

Abstract
MECP2, an X-linked gene encoding the epigenetic factor methyl-CpG-binding protein-2, is mutated in Rett syndrome (RTT) and aberrantly expressed in autism. Most children affected by RTT are heterozygous Mecp2−/+ females whose brain function is impaired postnatally due to MeCP2 deficiency. Recent studies suggest a role of glia in causing neuronal dysfunction via a non-cell-autonomous effect in RTT. Here we report a potent neurotoxic activity in the conditioned medium (CM) obtained from Mecp2-null microglia. Hippocampal neurons treated with CM from Mecp2-null microglia showed an abnormal stunted and beaded dendritic morphology, and signs of microtubule disruption and damage of postsynaptic glutamatergic components within 24 h. We identified that the toxic factor in the CM is glutamate, because (1) Mecp2-null microglia released a fivefold higher level of glutamate, (2) blockage of microglial glutamate synthesis by a glutaminase inhibitor abolished the neurotoxic activity, (3) blockage of microglial glutamate release by gap junction hemichannel blockers abolished the neurotoxic activity, and (4) glutamate receptor antagonists blocked the neurotoxicity of the Mecp2-null microglia CM. We further identified that increased levels of glutaminase and connexin 32 in Mecp2-null microglia are responsible for increased glutamate production and release, respectively. In contrast, the CM from highly pure Mecp2-null astrocyte cultures showed no toxic effect. Our results suggest that microglia may influence the onset and progression of RTT and that microglia glutamate synthesis or release could be a therapeutic target for RTT.
Introduction
Rett syndrome (RTT), caused by loss-of-function mutations in the X-linked MECP2 encoding methyl-CpG-binding protein 2 (MeCP2), is a devastating neurodevelopmental disorder that primarily affects young girls (Chahrour and Zoghbi, 2007). RTT infants develop normally until 6–18 months of age but then develop progressive loss of neurodevelopmental milestones, a process called regression. Clinical features include deceleration of brain growth, loss of motor skills including purposeful hand movements, ataxia, loss of vocalization skills, loss of cognitive capability, autistic features, seizures, and respiratory dysfunction (Ellaway and Christodoulou, 1999). RTT and the more prevalent regressive-type autism share a substantial phenotypic overlap, including the regressive clinical course, autistic behaviors, and dendritic and synaptic abnormalities (Levitt and Campbell, 2009). Because MeCP2 regulation is also abnormal in a large subgroup of autistic individuals (Samaco et al., 2004, 2005; Nagarajan et al., 2006, 2008), understanding the neurobiological substrates of RTT may help elucidate the complex mechanisms leading to autism.
MeCP2 is a DNA-binding epigenetic modulator that can both activate and repress transcription (Nan et al., 1997; Yasui et al., 2007; Chahrour et al., 2008). MeCP2 also affects differential splicing (Young et al., 2005). How MeCP2 deficiency causes neurological deficits is not well understood. Recent evidence indicates that RTT is a disease of synaptic plasticity (Asaka et al., 2006; Moretti et al., 2006) and RTT-like neurological deficits in both immature and mature Mecp2 mutant mice are reversible by postnatal activation of MeCP2 expression (Giacometti et al., 2007; Guy et al., 2007; Jugloff et al., 2008). These results suggest that RTT is potentially curable if synaptic disruption can be ameliorated.
Microglia are the resident inflammatory cells of the CNS. They extend an extensive network of processes in the CNS parenchyma. Although the traditional view held that microglia are activated to become part of the neuroinflammatory process after injury, recent studies suggest that microglia provide extensive and continuous surveillance of their cellular environment even in their “resting” state; thus they actively and constantly interact with neurons and astrocytes (Davalos et al., 2005; Nimmerjahn et al., 2005). Interestingly, a very recent study showed that microglial processes make brief and direct contacts with neuronal synapses at a regular frequency and that microglia may regulate the turnover of synaptic connections (Wake et al., 2009). Therefore, emerging evidence suggests that microglia may regulate synaptic functions and synaptic turnover without the context of neuroinflammation (Bessis et al., 2007). It is possible that inherent microglia abnormalities may influence neuronal and synaptic functions via a non-cell-autonomous effect.
We previously hypothesized that neuronal function in RTT may be detrimentally influenced by MeCP2-deficient glia in a non-cell-autonomous manner, in addition to the cell-autonomous damage in vulnerable neurons (Maezawa et al., 2009). We and others showed such an influence of MeCP2-deficient astrocytes on dendritic integrity (Ballas et al., 2009; Maezawa et al., 2009). During our study on astrocytes, we observed that the conditioned medium (CM) derived from primary mixed glia cultures established from Mecp2-null mice resulted in dendritic damage to differentiated wild-type (wt) neuronal cultures, while CM from highly pure Mecp2-null astrocyte cultures had no effect on neuronal cultures. Because one significant cell component in the mixed glial cultures is microglia, our result suggested a possible soluble factor(s) released by MeCP2-deficient microglia that compromise neuronal dendrites. Here we demonstrate that MeCP2-deficient microglia, without conventional signs of activation, release an abnormally high level of glutamate, causing excitotoxicity that may contribute to dendritic and synaptic abnormalities in RTT.
Materials and Methods
Mouse model of RTT.
Mecp2tm1.1Bird/+ mice originating from Dr. Adrian Bird's laboratory (Guy et al., 2001) were obtained from Jackson Laboratories. Mice were mated with C57BL/6J mice (Jackson Laboratories). Pups were immediately genotyped to determine the Mecp2 deletion according to the protocol provided by the Jackson Laboratory. The gender was determined using primers for the Sry gene on the Y chromosome, which were 5′-TGG GAC TGG TGA CAA TTG TC-3′ and 5′-GAG TAC AGG TGT GCA GCT CT-3′. The University of California Davis Institutional Animal Care and Use Committee approved all animal experiments.
Chemicals.
MK801 and NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide) were purchased from Calbiochem. 6-diazo-5-oxo-l-norleucine (DON), carbenoxolone disodium (CBX), and lanthanum chloride (Lan) were purchased from Sigma. All the connexin-mimetic peptides (Takeuchi et al., 2006) were purchased from American Peptide. The sequences of the peptides are as follows: for 32GAP24, GHGDPLHLEEVKC (intracellular loop, position 110–122 of connexin 32); for 32GAP27, SRPTEKTVFT (extracellular loop 2, position 182–191 of connexin 32); and for 43GAP27, SRPTEKTIFII (extracellular loop 2, position 201–210 of connexin 43).
Primary neural cultures.
Primary microglia cultures were prepared from mixed glia cultures with the “shaking-off” method as described previously (Suzumura et al., 1987). In the present study, our preparations were ≥99% pure for microglia as demonstrated by anti-CD11b immunostaining. Primary highly enriched astrocyte cultures were prepared as previously described (Maezawa et al., 2009). Cell viability was analyzed by LIVE/DEAD Reduced Biohazard Cell Viability Kit (Invitrogen) following the manufacturer's instruction.
To obtain CM, microglia and astrocytes were first cultured in 24-well culture plates for 24 h in DMEM with 10% fetal bovine serum (DMEM10). Cultures were washed extensively and changed to the Neurobasal medium with B27 supplement (NB/B27, Invitrogen) without serum and with or without indicated drug treatments. The NB/B27-based medium was conditioned for 24 h, collected, and briefly centrifuged. If not otherwise indicated, the medium contained 500 μmol/L glutamine. The concentrations of inhibitors used were as follows: DON, 1 mm; CBX, 200 μm; Lan, 50 μm; and connexin-mimetic peptides, 0.25 mg/L.
The hippocampal neuronal cultures were prepared from newborn wild-type C57BL/6J mice according to the method of Xiang et al. (1998). Neurons were cultured in NB/B27 at a density of 2.5 × 105 cells/well in 12-well plates or 8 × 105 cells/well in 6-well plates for at least 14 d before they were treated with microglia or astrocyte CM.
RT-PCR for Mecp2 transcript and quantitative RT-PCR for glutaminase and connexin 32.
Total RNA from microglia was isolated by RNeasy Mini kit (Qiagen) according to the manufacturer's instruction. Purified RNA was resuspended in RNase-free water and stored at −70°C until use. The forward and reverse primer sequences for wt Mecp2 transcript were 5′-GAC CCC TTG GGA CTG AAG TT-3′ and 5′-CCA CCC TCC AGT TTG GTT TA-3′ (Miralvès et al., 2007). RT-PCR was performed using SuperScript One-Step RT-PCR System (Invitrogen). Because these primer sets may also amplify the chromosomal DNA, the DNA content of the samples was further minimized by purifying RNA using the RNeasy MinElute Cleanup Kit (Qiagen) according to the manufacturer's instruction. Using the resulting RNA samples and the above primers, PCRs without prior reverse transcription yielded minimal detectable products.
We used a primer pair previously designed to quantify the murine glutaminase transcript in microglia (Takeuchi et al., 2006). The forward sequence was 5′-GTCACGATCTTGTTTCTCTGTG-3′ and the reverse sequence was 5′-GTCCAAAGAGCAGTGCTTCATCCATG-3′. We used a primer pair previously designed to quantify the murine connexin 32 transcript (Chanson et al., 1998). The forward sequence was 5′-AGTGCCAGGGAGGTGTGAAT-3′ and the reverse sequence was 5′-GGAACACCACACTGATGACA-3′. For GAPDH (glyceraldehyde 3-phosphate dehydrogenase), the forward sequence was 5′-ACTCACGGCAAATTCAACG-3′ and the reverse sequence was 5′-CCCTGTTGCTGTAGCCGTA-3′.
The cDNA was synthesized from 2 μg of total RNA using SuperScript First-stranded synthesis system (Invitrogen). Quantitative PCR was performed using the SYBR Green master mix in an ABI 7900HT Sequence Detection System (Applied Biosystems). The result was normalized to β-actin.
Immunofluorescence staining and quantification.
For immunofluorescence staining of brain tissue, we used paraformaldehyde-fixed frozen sections as previously described (Maezawa et al., 2009). Primary antibodies used were as follows: rabbit anti-MeCP2 IgG (1:250, UpState, for aa 465–478 of mouse MeCP2, detecting both MeCP2 isoforms, e1 and e2), chicken anti-MeCP2 IgY (1:1000, for the C terminal of MeCP2, detecting both e1 and e2 isoforms) (Yasui et al., 2007), and rat anti-CD11b IgG (1:100, Serotec).
For immunofluorescent staining of neurons in culture, neurons were fixed in 4% paraformaldehyde and stained with anti-PSD95 (1:200, Cell Signaling Technology), anti-MAP2 (microtubule-associated protein 2) (1:500, Millipore Bioscience Research Reagents), and anti-acetylated α-tubulin (1:250, Zymed) for overnight at 4°C followed by secondary Alexa488-conjugated anti-mouse or Alexa568-conjugated anti-rabbit antibody (1:700, Molecular Probes). Microglia cultures were similarly fixed and stained with anti-glutaminase (1:100, Frontier Science) or anti-connexin 32 (1:100, Zymed). Immunostained images were observed under a Nikon Eclipse E600 microscope and photographed by a digital camera (SPOT RTke, SPOT Diagnostics).
For quantification of immunofluorescent intensity, photomicrographs of MAP2-, PSD95-, and acetylated α-tubulin-immunostained cultures were randomly taken from each culture condition. The images were transformed to 8-bit grayscale and analyzed by the ImageJ program. The PSD95-immunoreactive puncta along the dendrites in each photomicrograph were counted manually and normalized by dendrite length. The photography and analysis of immunoreactivity were conducted in an investigator-blinded manner.
Western blot analysis.
To obtain cell lysates, cells were washed with ice-cold PBS and incubated with a buffer containing 50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 2% SDS, proteinase inhibitor mixture (Sigma), and phosphatase inhibitor mixture (Sigma). Lysates were briefly sonicated and cleared by centrifugation at 50,000 rpm for 10 min. Equivalent amounts of protein were analyzed by Tris-HCl gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes and probed with antibodies. Visualization was performed using enhanced chemiluminescence (ECL, GE Healthcare Pharmacia).
The following primary antibodies (dilutions) were used: anti-MeCP2 (1:4000, Aves), anti-CD11b (1:1000, Serotec), anti-GFAP (1:1000, Millipore Bioscience Research Reagents), anti-NeuN (1:1000, Millipore Bioscience Research Reagents), anti-synaptophysin (1:1000, Abcam), anti-PSD95 (1:1000, cell signaling), anti-GRIP1 (1:1000, UpState), anti-MAP2 (1:1000, Millipore Bioscience Research Reagents), anti-GluR1 (1:1000, Assay Designs), anti-GluR2/3 (1:1000, Millipore), anti-NR-1 (1:1000, Millipore), anti-GluR6/7 (1:1000, Millipore), anti-acetylated α-tubulin (1:2000, Zymed), and anti-β-actin (1:3000, Sigma). Secondary antibodies were HRP-conjugated anti-rabbit, anti-goat, or anti-mouse antibody (1:3000, GE Healthcare).
Preparation of synaptosomes.
To investigate the levels of synaptic proteins in brain, synaptosomes were prepared from the cerebral cortex of 9-week-old male wt and Mecp2-null mice by use of Percoll density gradients according to the methods described by Braun and Madison (2000). Briefly, fresh cortical samples were homogenized in Buffer A (0.32 m sucrose, 5 mm HEPES, pH 7.4, and 0.1 mm EDTA) with protease inhibitor using a Teflon glass homogenizer. The homogenate was centrifuged for 10 min at 1000 × g, and the supernatant was further centrifuged for 20 min at 12,000 × g. The pellet was resuspended in Buffer B (0.25 m sucrose, 5 mm HEPES, pH 7.4, and 0.1 mm EDTA) and layered onto a discontinuous 7.5–10–16% Percoll gradient. The gradient was centrifuged for 20 min at 15,000 × g in a Beckman SW41Ti rotor. Synaptosomes were collected from the 10–16% interface in the gradient. After isolation, synaptosomes were washed and resuspended in a balanced salt solution containing the following (in mm): (128 NaCl, 2.4 KCl, 1.2 MgSO4 1.2 KH2PO4, 10 HEPES, pH 7.4, and 10 d-glucose).
Glutamate assay.
Microglia were cultured in 48-well plates at a density of 7 × 104 cells/well in DMEM10 for 24 h. The cultures were washed and incubated in NB/B27 for another 24 h. The NB/B27-based conditioned medium was collected and briefly centrifuged. The glutamate level in the medium was measured as previously described (Montana et al., 2004) by using the glutamine/glutamate determination kit (Sigma) following the manufacturer's instruction.
Sandwich ELISA for cytokines.
To induce the innate immune response, microglia cultures were washed three times with serum-free Opti-MEM and cultured in Opti-MEM containing 100 ng/ml lipopolysaccharides (LPSs, Calbiochem) for 24 h. The activation was evaluated by measuring cytokines TNF-α and IL-6 in the medium using cytokine ELISA kits according to the manufacturer's instruction (R&D Systems and RayBiotec). To exclude the possibility that TNF-α is obscured by soluble receptors and other binding proteins, we heated the same samples at 95°C for 5 min in the presence of 1% SDS, diluted the samples 10-fold, and repeated the measurements by ELISA. The results were similar to those obtained without the heating procedure.
Statistical analysis.
Statistical analyses were performed using the SigmaPlot 11 software (Systat Software, Inc). ANOVA was used to compare quantitative values from cultures across groups. Tukey's studentized range test was used to adjust for multiple comparisons in post hoc pairwise tests.
Results
Microglia express MeCP2
Although MeCP2 had previously been thought to be absent in glia due to an apparent lack of immunoreactivity in glia compared with neighboring neurons (Shahbazian et al., 2002), recent data have shown MeCP2 expression in astrocytes, oligodendrocytes, and polydendrocytes (Schmid et al., 2008; Ballas et al., 2009; Maezawa et al., 2009). To determine whether microglia express MeCP2, we first examined the cerebral sections of littermates of an established RTT model with the Cre-mediated deletion of Mecp2 exons 3 and 4 (Mecp2tm1.1Bird/+ mice) (Guy et al., 2001). This Mecp2 deletion model shows RTT-like neuropathology and an earlier onset of neurological symptoms in males (Mecp2−/y, null) than in females (Mecp2−/+, mosaic heterozygous). Using this model, we previously demonstrated the expected strong neuronal MeCP2 immunoreactivity and the clearly positive but relatively weak astrocytic MeCP2 immunoreactivity (Maezawa et al., 2009). Using the CD11b antibody to specifically mark the microglia, we found that similar to astrocytes, microglia from wild-type male Mecp2+/y mice showed detectable but faint nuclear MeCP2 immunoreactivity. In contrast, microglia from male null (Mecp2−/y) mice (null microglia) showed no such immunoreactivity (Fig. 1A).
Figure 1.
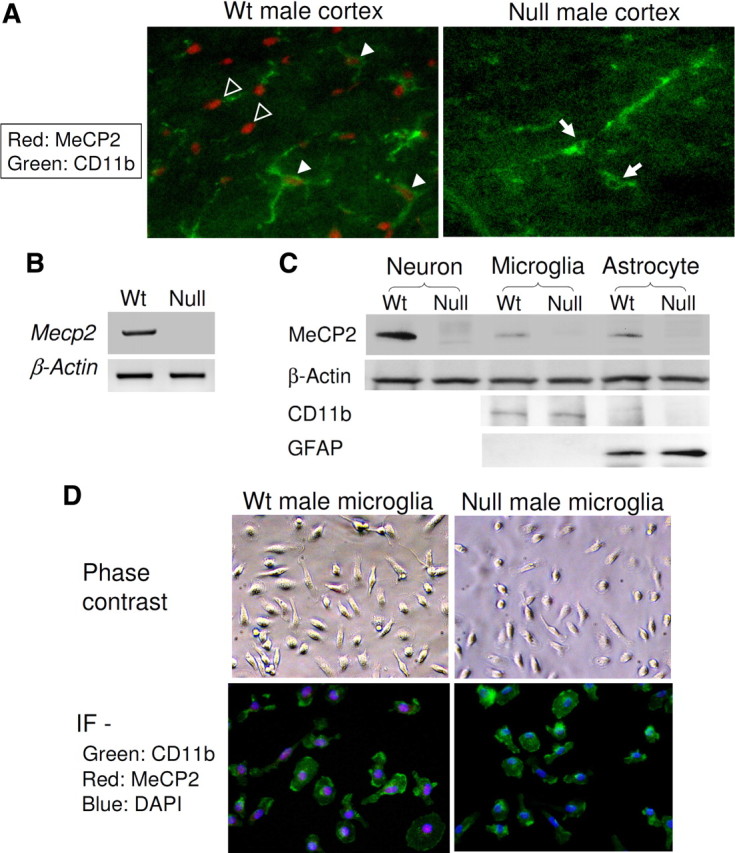
Microglia express MeCP2. A, Cortical sections from the indicated mice of 7 weeks of age were coimmunostained for MeCP2 (red nuclear stain) and the microglia marker CD11b (green cytoplasmic stain). Representative photomicrographs are shown. Filled arrowheads point to wt microglia with clear nuclear MeCP2 immunoreactivity, while arrows point to microglia without MeCP2 immunoreactivity in the null mouse. The brightly MeCP2-immunoreactive nuclei are likely those of neurons (empty arrowheads). B, Microglia derived from male mice of indicated Mecp2 genotypes (wt: Mecp2+/y; null: Mecp2−/y) were cultured and RNA was extracted. RT-PCR showed the presence of Mecp2 transcript in wt microglia but not in null microglia. C, Western blot analysis of lysates from indicated cells derived from wt or null mice, analyzed by antibodies to MeCP2, the protein loading control β-actin, microglia marker CD11b, and the astrocytic marker GFAP. D, Microglia cultured from mice of indicated Mecp2 genotypes were doubly immunofluorescently (IF) stained as in A and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
We next investigated cultured primary microglia derived from newborn mice. These cultures were typically ≥99% pure for microglia as assessed by anti-CD11b immunostain and were completely devoid of neurons (Maezawa et al., 2006). Western blot also confirmed the lack of the astrocytic marker GFAP in our microglia cultures and the lack of the microglia marker CD11b in our astrocyte cultures (Fig. 1C). RT-PCR and Western blot showed that wt microglia, but not null microglia, expressed MeCP2 transcript and protein, respectively (Fig. 1B,C). The MeCP2 protein level expressed by wt microglia was slightly less than that by wt astrocytes, and was ∼20% of that of neurons (Fig. 1C). wt microglia and Mecp2-null microglia did not show difference in viability or proliferation in cultures (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The morphology of Mecp2-null microglia was indistinguishable from wt microglia (Fig. 1D). Double immunofluorescent stains confirmed that almost all CD11b-immunoreactive wt microglia expressed MeCP2, while the CD11b-immunoreactive Mecp2-null microglia did not (Fig. 1D).
Conditioned media from MeCP2-deficient microglia are toxic to dendrites
Because we found a neurotoxic effect of CM from mixed glial cultures (data not shown), of which astrocytes were the major, and microglia the minor component cells, we compared the neurotoxicity between CM obtained from highly pure astrocyte and highly pure microglia cultures. It is estimated that in brain the ratio of astrocytes to microglia is approximately 3–4 to 1 (Streit, 2005; Pelvig et al., 2008). For pathophysiological relevance, therefore, we generated astrocyte CM from 2.0 × 105/cm3 astrocytes, four times more cells than the 0.5 × 105/cm3 microglia used for generating microglia CM. We transferred the CM from astrocyte or microglia cultures to hippocampal neurons that had been cultured for 14 d in vitro. Within 24 h, neurons treated with CM from Mecp2-null microglia cultures (null MCM), but not those treated with CM from wt microglia cultures (wt MCM), showed robust signs of dendritic damage (Fig. 2A). Dendrites of neurons treated with null MCM were thinner, shorter, with significantly stunted arborization and frequently fragmented or “beaded” appearance, best illustrated by sparsely plated neurons (Fig. 2B) (Takeuchi et al., 2006). The immunofluorescent stain for the dendritic marker MAP2 showed a dramatic reduction of staining intensity and the length of dendrites stained (Fig. 2A,B; for quantification, see Fig. 5B). The dendritic damage was further shown by immunofluorescence for acetylated tubulin (Ac-TN) (Fig. 2A; for quantification, see Fig. 5C). Although affecting dendrites, the null MCM did not affect the number of viable neurons (Fig. 2C). This effect of null MCM was dose dependent; null MCM added to the neuronal culture medium at levels >20% caused significant dendritic damage (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 2.
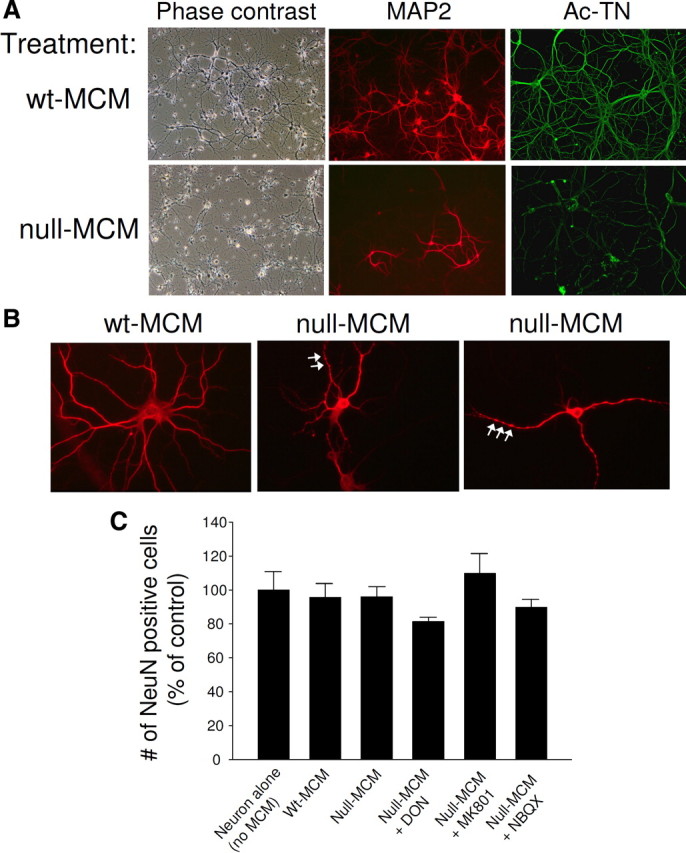
Conditioned medium from Mecp2-null microglia caused damage to dendrites. wt hippocampal neurons, at least 14 d in vitro, were incubated with 50% conditioned media from wt microglia (wt MCM) or null microglia (null MCM) for 24 h. A, Representative phase contrast images and immunofluorescent images in which the dendrites were demonstrated by immunostaining for MAP2 (red) and Ac-TN (green). B, Sparsely plated neurons immunostained for MAP2. Shown are examples of neurons damaged by null MCM showing stunted dendritic morphology and frequent beaded appearance (arrows). C, NeuN-immunoreactive neurons in each treatment condition were counted and averaged from three independent experiments to demonstrate that neuronal viability was not affected by MCM treatment or the drug treatment shown in Figures 3 and 5. The drugs shown here are glutaminase inhibitor DON, to reduce the glutamate production by microglia, and glutamate receptor antagonists MK801 and NBQX, to block the neuronal action of glutamate.
Figure 5.
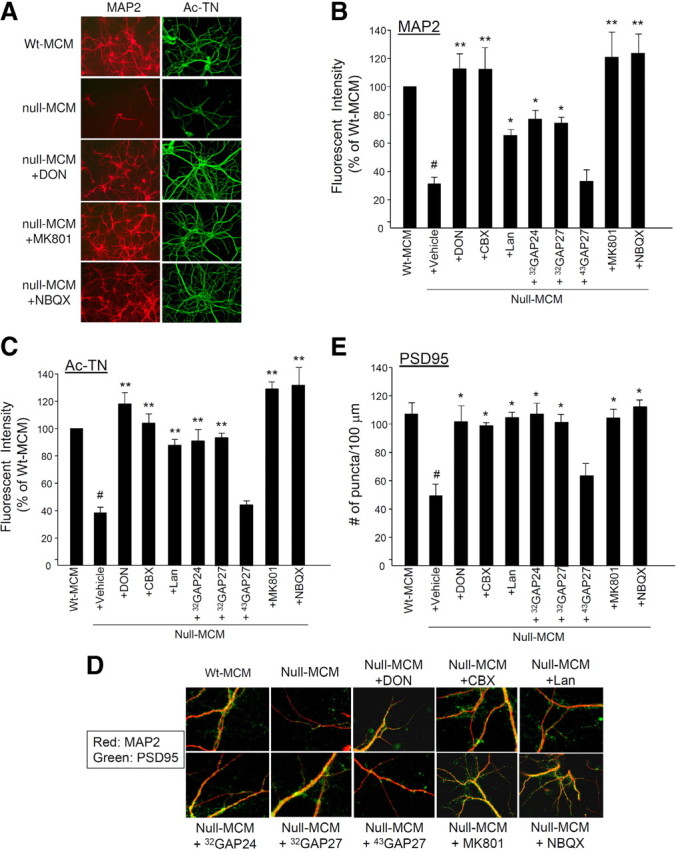
Inhibition of glutamate production/release or of glutamate action on receptors ameliorate the toxicity of null MCM. wt hippocampal neurons were treated with wt MCM or null MCM from indicated treatments as shown in Figure 4A. A–C, Neurons were treated as indicated and immunostained for MAP2 (red) and Ac-TN (green). Representative photomicrographs are shown in A. In B (MAP2) and C (Ac-TN), the immunoreactivities were quantified for each treatment and presented as percentages of control (wt MCM treatment group). n = 3, *p < 0.05 and **p < 0.001 compared with the null-MCM-plus-vehicle group; #p < 0.001 compared with the wt MCM group. There was no significant difference between the null-MCM-plus-vehicle and the null-MCM-plus-43GAP27 groups. D, Neurons were doubly immunostained for MAP2 (red) and PSD95 (green). Presented are representative merged images, in which the orange-yellow color represents colocalization of MAP2 and PSD95 immunoreactivities. The quantification of the count of PSD95-immunoreactive puncta per unit (100 μm) length of MAP2-positive dendrite is presented in E. n = 3, *p < 0.05 compared with the null-MCM-plus-vehicle group, #p < 0.05 compared with the wt MCM group. There was no significant difference between the null-MCM-plus-vehicle and the null-MCM-plus-43GAP27 groups.
In addition to the substantial reductions of MAP2 and Ac-TN indicating microtubule disruption, the number of PSD95-immunoreactive puncta, which represent the postsynaptic density (PSD) of excitatory synapses, and the overall PSD95 immunofluorescent intensity were also significantly diminished by null MCM (Fig. 3A; for quantification, see Fig. 5E). Western blots of neuronal extracts revealed that null-MCM-treated neurons, compared with wt MCM-treated neurons, showed reduced levels of MAP2, Ac-TN, PSD95, and another scaffolding protein in the postsynaptic compartment called glutamate receptor interacting protein 1 (GRIP1) (Hoogenraad et al., 2005), but not the presynaptic marker synaptophysin (Fig. 3B). This postsynaptic damage was also associated with decreased levels of NMDA, AMPA, and kainate glutamate receptor subunits NR1, GluR2/3, and GluR6/7, respectively (Fig. 3B). These results suggest that null MCM caused damage to the postsynaptic elements of excitatory synapses. Resembling this in vitro pattern of synaptic markers, synaptosomes prepared from 9 week-old Mecp2-null mice showed ∼30–40% decreases of PSD95, NR1, GluR2/3, and GluR6/7 levels compared with age-matched male wt littermates, although the differences for NR1 and GluR2/3 did not reach statistical significance. The level of the synaptophysin change is not remarkable (Fig. 3C,D). These results suggest that neurotoxicity from Mecp2-null microglia may contribute to synaptic changes in vivo.
Figure 3.

Null MCM caused damage to the postsynaptic elements of excitatory synapses. wt hippocampal neurons were cultured and treated as shown in Figure 2. A, Neurons were doubly immunofluorescently stained with antibodies to MAP2 (red) and PSD95 (green). Representative photomicrographs showing MAP2-positive dendrites are presented to illustrate the decreased abundance of PSD95-immunoreactive puncta along the dendrites of null-MCM-treated neurons. B, Western blot analysis of lysates from wt neurons with indicated treatment, analyzed by antibodies to dendritic proteins MAP2 and Ac-TN, postsynaptic protein PSD95 and GRIP1, presynaptic protein synaptophysin, and glutamate receptor subunits NR1, GluR2/3, and GluR6/7. The drugs shown here are glutaminase inhibitor DON and glutamate receptor antagonists MK801 and NBQX (see text). C, The levels of indicated synaptic proteins in synaptosomes prepared from 9-week-old Mecp2-null mice and their male wt littermates (n = 3 in each group) were evaluated by Western blot. D, The band intensities were quantified and expressed as percentages of values obtained from wt. *p < 0.05 and **p < 0.001 compared with the wt values.
In contrast, astrocyte CM (ACM) from both wt and null (Mecp2−/y) cultures induced no dendritic or synaptic damage, even when neurons were treated with 100% ACM (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and incubated for 3–6 d. Considering that our ACM samples were conditioned by four times more cells than the MCM samples, the MeCP2-deficient microglia, but not the astrocytes, appear to release highly active soluble factors that damage dendrites.
MeCP2-deficient microglia release a high level of glutamate
Numerous studies have shown that microglia activation could be neurotoxic by releasing proinflammatory and potentially toxic molecules including cytokines, chemokines, reactive oxygen and nitrogen species, and prostaglandin E2 (Block et al., 2007). However, the release of these neurotoxic molecules appears to be kept to a very low level when the cultured microglia are not activated by exogenous stimuli. We speculated that null microglia may over-release some of these neurotoxic molecules, despite the fact that our cultured null microglia, with morphology indistinguishable from wt microglia, did not assume an activated morphology and did not show enhanced proliferation.
The beaded and stunted dendritic morphology of neurons treated with null MCM is similar to that previously described for neurotoxicity caused by LPS-activated microglia (Maezawa et al., 2006; Takeuchi et al., 2006). In this model, tumor necrosis factor-α (TNF-α) is released by microglia and further upregulates in an autocrine manner the synthesis and release of a large amount of glutamate, thus causing excitotoxicity of neurons. Furthermore, it was previously shown that microtubule disruption and PSD remodeling involving the degradation of PSD95 and GRIP1 can be induced by glutamate stimulation (Guo and Wang, 2007; Hoskison et al., 2007; Forder and Tymianski, 2009). Therefore we reasoned that TNF-α and glutamate were two candidate soluble neurotoxic factors released by null microglia. We found that null microglia, similar to wt microglia, released an almost undetectable level of TNF-α when not stimulated. After stimulation by LPS, null microglia released substantially less TNF-α than wt microglia (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This aberrant regulation of cytokine release after activation appears to be specific to TNF-α because the release of the other proinflammatory cytokine interleukin 6 was comparable between wt and null microglia (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Interestingly, this under-responsive cytokine pattern resembles that manifested by MeCP2-deficient astrocytes (Maezawa et al., 2009).
We further showed that there was no increase in the release of nitric oxide and prostaglandin E2 by null microglia (data not shown). Together with the low levels of proinflammatory cytokines, these data support a lack of activated phenotype in null microglia. Despite this, the level of glutamate released by null microglia was approximately fivefold of that by wt microglia (Fig. 4A). Therefore, glutamate could be the active neurotoxic factor released by null microglia.
Figure 4.
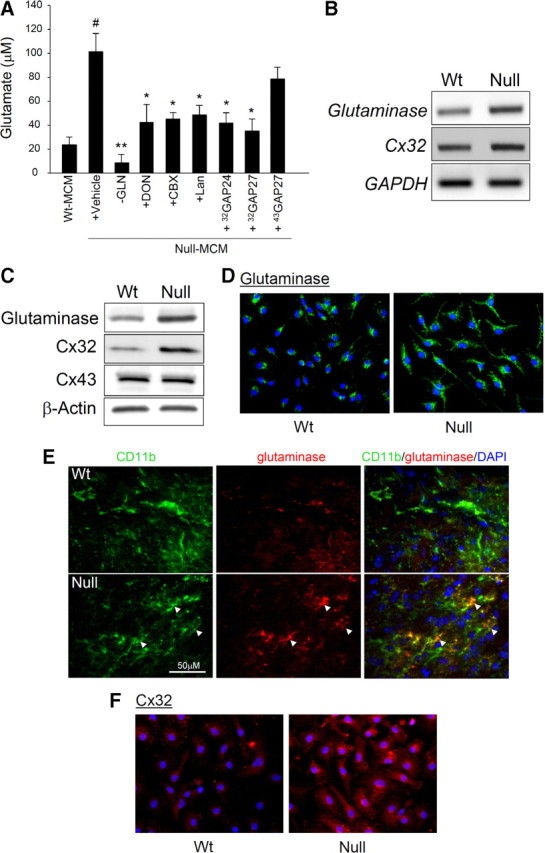
Mecp2-null microglia released a high level of glutamate due to increased production by glutaminase and increased release through Cx32 hemichannels. A, The glutamate concentrations in the 24 h conditioned medium of wt microglia (wt MCM), or Mecp2-null microglia (null MCM) with indicated treatments of microglia were measured. The treatments included culturing in medium lacking glutamine (−GLN) or in the presence of glutaminase inhibitor DON, gap junction blocker CBX, gap junction hemichannel blocker Lan, Cx32-mimetic peptides 32GAP24 and 32GAP27, and Cx43-mimetic peptide 43GAP27. The vehicle treatment control was DMSO diluted 500-fold into the medium. n = 3, *p < 0.05 and **p < 0.001 compared with the null-MCM-plus-vehicle group, #p < 0.05 compared with the wt MCM group. There was no significant difference between the null-MCM-plus-vehicle and the null-MCM-plus-43GAP27 groups. B, The levels of indicated transcripts were measured by quantitative RT-PCR (see text for results). Shown are representative PCR products in gel. C, Western blot analysis of cell lysates from microglia derived from mice of indicated Mecp2 genotype. See text for quantitative data. D, Microglia derived from mice of indicated Mecp2 genotypes were cultured and immunostained for glutaminase (green) and counterstained with DAPI (blue). E, Sections from the frontal brain region of 7-week-old male littermates of indicated Mecp2 genotypes were coimmunostained for CD11b (green) and glutaminase (red) and counterstained with DAPI (blue). Right, Merged images, in which the CD11b-immunoreactive microglia with high levels of glutaminase are indicated by orange-yellow immunoreactive areas (arrowheads). F, Microglia derived from mice of indicated Mecp2 genotypes were cultured and immunostained for Cx32 (red) and counterstained with DAPI (blue).
The proximate mechanism of elevated release of glutamate by MeCP2-deficient microglia
To reach a high extracellular glutamate level, MeCP2-deficient microglia may have a higher rate of production, a higher rate of release, or a lower rate of uptake of glutamate. To investigate the possibility of a higher rate of production, we focused on glutaminase, which produces glutamate from glutamine and is a major enzyme for glutamate synthesis in microglia. Previous reports showed that in TNF-α-activated microglia and HIV-infected macrophages/microglia, glutaminase is responsible for generating excessive glutamate, causing toxicity to cultured neurons (Zhao et al., 2004; Takeuchi et al., 2006). We found that the excessive glutamate generation by MeCP2-deficient microglia was blocked when cultured in medium lacking glutamine or in the presence of 6-diazo-5-oxo-l-norleucine (DON), an inhibitor of glutaminase (Takeuchi et al., 2006) (Fig. 4A). These results indicate that glutaminase is responsible for excessive glutamate production by Mecp2-null microglia. In addition, glutaminase expression in microglia is upregulated by MeCP2 deficiency. Quantitative RT-PCR showed that the transcript level of glutaminase in cultured Mecp2-null microglia was 152 ± 6% (mean ± SE) of the level in wt microglia (n = 3, p < 0.05) (Fig. 4B). Western blots showed that the protein level of glutaminase in cultured Mecp2-null microglia was 218 ± 27% of the level in wt microglia (n = 4, p < 0.05) (Fig. 4C). Immunocytochemistry also confirmed the more pronounced glutaminase immunoreactivities in Mecp2-null microglia, both in culture (Fig. 4D) and in vivo (Fig. 4E). The Mecp2-null microglia, in contrast to wt microglia, showed diffuse cytoplasmic immunoreactivities (Fig. 4D), which might be consistent with the notion that glutaminase released into cytosol from its normal mitochondria localization becomes more potent in generating glutamate (Erdmann et al., 2009). The increase in glutaminase was not observed in astrocytes (supplemental Fig. 5, available at www.jneurosci.org as supplemental material), consistent with our previous observation of the lack of increased production of glutamate by Mecp2-null astrocytes (Maezawa et al., 2009).
Glutamate is generally considered to be released through exocytosis or glutamate transporter, such as excitatory amino acid transporters and transporter system Xc− (Nicholls and Attwell, 1990; Barger and Basile, 2001; Barger et al., 2007). Recently, it was shown in the TNF-α-induced neurotoxicity model that the pool of glutamate generated by microglial glutaminase is released mainly via connexin 32 (Cx32) hemichannels of the gap junction (Takeuchi et al., 2006). Because our previous study on MeCP2-deficient astrocytes implicates a pathological role of gap junction hemichannels, here we focused on whether dysregulation of microglial hemichannels mediates the release of excessive glutamate. To evaluate this possibility, we tested carbenoxolone (CBX), a widely used gap junction blocker that also blocks the unopposed connexin hemichannel, and Lan, a hemichannel blocker that does not affect gap junctions when applied extracellularly (Anselmi et al., 2008). Both blockers reduced the level of glutamate released by Mecp2-null microglia (Fig. 4A). Next, we tested the effect of mimetic peptides that are identical to a short amino-acid sequence on the connexin subunit and have been shown to specifically block hemichannels made of their targeted connexin subunits (De Vuyst et al., 2006; Takeuchi et al., 2006). Blockade of Cx32 with 32GAP24 or 32GAP27 substantially diminished the excessive glutamate release, whereas blockade of connexin 43 (Cx43) with 43GAP27 did not. Although CBX is also an effective blocker of VRAC (volume-regulated anion channel)-mediated glutamate release (Ye et al., 2009), the above results using more specific hemichannel blockers suggest that the unopposed Cx32 hemichannel is the key mediator of abnormal glutamate release by Mecp2-null microglia. Interestingly, there was a 3.3 ± 0.43-fold increase of the level of Cx32 protein in Mecp2-null microglia compared with wt microglia (Fig. 4C) (p < 0.001, n = 3), although quantitative RT-PCR did not show an increase of the Cx32 transcript level (Fig. 4B). Immunocytochemistry also showed pronounced Cx32 immunoreactivities in Mecp2-null microglia, while wt microglia only showed background level immunoreactivities (Fig. 4F). Consistent with the lack of Cx43 involvement, the expression of Cx43 showed no changes by MeCP2 deficiency (Fig. 4C).
Together, our results indicate that the proximate mechanism of over-release of glutamate by Mecp2-null microglia involves the increased production of glutamate by glutaminase and increased release by Cx32 hemichannels. These functional increases can be explained in part by the upregulation of glutaminase at the transcript and protein levels and of Cx32 at the protein level.
The dendritotoxicity and synaptotoxicity can be attributed to elevated glutamate released by MeCP2-deficient microglia
To determine whether glutamate is responsible for the dendritotoxic and synaptotoxic activity in the null MCM, we used the above blockers to determine whether selectively reducing the glutamate level in null MCM also ameliorates neurotoxicity. We treated neurons with null MCM of which the glutamate level was reduced due to glutaminase inhibition by DON, or due to hemichannel blockade by CBX, Lan, 32GAP24 or 32GAP27. Neurons thus treated showed well preserved dendritic morphology. (Representative photomicrographs are shown in Fig. 5A.) Both immunofluorescence staining and Western blotting showed that the levels of dendritic proteins MAP2 and Ac-TN (Figs. 3B, 5A–C), postsynaptic density proteins PSD95 and GRIP1 (Figs. 3B, 5D,E), and glutamate receptor subunits NR1, GluR2/3, and GluR6/7 (Fig. 3B) were also preserved.
To further confirm the role of glutamate, we determined whether neurons could be protected from dendritotoxicity and synaptotoxicity by the presence of NMDA receptor antagonist MK801 or the AMPA receptor antagonist NBQX. Interestingly, both compounds provided full protection of neurons as shown by full preservation of dendrites (Fig. 5A). Both MK801 and NBQX also substantially preserved the levels of dendritic proteins MAP2 and Ac-TN (Figs. 3B, 5A–C), postsynaptic density proteins PSD95 and GRIP1 (Figs. 3B, 5D,E), and glutamate receptor subunits NR1, GluR2/3, and GluR6/7 (Fig. 3B). The protective effect was not due to an enhancement of neuronal survival in culture because there was no change in the count of NeuN-immunoreactive cells (Fig. 2C). Collectively, our results indicate that glutamate is the major neurotoxic factor released by Mecp2-null microglia and suggest that both neuronal NMDA and AMPA receptors are required for this mode of toxicity.
Discussion
RTT has been considered to be the result of cell-autonomous neuronal Mecp2 mutations, and glia have been excluded from prior investigations because of the reported absence of MeCP2 (Shahbazian et al., 2002). However, emerging evidence, including the results presented here, strongly suggest that the expression of MeCP2 in glia is more ubiquitous than originally thought, and that MeCP2 deficiency in glia may have a profound impact on brain function. The current study focuses on a neurotoxic activity released by MeCP2-deficient microglia. We found that this activity comes from the excessively released glutamate, as a consequence of enhanced glutaminase generation and Cx32 hemichannel-mediated release.
Previous studies on astrocytes by us (Maezawa et al., 2009) and by Ballas et al. (2009) demonstrated that MeCP2-deficient astrocytes detrimentally influence the neuronal dendrite formation in a non-cell-autonomous manner, although the neurotoxic mechanisms suggested by us and by Ballas et al. were different. We suggested a loss-of-function mechanism via which MeCP2-deficient astrocytes fail to provide adequate support for dendritic arborization and maturation, while Ballas et al. suggested a gain-of-function mechanism in which MeCP2-deficient astrocytes release a soluble neurotoxic activity into the culture medium to damage the dendrites. This released soluble activity found by Ballas et al. appeared rather slow-acting, requiring long incubations of at least 3 d in culture. The responsible soluble neurotoxic factors have not been identified (Ballas et al., 2009). Multiple experiments performed in our laboratory, however, could not demonstrate any such neurotoxicity induced by the CM from highly pure cultures of MeCP2-deficient astrocytes. Rather, we observed a source of potent neurotoxicity from microglia. Despite the fact that we generated astrocyte CM using four times more cells than those used for generating microglia CM, CM from Mecp2-null astrocytes at 5 to 100% did not induce any apparent morphological aberrations of neurons. In contrast, CM generated from Mecp2-null microglia induced robust neuronal damage within 24 h, even when it was titrated down to 20%. We conclude that microglia, but not astrocytes, are the major source of the soluble neurotoxic activity released by RTT glia.
Based on several lines of evidence as follows, we identified glutamate as the major soluble toxic factor released by null microglia: (1) Mecp2-null microglia released an abnormally high level (fivefold of the wt level) of glutamate, (2) the reduction of the microglial glutamate production or release rendered the null MCM much less toxic, and (3) specific glutamate receptor antagonists blocked the neurotoxicity of null MCM. Although not studied here, it would be interesting to determine whether other key neurotoxic glutamate receptor agonists such as quinolinic acid (Heyes et al., 1996) or d-serine (Wu et al., 2004) are also abnormally released by Mecp2-null microglia. It has been shown that these coagonists are released by microglia in proinflammatory conditions and can accentuate the actions of other excitatory amino acids (Wu et al., 2004, 2005; Yamada et al., 2009).
The proximate mechanism of excessive glutamate release by Mecp2-null microglia is strikingly similar to the previously reported mechanism mediating the TNF-α-induced neurotoxicity seen in neurodegenerative disorders (Takeuchi et al., 2006). In this model, microglia, upon activation (for example, by LPS), release TNF-α, which then stimulates excessive microglial glutamate release in an autocrine manner to cause excitatory neurotoxicity. TNF-α was shown to upregulate glutaminase to generate more glutamate, which was then released mainly through Cx32 hemichannels (Takeuchi et al., 2006). Despite this similarity in mechanism, however, our Mecp2-null microglia cultures, if not stimulated, showed no conventional evidence of an activated phenotype. There is no evidence of abnormal proliferation or increases in the level of TNF-α, interleukin 6, nitric oxide, and prostaglandin E2. When stimulated with LPS, Mecp2-null microglia responded with significantly less TNF-α release. Indeed, in RTT brains, there is no neuropathological manifestation of microgliosis (Jellinger, 2003; Armstrong, 2005), unlike most neurodegenerative disorders. While an excessive action of TNF-α can be excluded, it is possible that Mecp2-null microglia may have constitutively high activities of the signaling pathways that normally mediate the TNF-α action. Our findings suggest future studies to explore the epigenetic mechanisms regulated by MeCP2 for maintaining glutamate homeostasis, including the expression of glutaminase and Cx32. Other mechanisms of glutamate release, such as those mediated by exocytosis or glutamate transporter, remain to be studied (Nicholls and Attwell, 1990; Barger and Basile, 2001; Barger et al., 2007).
Our results are consistent with several in vivo studies showing increased glutamate levels in RTT brains. Using magnetic resonance spectroscopy (MRS) to evaluate the glutamate level, a recent study using a large sample size concluded an increase in the glutamate and glutamine/creatine in young patients with RTT (Horská et al., 2009). Studies measuring CSF glutamate concentrations also consistently showed a significant elevation in patients with RTT (Hamberger et al., 1992; Lappalainen and Riikonen, 1996). However, our findings are perhaps most relevant to local microglia-neuron interactions. It is evident that glutamatergic neurotransmission is impaired in RTT (Dani et al., 2005; Chao et al., 2007; Wood et al., 2009), which is likely to play a crucial role in the pathophysiology of epilepsy, movement disorders, aberrant control of respiration, as well as attenuation of LTP that underlie cognitive deficits—all present to varying degrees in patients with RTT or RTT mouse models (Chahrour and Zoghbi, 2007). It was found that in RTT the reduction in the excitatory synaptic pathway can be attributed to postsynaptic defects, as shown by electrophysiological evidence obtained from Mecp2 knockdown in individual postsynaptic cortical pyramidal neurons (Wood et al., 2009), as well as evidence showing decreased number of glutamatergic synapses in hippocampal neurons (Chao et al., 2007), reduced level of PSD95 and reduced dendritic spine density (Tropea et al., 2009; Belichenko et al., 2009a,b). Although this glutamatergic aberration may be a direct consequence of MeCP2 deficiency in neurons, our results suggest the possibility that a focal increased release of glutamate in the vicinity of synapses by MeCP2-deficient microglia might contribute to the postsynaptic defects, causing decreased excitatory synaptic strength. This notion is supported by our finding that the levels of glutamate receptor subunit proteins and postsynaptic density proteins of neurons were decreased by null MCM, and that this synaptotoxic effect was prevented when the glutamate release by microglia was reduced or the glutamate action on neurons was blocked (Fig. 3B). Indeed, it was shown that microglia processes make regular direct contact with synapses and a prolonged contact increases the turnover of synapses (Wake et al., 2009). Aggravating this situation, Mecp2−/y neurons were shown to be more vulnerable to NMDA- and AMPA-induced excitotoxicity (Russell et al., 2007), therefore more susceptible to a small increase of glutamate released by microglia. Interestingly, both MK801 and NBQX showed full neuroprotection from RTT microglia-induced neurotoxicity, suggesting that a convergence or mutual facilitation of specific neurotoxic pathways linked to the activation of NMDA and AMPA receptors is required for this mode of toxicity. It is generally accepted that NMDA receptors play a key role in mediating at least certain aspect of glutamate neurotoxicity, because of their high Ca2+ permeability. However, the role of AMPA receptor in triggering excitotoxicity is also important as AMPA receptor antagonists apparently provide better protection than NMDA receptor antagonists in animal models of global cerebral ischemia (Sattler and Tymianski, 2001). One possible explanation for our results is that activation of the AMPA receptors is required to achieve the depolarization necessary for full NMDA receptor activation.
Notably, dendritic spine defects and altered dendritic postsynaptic proteins in excitatory neurons are shared key features of neurodevelopmental and neurodegenerative disorders that manifest abnormal synaptic plasticity, such as autism, fragile X syndrome, X-linked mental retardation, Down syndrome, and Alzheimer's disease (AD) (Fiala et al., 2002; Dierssen and Ramakers, 2006; Zhao et al., 2006). Our present results add an additional mechanistic link between these disorders of synaptic plasticity because glutamate excesses have also been implicated in several neurodegenerative disorders. For example, Aβ oligomers, a proximate neurotoxin in AD pathogenesis, inhibit glutamate uptake at the synapse and significantly increase the extracellular glutamate concentration (Li et al., 2009). In HIV dementia, human immunodeficiency virus-infected microglia, through the increased activity of glutaminase, release excessive glutamate to damage neurons and synapses (Zhao et al., 2004). In addition, it has become increasingly evident that the epigenetic regulatory mechanisms, the perturbation of which causes RTT, play an important role in modulation of susceptibility to such disorders of synaptic plasticity (LaSalle et al., 2005; Siegmund et al., 2007; Eiges et al., 2007; Gräff and Mansuy, 2009). A strong support for this notion is that the methylation status of CpG sites, therefore the regulation of these genes by epigenetic factors such as MeCP2, has been linked to the development of AD because hypomethylation of these sites was found in the AD subject of a pair of monozygotic twins discordant for AD (Mastroeni et al., 2009).
Almost all patients with RTT are girls (Mecp2−/+) who are mosaics for MeCP2 expression. It is critical to identify factors triggering the clinical regression in girls with RTT. It appears that the MeCP2-related functions are well compensated in early life despite the fact that only ∼50% of cells express MeCP2. However, unknown disruption by certain age-dependent accumulative events in early life must occur to trigger the clinical regression. Previous studies, including ours, on the Mecp2−/+ tissues have revealed non-cell-autonomous effects of MeCP2− cells on MeCP2+ cells that might progressively disrupt the well compensated structural and functional integrity, thereby triggering regression (Braunschweig et al., 2004; Belichenko et al., 2009a; Maezawa et al., 2009). Belichenko et al. (2009a) found that in Mecp2−/+ mice, the abnormalities of dendritic spines affected both MeCP2+ and MeCP2− neurons, suggesting an impact from neighboring MeCP2− cells on the structure of MeCP2+ neurons. One explanation for this observation is that the abnormal MeCP2-deficient astrocytes provide poor support to both MeCP2− and MeCP2+ neurons, as we previously proposed (Maezawa et al., 2009). Our findings here support an additional possibility that the excessive release of glutamate by MeCP2-deficient microglia may damage MeCP2+ and MeCP2− neurons indiscriminately. Interestingly, Belichenko et al. (2009b) also reported that the abnormal RTT dendritic phenotype resembles that present in cultured hippocampal dendrites in response to excessive synaptic activation produced by high NMDA exposure, which is consistent with our present data. Therefore, microglia-released glutamate could be a factor contributing to clinical regression in patients with RTT.
Although our data demonstrate the neuroprotective effect of glutamate receptor antagonists, they are not suitable candidates for therapy of RTT because they can further perturb the already reduced glutamatergic neurotransmission. The unique microglial mechanism shown here implies the potential use of glutaminase inhibitors or gap junction hemichannel blockers to diminish microglial glutamate release without affecting the physiological glutamate level (Takeuchi et al., 2006). Notably, gap junctions/hemichannels are also implicated in mediating the spread of the MeCP2-deficincy state between astrocytes in our previous study (Maezawa et al., 2009). Therefore, gap junction hemichannel blockers might provide dual benefits for blocking harmful effects from both astrocytes and microglia in RTT.
Footnotes
This work was funded by the University of California Davis M.I.N.D. (Medical Investigation of Neurodevelopmental Disorders) Institute. We thank Dr. Janine LaSalle for helpful comments on this manuscript.
References
- Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA, Monyer H, Mammano F. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008;105:18770–18775. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Mobley WC. Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol Dis. 2009a;34:71–77. doi: 10.1016/j.nbd.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009b;514:240–258. doi: 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Braun JE, Madison DV. A novel SNAP25-caveolin complex correlates with the onset of persistent synaptic potentiation. J Neurosci. 2000;20:5997–6006. doi: 10.1523/JNEUROSCI.20-16-05997.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanson M, Fanjul M, Bosco D, Nelles E, Suter S, Willecke K, Meda P. Enhanced secretion of amylase from exocrine pancreas of connexin32-deficient mice. J Cell Biol. 1998;141:1267–1275. doi: 10.1083/jcb.141.5.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- De Vuyst E, Decrock E, Cabooter L, Dubyak GR, Naus CC, Evans WH, Leybaert L. Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J. 2006;25:34–44. doi: 10.1038/sj.emboj.7600908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5(Suppl 2):48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Ellaway C, Christodoulou J. Rett syndrome: clinical update and review of recent genetic advances. J Paediatr Child Health. 1999;35:419–426. doi: 10.1046/j.1440-1754.1999.355403.x. [DOI] [PubMed] [Google Scholar]
- Erdmann N, Tian C, Huang Y, Zhao J, Herek S, Curthoys N, Zheng J. In vitro glutaminase regulation and mechanisms of glutamate generation in HIV-1-infected macrophage. J Neurochem. 2009;109:551–561. doi: 10.1111/j.1471-4159.2009.05989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. Eur J Neurosci. 2009;30:1–8. doi: 10.1111/j.1460-9568.2009.06787.x. [DOI] [PubMed] [Google Scholar]
- Guo L, Wang Y. Glutamate stimulates glutamate receptor interacting protein 1 degradation by ubiquitin-proteasome system to regulate surface expression of GluR2. Neuroscience. 2007;145:100–109. doi: 10.1016/j.neuroscience.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberger A, Gillberg C, Palm A, Hagberg B. Elevated CSF glutamate in Rett syndrome. Neuropediatrics. 1992;23:212–213. doi: 10.1055/s-2008-1071344. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci. 2005;8:906–915. doi: 10.1038/nn1487. [DOI] [PubMed] [Google Scholar]
- Horská A, Farage L, Bibat G, Nagae LM, Kaufmann WE, Barker PB, Naidu S. Brain metabolism in Rett syndrome: age, clinical, and genotype correlations. Ann Neurol. 2009;65:90–97. doi: 10.1002/ana.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskison MM, Yanagawa Y, Obata K, Shuttleworth CW. Calcium-dependent NMDA-induced dendritic injury and MAP2 loss in acute hippocampal slices. Neuroscience. 2007;145:66–79. doi: 10.1016/j.neuroscience.2006.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Rett syndrome—an update. J Neural Transm. 2003;110:681–701. doi: 10.1007/s00702-003-0822-z. [DOI] [PubMed] [Google Scholar]
- Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet. 2008;17:1386–1396. doi: 10.1093/hmg/ddn026. [DOI] [PubMed] [Google Scholar]
- Lappalainen R, Riikonen RS. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996;15:213–216. doi: 10.1016/s0887-8994(96)00218-4. [DOI] [PubMed] [Google Scholar]
- LaSalle JM, Hogart A, Thatcher KN. Rett syndrome: a Rosetta stone for understanding the molecular pathogenesis of autism. Int Rev Neurobiol. 2005;71:131–165. doi: 10.1016/s0074-7742(05)71006-0. [DOI] [PubMed] [Google Scholar]
- Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest. 2009;119:747–754. doi: 10.1172/JCI37934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Nivison M, Montine KS, Maeda N, Montine TJ. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 2006;20:797–799. doi: 10.1096/fj.05-5423fje. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease. PLoS One. 2009;4:e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralvès J, Magdeleine E, Joly E. Design of an improved set of oligonucleotide primers for genotyping MeCP2tm1.1Bird KO mice by PCR. Mol. Neurodegener. 2007;2:16. doi: 10.1186/1750-1326-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V, Ni Y, Sunjara V, Hua X, Parpura V. Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci. 2004;24:2633–2642. doi: 10.1523/JNEUROSCI.3770-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz-Picciotto I, Hansen RL, Van de Water J, Pessah IN, Jiang R, Robinson WP, LaSalle JM. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 2008;1:169–178. doi: 10.1002/aur.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008;29:1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Russell JC, Blue ME, Johnston MV, Naidu S, Hossain MA. Enhanced cell death in MeCP2 null cerebellar granule neurons exposed to excitotoxicity and hypoxia. Neuroscience. 2007;150:563–574. doi: 10.1016/j.neuroscience.2007.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24:107–129. doi: 10.1385/MN:24:1-3:107. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Tsujimoto N, Qu Q, Lei H, Li E, Chen T, Blaustein CS. A methyl-CpG-binding protein 2-enhanced green fluorescent protein reporter mouse model provides a new tool for studying the neuronal basis of Rett syndrome. Neuroreport. 2008;19:393–398. doi: 10.1097/WNR.0b013e3282f5661c. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglia cells. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford UP; 2005. [Google Scholar]
- Suzumura A, Mezitis SG, Gonatas NK, Silberberg DH. MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol. 1987;15:263–278. doi: 10.1016/0165-5728(87)90121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci. 2009;29:12440–12448. doi: 10.1523/JNEUROSCI.3321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Bodles AM, Porter MM, Griffin WS, Basile AS, Barger SW. Induction of serine racemase expression and d-serine release from microglia by amyloid beta-peptide. J Neuroinflammation. 2004;1:2. doi: 10.1186/1742-2094-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Jiang S, Sims TJ, Barger SW. Schwann cells exhibit excitotoxicity consistent with release of NMDA receptor agonists. J Neurosci Res. 2005;79:638–643. doi: 10.1002/jnr.20401. [DOI] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Akimoto H, Kagawa S, Guillemin GJ, Takikawa O. Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1–42: implications for the pathogenesis of Alzheimer's disease. J Neurochem. 2009;110:791–800. doi: 10.1111/j.1471-4159.2009.06175.x. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Oberheim N, Kettenmann H, Ransom BR. Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia. 2009;57:258–269. doi: 10.1002/glia.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lopez AL, Erichsen D, Herek S, Cotter RL, Curthoys NP, Zheng J. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: linkage to HIV-1 associated dementia. J Neurochem. 2004;88:169–180. doi: 10.1046/j.1471-4159.2003.02146.x. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]