated using MassLynx 4.1 (Waters Inc.) were normalized to the respective protein concentrations and the resultant peak areas were subjected to relative quantification analyses by utilizing Metaboanalyst 3.0 (www.metaboanalyst.ca) (Xia and Wishart, 2016).
Lentiviral knockdown/knockout
shRNA-mediated knockdown of HIF1A and HIF2A in WT and Gem-R (Capan-1 and T3M4), and GLS and GLS2 in T3M4 Gem-R cells were generated using targeted sequences described previously (Chaika et al., 2012a; Cheng et al., 2011). Single vectors carrying Cas9 nuclease and CRISPR sgRNA targeting control or different regions of HIF1A were utilized to establish HIF1A knockout. CRISPR sgRNA-Cas9 cassettes were lentivirally transduced to WT and Gem-R cells. Cells were selected against puromycin.
FACS sorting for GLUT1-high and -low cells
2.5×106 Capan-1 Gem-R cells plated in a 10 cm dish were gently trypsinized and washed in cold PBS with 1% BSA twice. Cells were incubated with primary antibody for GLUT1 (Abcam, 1:200) for 45 min on ice. The cells were then washed thrice with cold PBS containing 1% BSA, and incubated with Alexa–488-conjugated secondary Ab for 30 min. The cells were finally washed thrice and sorted by flow cytometry for high- and low-GLUT1 expression.
MTT cytotoxicity assays
A total of 6,000 cells/well were plated in 96 well plates 12 hr before the treatment. The cells were then treated with indicated agents for 72 hr. At the end of the treatment, 10% v/v of 5-mg/ml solution of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) agent was added for 2 hr. The medium was then removed and the cells were dissolved in DMSO (Sigma, St Louis, MO). Relative cytotoxicity was determined by measuring the absorbance at 570 nm using a BMG Labtek plate reader. All experiments were done in triplicates and a mean with SEM was calculated.
Clonogenic assays
Capan-1 and T3M4, WT and Gem-R cells were treated with gemcitabine (concentrations indicated) for 72 hr, followed by plating 500 cells per well in 6 well plates. Colonies were stained and imaged 18 days later using 0.2% crystal violet in 80% methanol.
Soft agar assays
5000 cells were suspended in 1 ml top agar made of 0.4% NuSieve GTG agarose in DMEM and seeded over 2 ml of bottom agar composed of 0.8% NuSieve GTG agarose in DMEM. Colonies were counted after 15 days and colony size measured by pictomicrography.
Glutamine uptake assay
Pancreatic cancer cells were seeded in 24 well plates. At the end of the treatment, the control wells were treated with 4 mM of glutamine for 10 min, followed by addition of 4 μl of [3H]-glutamine (Perkin Elmer). The cells were washed and lysed in 1% SDS followed by measurement of radiolabel using a scintillation counter. The raw values were normalized to cell counts.
XF24 extracellular flux analysis
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) analyses were performed with an XF24 extracellular flux analyzer (Seahorse Biosciences, North Billerica, MA) as described previously (Wu et al., 2007). Briefly, 3.5×104 cells were seeded per well in 24-well cell culture plates (Seahorse Biosciences, North Billerica, MA) in DMEM with 10% FBS and incubated at 37°C, overnight in 5% CO2 incubator. Next day, growth medium was replaced with bicarbonate free DMEM and cells were incubated at 37°C for 1 hr in CO2 free incubator to equilibrate media temperature and pH. By utilizing a Seahorse XF24 analyzer, ECAR and OCR were measured in baseline conditions and under treatment with 2,4-dinitrophenol (2,4 DNP; 100 μM), 2-deoxy glucose (2-DG; 100 mM) and rotenone (1 μM). Values ae presented as mean ± standard error of mean.
1-14C- and 6-14C-glucose CO2 release assay
CO2 release assay was performed as previously described (Ying et al., 2012). Briefly, cells were treated with 1 μCi of 1-14C-and 6-14C-glucose followed by incubation at 37°C for the indicated durations. At the end, 150 μl of 3M perchloric acid was added and the wells were immediately covered with Whatmann’s filterpaper dipped in phenylethylamine. The released radiolabeled CO2 captured on the filter paper was measured with a scintillation counter.
Lactate release assay
Lactate levels secreted into the media were analyzed by colorimetric assays. The assay was performed as per the manufacturer’s protocol utilizing Lactate Assay Kit II (Eton Bioscience Inc.).
Glucose-6-phosphate dehydrogenase assay
Glucose 6-phosphate dehydrogenase activity was measured by flurometric measurement. The assay was performed as per manufacturer’s protocol by utilizing Glucose–6-Phosphate Dehydrogenase Assay Kit (Cayman Chemicals).
Gene expression analysis by real-time PCR (qPCR)
Total RNA was isolated by using RNAeasy columns (Quiagen) as per manufactures protocol. Total RNA (5 μg) was reverse transcribed by using Verso-cDNA synthesis kit (Thermo-Scientific) according to the manufacturer’s guidelines. qPCR was performed with gene specific primers at 95°C for 10 sec, 60°C for 60 sec (40 cycles) in 10 μl reaction mix containing 3μl cDNA, 2 μl primers and 5 μl SYBR Green master mix (Applied Biosystems) using an ABI 7900 thermocycler. Beta-actin was used as an internal control. Quantification was performed with the ΔΔCt method (Schmittgen and Livak, 2008). See Table S3 for the primer sequences used for qPCR analysis. All the mRNA correlations with gemcitabine IC50 were performed by utilizing the following cell lines: Capan-1, PANC-1, CFPAC-1, BxPC-3, HPAF-II, AsPC-1, SUIT-2, Colo 357, MIA PaCa-2, S2-013, T3M4, S2-007, Panc 03.27, Capan-2, HuP-T3, PaTu 8902, and QGP-1.
Chromatin Immunoprecipitation analysis
ChIP assays were performed as described previously (Behrens et al., 2010). 3.0 μl purified chromatin for ChIP analysis were used with Sybr green master mix (Applied Biosystems, Foster City, CA, USA) and subjected to qPCR analysis using ABI 7900 thermocycler. Each reaction was repeated in triplicate and the experiments were repeated at least twice to confirm reproducibility. Values were obtained for the threshold cycle (Ct) for each gene or genomic region and data were analyzed using the standard curve method. For ChIP qPCR analysis, values were normalized to an input control and expressed as a fold increase over enrichment detected using IgG, as published previously (Behrens et al., 2010). The average expression ± S.E.M. was reported. See the Key Resources Table for primer sequences.
Immunoblotting
Cell lysates were prepared by scraping cells (225 cm2, 80–90% confluent) into 1.5 mL of lysis buffer (50 mM Tris-HCl pH 8.0 containing 1% NP-40, 150 mM NaCl, 5 mM EDTA and 1 mM phenylmethylsulphonyl fluoride). Western blottings were performed as previously described (Singh et al., 2008). The membranes were probed with primary antibodies against HIF1α (BD Biosciences), MUC1 (Abcam), GLUT1 (Abcam) and LDHA (Abcam).
Immunohistochemistry
Immunohistochemistry was performed by utilizing goat anti-rabbit IgG-AP with Fast-Red to produce red stain (PicTure™-Double Staining kit, Invitrogen), as per the manufacturer’s instructions. Following primary antibodies were utilized: Ki67 (Thermo Fisher), CTPS (Sigma) and TKT (Sigma). The stained sections were imaged at 200 X under an inverted microscope. Cells with Ki67 positive nuclear staining were counted in 3 random fields at 200 X magnification. The intensity score was given by evaluating staining intensity of positive staining (0 = none; 1 = weak; 2 = intermediate, 3 = strong). The proportion score representing the percentage of positively stained cell (0 = none; 1 = less than 5%; 2 = 5–25%; 3 = 26–50% 4 = 51–75% 5 = above 75%). The overall protein expression in each sample is expressed as histoscore, which is the multiplication product of the proportion (0–5) and intensity scores (0–3) and is between 0–15, with a maximum of 15. The staining score was evaluated by two independent pathologist-trained observers.
Hypoxia imaging and co- localization with CTPS and TKT
Hypoxia imaging in tumor tissues and co-localization of CTPS or TKT with EF5 or CA IX by immunofluorescence microscopy was performed as described previously (Chaika et al., 2012a). Briefly, tumor-bearing mice were injected with100 micro liter EF5 solution (3 mg/ml) three hours before necropsy. After necropsy tumor pieces were flash-frozen. Seven micrometer thick Flash-frozen tumor sections were then first stained with anti-CTPS/ anti-TKT antibodies followed by staining with Cy3-conjugated anti-EF5 monoclonal antibody along with Alexa-488 anti-Rabbit polyclonal antibodies. Tissue sections were stained with mouse anti-CA IX antibody along with rabbit anti-CTPS or anti-TKT antibodies where applicable. Sections were counterstained with DAPI. Immunofluorescence images were capture by using Leica DMI6000B microscope at 20X magnification.
Tumor growth studies
Congenitally athymic female nude mice (NCr-nu/nu) were purchased from the Charles River Laboratories, USA. We orthotopically implanted 0.5×106 WT or Gem-R cells in the pancreas of athymic nude mice. Starting day 7 post-implantation, the mice were treated with digoxin (2 mg/kg, daily), leflunomide (10 mg/kg/day), gemcitabine (50 mg/kg, biweekly), YC1 (15 mg/kg, daily), PBS (100 μl, daily), saline control or combinations. Mice were monitored for a period of 30 days and were sacrificed when the tumor reached 1000 mm3 in dimension. For the macroscopic evaluation of metastasis, we considered occurrence of metastasis if at least one metastatic nodule was present in an organ.
Patient-Derived Xenograft (PDX) studies
Six to eight-week-old female athymic nude mice were used to implant PDX tumors. Tumor samples were cut in to 3–4 mm pieces and immediately placed in F-12 HAM (SIGMA-ALDRICH, MO, USA) medium supplemented with 50% fetal calf serum and 50 units/ml penicillin and 50 microgram/ml streptomycin. Tumor tissue pieces were embedded into Matrigel before subcutaneous implantation into mice. After implantation of tumor tissue, xenografts were allowed to grow for 4 weeks. When tumor size reached 150 mm3, mice were divided into four groups (vehicle control, gemcitabine (50 mg/kg, twice per week), digoxin (2 mg/kg, per day) and digoxin with gemcitabine), and treated for 18 days. Tumor volume and body weight were recorded regularly during the treatment. After 18 days of treatment mice were sacrificed and tumor volume, tumor weight etc. were measured. Patient-derived xenograft PATX162 was implanted in nude mice with previously published method (Kim et al., 2009) at MD Anderson Cancer Center. Briefly, tumor pieces were divided in 4–5 mm pieces and placed into Matrigel. Matrigel-embedded tumor pieces were then subcutaneously implanted to both flanks of athymic nude mice. Six to eight-week-old athymic female nude mice were utilized for subcutaneous implantation. When tumors reach the size of ~100 mm3 mice divided into 4 groups with 5 mice in each group. Mice were treated for 6 weeks with gemcitabine at 50 mg/kg twice a week, digoxin at 2 mg/kg daily, through intraperitoneal injection, or the combination of both agents. The mice were sacrificed when their tumors reached the size of 15 mm in diameter.
In vivo glucose uptake assays
Three female athymic Foxn1nu/Foxn1nu mice per group were intraperitonially injected with 10 nmoles of the XenoLight RediJect-2DG 750 optical probe (Perkin Elmer Inc.) in 100 μl PBS per mouse (as per the manufacturer’s recommendations). Animals were then imaged using IVIS Spectrum small animal imaging system.
NMR Metabolite extraction
Cells were harvested at 70–80% confluence. After removing the media by aspiration and washing twice with 1X PBS to remove remnants of the media, metabolites were extracted with 1 ml of cryogenically cold 80% methanol. The plates were then incubated at −80°C for at least 15 min. Scrapers were used to scrap the cells from the plate and transferred in to an Eppendorf tube. The tube centrifuged at 13,000 rpm for 5 min to separate the cell extract from the cell debris. The supernatant was then transferred to a clean tube and 250 μl of Milli-Q water (Millipore, Billerica, MA, USA) was added to the remaining cell debris for re-extraction. The cell debris were mixed with the water by pipetting. The samples were centrifuged to collect the aqueous extract and the collected water extract was mixed with the 80% methanol extract. Vacuum evaporator (SpeedVac® Plus, Savant, Thermo Scientific, Waltham, MA) and lyophilizer (Labconco, Kansas City, MO) were utilized to evaporate the methanol and lyophilize the water, respectively. The dried samples were dissolved in 600 μl μL of 50 mM phosphate buffer in 99.8% D2O (Isotec, St. Louis, MO) at pH 7.2 (uncorrected) with 50 μM 3-(tetramethysilane) propionic acid-2,2,3,3-d4 (TMSP) for spectral referencing. The solution was centrifuged down at 13000 for 5 min and the supernatant was transferred to 5 mm NMR tube.
NMR experiment
Bruker AVANCE DRX 500 MHz spectrometer equipped with 5 mm triple-resonance cryogenic probe (1H, 13C and 15 N) with a Z-axis gradient was utilized to acquire the NMR data. The experiment was automated using BACS-120 sample changer, ATM (automatic tuning and matching) and Bruker IconNMR™ software. The one-dimensional (1D) proton nuclear magnetic resonance (1H NMR) data were collected at 300 K with 32 K data points, 128 scans, 16 dummy scans and a spectral width of 5,483 Hz using an excitation sculpting pulse sequence (Nguyen et al., 2007). The 2D 1H-13C hetero-nuclear single quantum coherence (HSQC) NMR spectra were collected with 2 K data points and a spectrum width of 4,735 Hz in the direct dimension and 64 data points and a spectrum width of 17,607 Hz in the indirect dimension at 300 K with 64 scans, 16 dummy scans and a 1.5 s relaxation delay.
NMR data analysis
The 1D-1H NMR data were analyzed using MVAPACK software (http://bionmr.unl.edu/mvapack.php) (Worley and Powers, 2014). The raw NMR data was Fourier transformed, automatically phased and binned using adaptive intelligent binning (De Meyer et al., 2008). The spectra were then normalized using standard normal variate (SNV) and the noise was removed. This data was used to generate principal component analysis (PCA) model. The intact spectrum was normalized using SNV, noised was removed and scaled using Pareto scaling to generate orthogonal projections to latent structures discriminant analysis (OPLS-DA) scores and backscaled loadings. Metabolite identification from the 1D 1H NMR spectra and backscaled loading were accomplished using the Chenomx NMR Suite 7.6 (http://www.chenomx.com/). NMRPipe (NIH, Bethesda, Maryland) and NMRViewJ Version 8.0.3 were used to process and analyze the 2D 1H-13C HSQC NMR spectra respectively. 2D-1H-13C HSQC NMR spectra peak intensities were normalized by the average peak intensity for a given spectrum. The Human Metabolomics Database (Wishart et al., 2013), Madison Metabolomics Consortium Database (Cui et al., 2008) and Platform for RIKEN Metabolomics (Akiyama et al., 2008) were used for peak assignment.
[18F]FDG-PET, Magnetic Resonance Imaging
[18F]FDG-PET imaging was performed as described previously (Schlaepfer et al., 2015). We performed single-position, whole-body imaging by using a Siemens Inveon microPET scanner at the University of Colorado Cancer Center Animal Imaging Shared Resources (AISR). Mice were fasted for 6 h before [18F]FDG injection, and blood glucose levels was assessed prior to the [18F]FDG injection (all animals had < 80 mg/ml of circulating glucose). Approximately 250 μCi of [18F]FDG, obtained through University of Colorado Hospital (PetNet solutions), was administered by tail vein injection to conscious animals (the precise dose was assessed by measuring the syringe before and after the injection) (Morelli et al., 2012). Animals were maintained in temperature-controlled cages for 1 h to allow for awake [18F]FDG uptake in tumors. Under isoflurane anesthesia (2.5 %), animals were placed on a warm pad (m2m Imaging), and a 10-min emission scan was acquired. [18F]FDG uptake in mouse orthototpic tumors, as well as control tissues (muscle) was performed by analyzing the micro-PET images with the ASIProVM (Concorde Microsystems) software. Regions of interest were drawn with the trace command around the tumors on scan slices, and the total activity of all tumor slices was summed. The normalized uptake values (NUVs) were obtained by dividing the total activity of the tumor by the time-corrected dose-delivered [time-corrected dose = dose injected × exp(−0.006317 × t)], where t is the time between the injection and scan time, and it is shown as the fold change of the baseline scan of each respective tumor. Representative images were generated using the Siemens Inveon Research Workplace software (v3.1.2).
Detection of gemcitabine and dCTP
Relative quantification of Gemcitabine in the cell lysates was performed using LC-MS/MS-based method. Dried cell extracts were reconstituted in mass spec grade water followed by chromatography through Synergi Hydro-Reverse Phase column (150×2.0 mm, Phenomenex) maintained at room temperature. The chromatography method included a gradient with increase of Buffer B (100% methanol) linearly with simultaneous decrease in the content of Buffer A (20 mM Ammonium acetate, pH 5.0) till 10 min and returning to initial concentration at 12 min. Gemcitabine elution and detection were performed using Waters Acquity UPLC coupled to Xevo TQ-S mass spectrometer. For detecting dCTP dried cell extracts were reconstituted in mass spec grade water followed by chromatography using HSS-T3 column (100×2.0 mm, Waters). Chromatography method constituted a linear gradient with increase of Buffer A (10 mM Ammonium formate, pH 6.5) from 0 to 50% in 5 min, hold at 50% for 3 min, followed by a decrease to 0% by 13 min and final rinse of column at 100% B (100% Acetonitrile) till 15 min. The dCXP correlations with gemcitabine IC50 (Figure 4E) were performed by utilizing the following cell lines: Capan-1, PANC-1, CFPAC-1, BxPC-3, HPAF-II, AsPC-1, SUIT-2, Colo 357, MIA PaCa-2, S2-013, T3M4, S2-007, Panc 03.27, Capan-2, HuP-T3, PaTu 8902, and QGP-1.
LC-MS/MS for polar metabolites and 13C-glucose based flux analysis
Relative quantification of polar metabolites including deoxycytidine and 13C-labeled metabolites was performed using selected reaction monitoring and scheduled reaction monitoring based mass spectrometry methods, respectively. Liquid chromatography method used for separating unlabeled and labeled metabolites constituted an UPLC-based BEH amide column (150×2.1 mm, Waters) eluted with a gradient buffer composition consisting of Buffer A (100% Acetonitrile) and Buffer B (20 mM Ammonium acetate, pH 9.0) (Gunda et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Nonparametric one-way ANOVA (Kruskal-Wallis tests) were utilized to compare differences between animal groups. One-way ANOVA with Dunnett’s test was performed to determine the effect of inhibitors on treated and Gem-R cells, as compared to the WT cells. One-way ANOVA alone was performed to determine the effect of different inhibitors on one cell type (WT or Gem-R). Patient survival data was compared by Kaplan Meier survival analysis using Log-rank (Mantel-Cox) test. Student’s t-test was used when appropriate. P < 0.05 was considered significant. All statistical tests were performed by utilizing GraphPad Prism5 and SPSS 16.0 software. For all experiments * p < 0.05, ** p < 0.01, and *** p < 0.001.
Supplementary Material
Table S3, related to STAR methods. Primers for qPCR
SIGNIFICANCE.
Multiple mechanisms have been proposed that lead to reduced effectiveness of gemcitabine in pancreatic cancer and yet resistance to gemcitabine still remains a challenge in the clinics. Aberrant metabolism is one of the important hallmarks of cancer that facilitates cancer cell survival and proliferation. The present study demonstrates existence of a widely prevalent metabolic mechanism that mediates chemotherapy resistance in pancreatic cancer. Utilizing cell-based models of acquired and intrinsic gemcitabine resistance, animal models and patient-derived specimens, we identify key metabolic alterations that mediate gemcitabine resistance. Our studies provide potential targets to improve the efficacy of gemcitabine in patients.
HIGHLIGHTS.
Gemcitabine resistance is associated with increased glucose uptake in PDAC
HIF-1α upregulates non-oxidative PPP and pyrimidine biosynthesis
HIF-1α is stabilized by MUC1 in gemcitabine resistant tumors
Targeting HIF-1α or metabolic pathways can sensitize PDAC tumors to gemcitabine
Acknowledgments
We would like to thank Dr. Prakash Radhakrishnan for technical assistance with initial animal studies. This work was supported in part by funding from the National Institutes of Health grant (R01 CA163649, R01 CA210439, and R01 CA216853, NCI) to PKS and (P01CA117969) to LCC; American Association for Cancer Research (AACR)—Pancreatic Cancer Action Network (PanCAN) Career Development Award (30-20-25-SING) to PKS; the Specialized Programs for Research Excellence (SPORE, 2P50 CA127297, NCI) to PKS, JLG, and MAH; Pancreatic Tumor Microenvironment Research Network (U54, CA163120, NCI) to PKS and MAH; and Lustgarten Foundation award to LCC. JMM thanks to SAF2015## 64501 ## R. We would also like to acknowledge the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727, NCI) and NCCS COBRE (P30 GM106397, NIGMS) for supporting shared resources and the Animal Imaging Shared Resources of the University of Colorado Cancer Center (P30CA046934, NCI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION: Supplementary information-6 Figures and two tables, can be found online.
AUTHOR CONTRIBUTIONS: S.K.S, V.P., K.M., V.G., N.J.S., J.B.F., C.A.L., L.B., and P.K.S. designed the research; S.K.S., V.P., K.M., V.G., N.V.C., E.V., R.J.K., J.A., G.D.G., A.D., A.L.I., T.G., B.D., J.J.A., D.M., K.S.A., O.M., J.M.A., N.J.S., and C.A.L. performed research; K.M., V.P., S.K.S., V.G., N.V.C., R.J.K., P.M.G., R.P., Q.P.L., J.L.G., J.M.M, J-W.K., J.H.H., C.W., M.A.H., C.A.L., A.R.S., J.B.F., J.M.O., L.C.C. and P.K.S. contributed reagents/analytic tools; S.K.S., V.P., K.M., V.G., N.V.C., T.G., A.J.L., F.Y., J.B.F., J.M.O., C.A.L., L.B., and P.K.S. analyzed data; and S.K.S., V.P., K.M., V.G., N.J.S., J.B.F., C.A.L. and P.K.S. wrote the paper.
COMPETING INTERESTS: The authors have declared that no competing interests exist.
References
- Akiyama K, Chikayama E, Yuasa H, Shimada Y, Tohge T, Shinozaki K, Hirai MY, Sakurai T, Kikuchi J, Saito K. PRIMe: a Web site that assembles tools for metabolomics and transcriptomics. In silico biology. 2008;8:339–345. [PubMed] [Google Scholar]
- Behrens ME, Grandgenett PM, Bailey JM, Singh PK, Yi CH, Yu F, Hollingsworth MA. The reactive tumor microenvironment: MUC1 signaling directly reprograms transcription of CTGF. Oncogene. 2010;29:5667–5677. doi: 10.1038/onc.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris H, Storniolo AM. Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer. 1997;33(Suppl 1):S18–22. doi: 10.1016/s0959-8049(96)00324-3. [DOI] [PubMed] [Google Scholar]
- Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012a;109:13787–13792. doi: 10.1073/pnas.1203339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaika NV, Yu F, Purohit V, Mehla K, Lazenby AJ, DiMaio D, Anderson JM, Yeh JJ, Johnson KR, Hollingsworth MA, Singh PK. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS One. 2012b;7:e32996. doi: 10.1371/journal.pone.0032996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- Cui Q, Lewis IA, Hegeman AD, Anderson ME, Li J, Schulte CF, Westler WM, Eghbalnia HR, Sussman MR, Markley JL. Metabolite identification via the Madison Metabolomics Consortium Database. Nature biotechnology. 2008;26:162–164. doi: 10.1038/nbt0208-162. [DOI] [PubMed] [Google Scholar]
- Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010;70:859–862. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer T, Sinnaeve D, Van Gasse B, Tsiporkova E, Rietzschel ER, De Buyzere ML, Gillebert TC, Bekaert S, Martins JC, Van Criekinge W. NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm. Analytical chemistry. 2008;80:3783–3790. doi: 10.1021/ac7025964. [DOI] [PubMed] [Google Scholar]
- de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. European journal of pharmacology. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Fanciulli M, Bruno T, Giovannelli A, Gentile FP, Di Padova M, Rubiu O, Floridi A. Energy metabolism of human LoVo colon carcinoma cells: correlation to drug resistance and influence of lonidamine. Clin Cancer Res. 2000;6:1590–1597. [PubMed] [Google Scholar]
- Gunda V, Yu F, Singh PK. Validation of Metabolic Alterations in Microscale Cell Culture Lysates Using Hydrophilic Interaction Liquid Chromatography (HILIC)-Tandem Mass Spectrometry-Based Metabolomics. PLoS One. 2016;11:e0154416. doi: 10.1371/journal.pone.0154416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heinemann V, Wilke H, Mergenthaler HG, Clemens M, Konig H, Illiger HJ, Arning M, Schalhorn A, Possinger K, Fink U. Gemcitabine and cisplatin in the treatment of advanced or metastatic pancreatic cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2000;11:1399–1403. doi: 10.1023/a:1026595525977. [DOI] [PubMed] [Google Scholar]
- Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I, Bapiro TE, Frese KK, Gopinathan A, Richards FM, et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 2017 doi: 10.1136/gutjnl-2016-311954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nature protocols. 2009;4:1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M. Pancreatic tumors show high levels of hypoxia. International journal of radiation oncology, biology, physics. 2000;48:919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- Lin J, Denmeade S, Carducci MA. HIF-1alpha and calcium signaling as targets for treatment of prostate cancer by cardiac glycosides. Current cancer drug targets. 2009;9:881–887. doi: 10.2174/156800909789760249. [DOI] [PubMed] [Google Scholar]
- Lin J, Zhan T, Duffy D, Hoffman-Censits J, Kilpatrick D, Trabulsi EJ, Lallas CD, Chervoneva I, Limentani K, Kennedy B, et al. A pilot phase II Study of digoxin in patients with recurrent prostate cancer as evident by a rising PSA. American journal of cancer therapy and pharmacology. 2014;2:21–32. [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Chien CW, Lee JC, Yeh YC, Hsu KF, Lai YY, Lin SC, Tsai SJ. Suppression of dual-specificity phosphatase-2 by hypoxia increases chemoresistance and malignancy in human cancer cells. The Journal of clinical investigation. 2011;121:1905–1916. doi: 10.1172/JCI44362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla K, Singh PK. MUC1: a novel metabolic master regulator. Biochim Biophys Acta. 2014;1845:126–135. doi: 10.1016/j.bbcan.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MP, Tentler JJ, Kulikowski GN, Tan AC, Bradshaw-Pierce EL, Pitts TM, Brown AM, Nallapareddy S, Arcaroli JJ, Serkova NJ, et al. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer Res. 2012;18:1051–1062. doi: 10.1158/1078-0432.CCR-11-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BD, Meng X, Donovan KJ, Shaka AJ. SOGGY: solvent-optimized double gradient spectroscopy for water suppression. A comparison with some existing techniques. J Magn Reson. 2007;184:263–274. doi: 10.1016/j.jmr.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Ruckemann K, Fairbanks LD, Carrey EA, Hawrylowicz CM, Richards DF, Kirschbaum B, Simmonds HA. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. The Journal of biological chemistry. 1998;273:21682–21691. doi: 10.1074/jbc.273.34.21682. [DOI] [PubMed] [Google Scholar]
- Schlaepfer IR, Glode LM, Hitz CA, Pac CT, Boyle KE, Maroni P, Deep G, Agarwal R, Lucia SM, Cramer SD, et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[(18)F]Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol Imaging Biol. 2015;17:529–538. doi: 10.1007/s11307-014-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, Mehla K, Pipinos II, Powers R, Yu F, Singh PK. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer & metabolism. 2014;2:18. doi: 10.1186/2049-3002-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Behrens ME, Eggers JP, Cerny RL, Bailey JM, Shanmugam K, Gendler SJ, Bennett EP, Hollingsworth MA. Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. J Biol Chem. 2008;283:26985–26995. doi: 10.1074/jbc.M805036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Liu X, Chi W, Liu Y, Wei L, Wang X, Yu J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1alpha gene. Cancer chemotherapy and pharmacology. 2006;58:776–784. doi: 10.1007/s00280-006-0224-7. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, Gil Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33:3812–3819. doi: 10.1038/onc.2013.357. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. HMDB 3.0–The Human Metabolome Database in 2013. Nucleic acids research. 2013;41:D801–807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley B, Powers R. MVAPACK: a complete data handling package for NMR metabolomics. ACS chemical biology. 2014;9:1138–1144. doi: 10.1021/cb4008937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Xia J, Wishart DS. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics. 2016;55:14 10 11–14 10 91. doi: 10.1002/cpbi.11. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi K, Fidler IJ. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:2299–2306. doi: 10.1158/1078-0432.ccr-03-0488. [DOI] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, Schito L, Chen J, Krishnamachary B, Winnard PT, Jr, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–1770. doi: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV, et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene. 2010;29:2962–2972. doi: 10.1038/onc.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3, related to STAR methods. Primers for qPCR