

Abstract
Cytochrome P450 epoxygenase (CYP450)-derived epoxyeicosatrienoic acids (EETs) are important regulators of cardiac remodeling; but the underlying mechanism remains unclear. The present study aimed to elucidate how EETs regulated cardiac fibrosis in response to isoprenaline (Iso) or angiotensin (Ang) II. Cardiac-specific human CYP2J2 transgenic mice (Tr) and wild-type (WT) C57BL/6 littermates were infused with Iso- or Ang II. Two weeks after infusion, Tr mice showed more alleviative cardiac fibrosis and inflammation compared with WT mice. In vitro, we found Iso or Ang II induced nuclear transfer of NF-κB p65 and inflammatory cytokines expression in cardiomyocytes. Furthermore, inflammation response emerged in macrophages cultured in cardiomyocytes-conditioned medium. When pretreatment with 14,15-EET in cardiomyocytes, the inflammatory response was markedly suppressed and the transmission of inflammation from cardiomyocytes to macrophages was reduced. In conclusion, CYP2J2 and EETs prevent cardiac fibrosis and cardiac dysfunction by suppressing transmission of pro-inflammation from cardiomyocytes to macrophages in heart, suggesting that elevation of EETs level could be a potential strategy to prevent cardiac fibrosis.
Keywords: Cardiac fibrosis, Heart failure, Inflammation, EETs, CYP2J2
1. Introduction
Cardiac fibrosis, an integral component of most cardiac pathologic conditions such as heart failure, is characterized by net accumulation of extracellular matrix (ECM) in the myocardium [1]. Cardiac fibrosis can be induced by various factors, such as the chronic activation of the sympathetic nervous system, myocar-dial hypoxia, ischemia, senescence, inflammation and hormones [2]. Consecutive administration with isoproterenol (Iso), a β-adrenergic receptors (β-AR) agonist, caused cardiac fibrosis in rats [3]. Emerging evidence suggests that activation of renin-angiotensin II (Ang II) system is the primary cause of cardiac fibrosis in hypertensive heart disease [4].
Recent studies demonstrated that inflammation plays a fundamental role in cardiovascular diseases, such as atherosclerosis, hypertension, and myocardial infarction [5]. Infiltration of inflammatory cells in the heart including macrophages is an early event. Fibrogenic growth factors secreted by macrophages induce cardiac remodeling. Accumulating evidences have suggested that myocardial infiltration of pro-inflammatory cells such as macrophages play pivotal roles in the initiation and development of cardiac fibrosis and dysfunction [6]. Upon activation, heart-infiltrated macrophages release various pro-inflammatory cytokines and chemokines, which interact with other cells, such as fibroblasts and cardiomyocytes, leading to cardiac remodeling [6–8]. Insults result in cardiomyocyte death and induce an intense inflammatory response; subsequent activation of pro-fibrotic pathways is an important component of the reparative process, but also plays a role in the adverse remodeling [1]. However, whether the stressed cardiomyocytes could influence the activation of macrophages is incompletely understood. Exposed to different stress, the cardiomyocytes induce inflammatory response and promote cardiac fibrosis that attenuates cardiomyocyte loss. Therefore, we proposed in this study that inflammation in cardiomyocytes participates in the fibrotic process, by the direct crosstalk between cardiomyocytes and myofibroblasts or by the intermediate mechanisms; if this is the case, inhibition of inflammatory response in cardiomyocytes by transgene or drugs would prevent cardiac fibrosis.
Cytochrome P450 epoxygenase 2J2 (CYP2J2), which is of human origin and dominantly expressed in cardiovascular system, metabolizes arachidonic acid to epoxyeicosatrienoic acids (EETs) [9]. EETs possess diverse biological functions, and observations revealed that EETs exert beneficial effects on various cardiovascular diseases, including atherosclerosis, hypertension and heart failure [10–17]. Previous studies showed that 14,15-epoxyeicosatrienoic acid (14,15-EET) protected cardiomyocytes from various injuries [18,19]. It has been reported that administration of soluble epoxide hydrolase (s-EH) inhibitor, which prevents EET hydration, and over-expression of CYP2J2 could prevent angiotensin (Ang) II-induced cardiac hypertrophy and heart failure in mice [20,21]. However, the effect of CYP2J2 on cardiac fibrosis is incompletely understood. We hypothesized that inflammatory cardiomyocytes could aggravate cardiac fibrosis, and EETs in heart could inhibit cardiac inflammation and attenuate cardiac dysfunction.
In the current study, we investigated the effects of overexpression of CYP2J2 on Iso-or Ang II-induced cardiac fibrosis in mice, and determined the impact of CYP2J2 on inflammation signaling pathways involved in cardiac fibrosis.
2. Materials and methods
2.1. Reagents
Materials were obtained from the following suppliers: antibodies against NF-κB p65, LaminB and β-actin were from santa cruz biotechnology Inc. (Santa cruz, CA); antibody against MOMA-2 was from Cell Signaling Technology (Beverly, MA); antibodies against collagen I, collagen III and alpha smooth muscle actin (β-SMA) were from Boster Bio-Engineering Limited Company (Wuhan, China); EETs and 14,15-EEZE were from Cayman Chemical. All other chemicals and reagents were purchased from Sigma–Aldrich (Sigma–Aldrich China Inc., Shanghai, China).
2.2. Ethics statement
All animal studies were approved by Tongji Medical College Animal Care and Use Committee.
2.3. Animals
Cardiac-specific human CYP2J2 transgenic mice driven by αMHC promoter on a pure C57BL/6 genetic background were gifts from Dr. Darryl Zeldin’s laboratory (NIEHS) and were identified as described previously [22]. All animals were housed at the animal care facility of Tongji Medical College at 25 °C with 12/12 h light/dark cycles and allowed free access to normal mice chow and water throughout the study period. All animal experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health.
Thirty male CYP2J2 transgenic mice and thirty C57BL/6 mice were used. Mini-osmotic pumps (Alzet model 1002; Durect, Cupertimo, California) were implanted as described previously [23]. The mice received a continuous subcutaneous infusion of Iso (dissolved in 0.002% ascorbic acid) [23,24] at a rate of 30 mg/kg/day for 14 days, while human Ang II was dissolved in 0.9% normal saline, and pumps were filled to deliver at the rate of 1.5 mg/kg/day over a period of 14 days. In control groups, vehicle (0.002% ascorbic acid or 0.9% normal saline) was used.
2.4. Analysis of cardiac function by echocardiography
Two weeks after infusion, echocardiography (Visualsonic Vevo 2100 System with a 40 MHz high resolution transducer) was used to detect the alterations of cardiac structure and functions. Mice were anaesthetized by isoflurane inhalation (1.5–2.5%). Measurements included IVS (interventricular septal wall thickness), LVID (left ventricular internal diameter), LVPW (left ventricular posterior wall), EF% and FS% under Long axis M-mode. All data and images were saved and analyzed by the Vevo 2100 Imaging System software version 1.0.0.
2.5. In vivo hemodynamics
In vivo LV function was assessed by Millar PV catheter as described previously [25]. Mice were anesthetized as described above and placed on heating pads with core temperature maintained at 37 °C. During the whole experiment, we monitored the vital signs (e.g. heart rate and blood pressure) under anesthesia. A microtip pressure–volume catheter (SPR-839; Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced into the left ventricle (LV) under pressure control. After stabilization for 20 min, the signals were continuously recorded at a sampling rate of 1000/s using an ARIA pressure-volume conductance system (Millar Instruments) coupled to a Powerlab/4SP analog-to-digital converter (AD Instruments, Mountain View, CA) and a personal computer. All pressure–volume loop data were analyzed using a cardiac pressure–volume analysis program (PVAN3.6; Millar Instruments), and HR, left ventricular end diastolic pressure (LVEDP), left ventricular end systolic pressure (LVESP), maximal slope of systolic pressure increment (dP/dtmax) and diastolic pressure decrement (dP/dtmin) were computed as described previously [26,27].
2.6. Cell culture
2.6.1. Primary cardiomyocytes and fibroblasts
All experiments involving animals were approved by Tongji Medical College Animal Care and Use Committee. 1–3 day old Sprague–Dawley rats were decapitated and the excised hearts placed in 1× PBS Buffer. The atria were carefully removed and the blood washed away. The ventricles were minced and incubated with 3 mL digestion buffer containing type II collagenase (0.025%) and trypsin (0.05%) at 37 °C while shaking at 80 RPM for 10 min. Then, stopped digestion with DMEM containing 20% newborn calf serum and stewed for 2 min. After that, discarded the supernatant and resuspended in 3 mL digestion buffer and incubated 37 °C with occasional agitation for 8 min. The steps for enzymatic digestion and isolation of myocytes were repeated 10–12 times to maximize yield. The myocytes were pooled and filtered in 200 mesh sieve and centrifuged at 1000 RPM for 8 min and resuspended in DMEM with 10% fetal bovine serum and penicillin–streptomycin (100 IU/ml) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. After culture for 1 h, transited the supernatant to another plates and continued to cultivate, and the adhered cells were fibroblasts and added fresh medium to culture.
2.6.2. Primary peritoneal macrophages
Peritoneal macrophages from 6 to 8 weeks old C57BL/6 mice were collected by infusing their peritoneal cavity with ice-cold sterile PBS as previously described with minor modifications. After a soft abdominal massage for 30 s, the peritoneal fluid was collected and centrifuged at 1000 rpm for 10 min, resuspended in DMEM media with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin. After 2 h, non-adherent cells were removed by washing twice with PBS and the adherent monolayer cells were used for experiments.
2.6.3. Cell lines
H9c2 (2–1), RAW 264.7 and NIH/3T3 cells were obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (100 IU/ml) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C.
2.7. Immunocytofluorescence
Cells grown in 96-well plates were washed with PBS and fixed in 4% paraformaldehyde in PBS for 10 min and then in 0.25% Triton-X 100 for 10 min. After blocking with 5% BSA for 30 min, cells were incubated in indicated antibody to NF-κB p65 at 4 °C overnight. The cells were then revealed with appropriate Cy3-conjugated secondary antibody for 1 h at 37 °C and DAPI (Sigma–Aldrich) for nuclear counter staining, and were subsequently visualized under a Nikon DXM1200 fluorescence microscope. Image-Pro Plus (Media Cybernetics, Bethesda, MD) was applied for image merging.
2.8. Histochemical analysis
Formalin fixed hearts were embedded in paraffin, sectioned into 4 μm slices, and stained with H&E and Sirius red (collagen stains red with Sirius red staining), as described previously [28]. The MOMA-2 representing the accumulation of macrophages was detected by immunohistochemical staining. Briefly, after deparaffinization and rehydration, slides were incubated in 3% hydrogen peroxide for 10 min, PBS containing 10% of goat serum for 30 min and with the rat monoclonal antibody to MOMA-2, diluted with PBS to 1:100 (overnight incubation at 4 °C). After washing, sections were stained with a secondary biotinylated antibody (Vector, CA) (room temperature; 2 h). Subsequently the slides were incubated in DAB chromogen for 5 min at room temperature. Then the sections were counterstained with hematoxylin, and covers lipped. Image-Pro Plus (Media Cybernetics) was applied to determine quantitative results. We counted the number of MOMA-2 positive cells (brown staining) per mm2 for 5 different visual fields in each animal, and the average number was calculated in each animal and each group.
For immunofluorescence visualization of collagen type I and III, sections were incubated with rabbit anti-collagen type I and mice anti-collagen III (Boster Bio-Engineering Limited Company, Wuhan, China), and then the slides were revealed with appropriate FITC, DyLight 488 or CY3-conjugated secondary antibodies for 1 h at 37 °C, and were subsequently visualized under a Nikon DXM1200 fluorescence microscope. Image-Pro Plus (Media Cybernetics, Bethesda, MD) was applied for image merging.
2.9. Immunoblotting
Lysates from cultured cells and heart tissues were prepared and immunoblotting was performed as described previously [28].
2.10. EMSA
Nuclear extracts of H9c2 cells or RAW 264.7 cells were prepared with the nuclear extraction reagent (Boster Bio-Engineering, Wuhan, China). EMSA was performed using the Light Shift Chemiluminescent EMSA Kit (Beyotime Institute Biotechnology, China) according to the manufacture’s instructions. In brief, 100 fmol of biotin end-labeled probe was incubated with 10 μg of nuclear extract, and chemiluminescent substrate was used for detection. The biotin-labeled NF-κB p65 oligonucleotides (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ and 3′-TCA ACT CCC CTG AAA GGG TCC G-5′) were purchased from Beyotime Institute Biotechnology.
2.11. ELISA analysis of inflammatory cytokines
The levels of IL-1β, IL-6, IL-10, MCP-1 and TGF-β1 in mice serum or heart tissue and in H9c2 or RAW 264.7 cultured media after indicated interventions were assayed by corresponding ELISA kits (Boster Bio-Engineering Limited Company, Wuhan, China) following the manufacturer’s instruction.
2.12. RNA extraction and real-time quantitative PCR
Total RNAs were extracted using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacture’s protocol. Reverse transcription of total mRNA were done starting from equal amounts of total RNA/sample (1 μg) using EasyScript First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) according to manufacturer’s instruction. Real-time quantitative PCRs using Trans StartTM SYBR Green qPCR Supermix (TransGen Biotech, Beijing, China) were performed for mRNA levels of TGFβ1, IL-1β, IL-6, IL-10, MCP-1 and collagen I and III, as well as GAPDH (as internal reference), using corresponding primers (Table 3). The qRT-PCR results were analyzed and expressed as relative mRNA levels of the CT (cycle threshold) value, which was then converted to fold change [29].
Table 3.
List of primers.
| Forward | Reverse | |
|---|---|---|
| TGF-β1 | 5′-CCTGCA AGACCATCGACATG-3′ | 5′-ACAGGATCTGGCCACGGAT-3′ |
| IL-1β | 5′-TGGTGTGTGACGTTCCCATT-3′ | 5′-CAGCACGAGGCTTTTTTGTTG-3′ |
| IL-6 | 5′-TGATGCTGGTGACAACCACGG-3′ | 5′-TAAGCCTCCGACTTGT-3′ |
| MCP-1 | 5′-TTAAAAACCTGGATCGGAACCAA-3′ 5′-TTAAAAACCTGGATCGGAACCAA-3′ |
5′-GCATTAGCTTCAGATTTACGGGT-3′ |
| IL-10 | 5′-CCA AGCCTTATCGGA AATGA-3′ | 5′-TTTTCACAGGGGAGA AAT CG-3′ |
| Collagen I | 5′-GAGAGAGCATGACCGATGGATT-3′ | 5′-TGGACATTAGGCGCAGGA A-3′ |
| Collagen III | 5′-GAA AAA ACCCTGCTCGGA ATT-3′ | 5′-GGA TCA ACCCAGTATTCTCCA CT-3′ |
| gapdh | 5′-TGCCAAGTATGATGACATCAAGAAG-3′ | 5′-AGC CCA GGA TGC CCT TTA GT-3′ |
2.13. Determination of urine 14,15-DHET
14,15-dihydroxyeicosatrienoic acid (14,15-DHET), the stable 14,15-EET metabolite, was detected in the urine of mice using an enzyme-linked immunosorbent assay kit (Detroit R&D, Detroit, MI) according to the manufacturer’s instructions as previously described [30].
2.14. Statistical analysis
All values are expressed as mean ± S.E.M. unless noted otherwise. Differences between data groups were evaluated for significance using Student t-test of unpaired data or one-way analysis of variance (ANOVA) and Bonferroni post-test. P < 0.05 was accepted as statistically significant.
3. Results
3.1. CYP2J2 transgene prevents Iso- or Ang II-induced cardiac fibrosis in mice
To investigate the effect of CYP2J2 on cardiac fibrosis in vivo, we established two cardiac fibrosis models by continuous subcutaneous infusion of β-adrenergic agonist isoprenaline (Iso) or angiotensin (Ang) II using mini-osmotic pump for 2 weeks in WT and Tr mice. Similar to previous reports, cardiac specific expression of CYP2J2 prevented Iso- or Ang II-induced cardiac hypertrophy and cardiac dysfunction [21]. As shown in Table 1, Iso- or Ang II-infusion damaged cardiac function including systolic and diastolic functions in comparison with the vehicle-infused control animals as reflected by the markedly reduced dP/dtmax and dP/dtmin, and increased left ventricular end systolic pressure (LVESP) and left ventricular end diastolic pressure (LVEDP) (Table 1, P < 0.05). However, CYP2J2 overexpression in Tr mice markedly restored the systolic and diastolic functions of heart and decreased LVESP and LVEDP to normal levels compared with the controls (Table 1, P < 0.05). Echocardiographic analysis revealed increased thicknesses of the left ventricular dimension, intraventricular septal and posterior walls, and demonstrated a significant decrease in ejection fraction and in the percent fractional shortening of the left ventricle in Iso and Ang II mice, and these changes were normalized by CYP2J2 overexpression (Table 2, P < 0.05).
Table 1.
Hemodynamic measurement.
| Parameters | WT
|
Tr
|
||||
|---|---|---|---|---|---|---|
| Saline | Iso | Ang II | Saline | Iso | Ang II | |
| HR (bpm) | 394 ± 45 | 397 ± 39 | 363 ± 1 | 376 ± 23 | 372 ± 34 | 375 ± 23 |
| dP/dtmax (mmHg/s) | 15,210 ± 974 | 3964 ± 828* | 3128 ± 177* | 14,690 ± 318 | 12,123 ± 1678# | 9137 ± 167# |
| dP/dtmin (mmHg/s) | −11,083 ± 1257 | −3200 ± 332* | −2315 ± 88* | −8216 ± 1088 | −9590 ± 1121# | −8863 ± 199# |
| LVESP (mmHg) | 113 ± 3 | 190 ± 2* | 187 ± 1* | 117 ± 2 | 104 ± 3# | 105 ± 2# |
| LVEDP (mmHg) | 3.80 ± 1.07 | 13.23 ± 0.80* | 13.74 ± 1.51* | 4.01 ± 1.22 | 4.96 ± 0.09# | 5.12 ± 1.15# |
HR: heart rate; LVESP: left ventricular end systolic pressure; LVEDP: left ventricular end-diastolic pressure; dP/dtmax and dP/dtmin: left ventricular maximal and minimal indicate rates of cardiac contractility;
P < 0.05 vs. WT + saline;
P < 0.05 vs. WT + Iso or WT + Ang II. N = 5 for each group.
Table 2.
Echocardiography measurement.
| Parameters | WT
|
Tr
|
||||
|---|---|---|---|---|---|---|
| Saline | Iso | Ang II | Saline | Iso | Ang II | |
| LVEF (%) | 95 ± 5 | 41 ± 12* | 46 ± 0.05* | 88 ± 1 | 86 ± 13# | 70 ± 10# |
| LVFS (%) | 72 ± 12 | 16 ± 2* | 22 ± 0.1* | 57 ± 1.51 | 58 ± 19# | 42 ± 9# |
| LVID;d (mm) | 2.41 ± 0.23 | 3.72 ± 0.34* | 3.43 ± 0.24* | 3.52 ± 0.33 | 2.42 ± 0.12# | 2.84 ± 0.23# |
| LVID;s (mm) | 0.73 ± 0.13 | 3.02 ± 0.51* | 2.43 ± 0.39* | 1.43 ± 0.21 | 1.12 ± 0.34# | 1.79 ± 0.14# |
| IVS;d (mm) | 0.91 ± 0.13 | 1.33 ± 0.15* | 1.71 ± 0.03* | 0.98 ± 0.21 | 1.22 ± 0.22# | 1.02 ± 0.13# |
| IVS;s (mm) | 1.42 ± 0.20 | 2.01 ± 0.35* | 2.11 ± 0.31* | 1.32 ± 0.32 | 1.21 ± 0.23# | 1.37 ± 0.17# |
| LVPW;d (mm) | 0.84 ± 0.15 | 0.99 ± 0.02* | 0.98 ± 0.03* | 0.73 ± 0.01 | 1.01 ± 0.02# | 0.93 ± 0.10# |
| LVPW;s (mm) | 1.19 ± 0.04 | 1.34 ± 0.07* | 1.51 ± 0.05* | 1.05 ± 0.01 | 1.03 ± 0.04# | 0.99 ± 0.03# |
HR: heart rate; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; LVID;d and LVID;s: left ventricular dimension at diastole and systole; IVS;d and IVS;s: interventricular septal wall thickness (diastole and systole); LVPW;d and LVPW;s: posterior wall thickness at diastole and systole.
P < 0.05 vs. WT + saline;
P < 0.05 vs. WT + Iso or WT + Ang II. n = 5 for each group.
To investigate the effects of CYP2J2 on myocardial fibrosis, we firstly analyzed total collagen contents in Iso- or Ang II-induced failing hearts. As expected, Iso and Ang II significantly increased total collagen deposition in Iso or Ang II-infused WT mouse hearts (Fig. 1A). Overexpression of CYP2J2 reduced Iso-induced collagen deposition (Fig. 1A). When stained by Picric acid–Sirius red and detected using polarization microscopy, the heart sections present the different colors in the regions. Collagen I presents a yellow, orange or red color, while collagen III appears green. And results showed that overexpression of CYP2J2 substantially lowered collagen I and III (predominant collagen isoforms in the heart) accumulation induced by Iso or Ang II (Fig. 1B, Supplemental Figure 1A and 1B), and consistently, markedly reduced the protein and mRNA the levels of collagen I and III in hearts (Fig. 1C and D). TGF-β is the most important cytokine of cardiac fibrogenesis and we found overex-pression of CYP2J2 inhibited the up-regulation of TGF-β1 induced by Iso and Ang II infusion in WT mice, as assessed by reduced protein and mRNA levels in hearts and serum concentration of TGF-β1 (Fig. 1C, E and F). These data suggest that overexpression of CYP2J2 reduces cardiac fibrosis induced by Iso or Ang II.
Fig. 1.

CYP2J2 prevents Iso or Ang II-induced cardiac fibrosis in mice. (A) Representative staining for collagen deposition in LV is presented. Collagen deposition is stained with a saturated solution of picric acid containing 1% Sirius red as red color and was quantified as percent of cardiac area. Scale bar, 100 μm. (B) Collagen types were examined by Picric acid-Sirius Red staining with polarization microscopy. Under a polarization microscope, collagen appears bright yellow-red (mainly type I collagen), and/or bright green (mainly type III collagen). Scale bar, 100 μm. (C) Immunoblotting analysis showed collagen I, III and TGF-β1 was increased in hearts of WT mice with Iso or Ang II infusion, and decreased in Tr mice. TGF-β1 was normalized to β-actin. (D) Expression of collagen I and III in LV, analyzed by real-time polymerase chain reaction. (E) Gene expression of TGF-β1 in LV analyzed by real-time polymerase chain reaction. (F) Expression of TGF-β1 in serum measured by ELISA. LV, left ventricle; WT, wild type mice; Tr, CYP2J2 transgenic mice; Iso, isoprenaline; Ang II, angiotensin II. n = 5 each group. *P < 0.05 vs. WT + saline, #P < 0.05 vs. WT + Iso or WT + Ang II.
Supplementary Figure 1 related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.prostaglandins.2015.01.004.
3.2. Overexpression of CYP2J2 attenuated inflammatory response induced by Iso or Ang II in mice
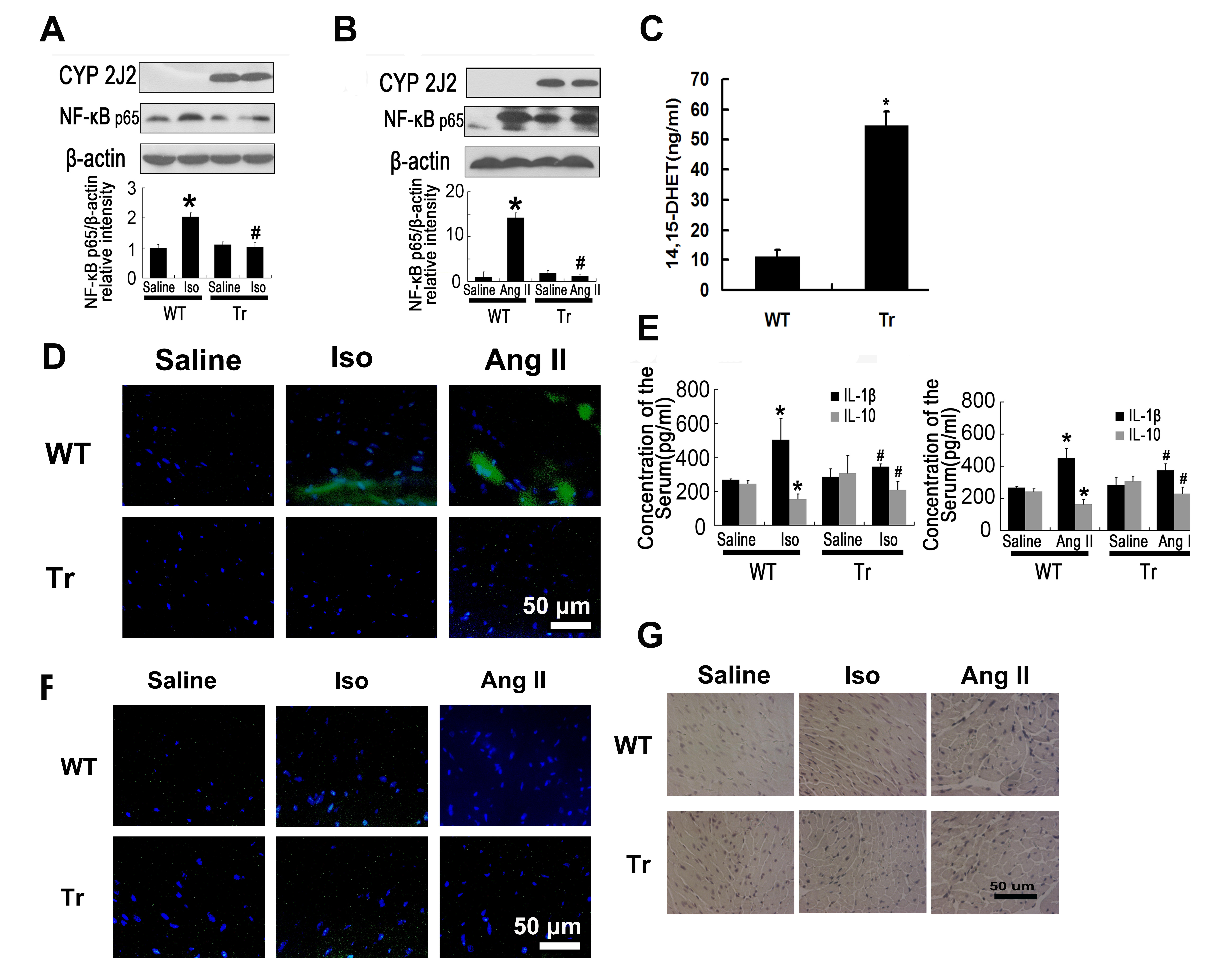
Inflammation plays a central role throughout the progression of cardiac fibrosis [31]. It has been suggested that Iso and Ang II could lead to the activation of NF-κB signaling to promote inflammation [32,33]. Firstly, we detected the CYP2J2 protein level by western blotting in the mice heart. As shown in supplemental Figure 1C, CYP2J2 protein was abundant in αMHC-2J2 Tr mice. Nearly no cross-reactivity of the antibody with endogenous murine CYP2J proteins was observed in WT mouse hearts. Meanwhile, CYP2J2 functionality was demonstrated by the nearly 5-fold increase in urinary 14,15-DHET levels in CYP2J2 Tr mice compared with WT mice (Supplemental Figure 1D). Consistently, Iso- or Ang II-infusion substantially increased the nuclear translocation activity and total expression of NF-κB p65 in WT mice. However, its nuclear translocation activity and total expression was markedly reduced in Iso or Ang II-infused CYP2J2 Tr mice (Fig. 2A and C; Supplemental Figure 1C). We found that Iso- or Ang II-infusion also increased cardiac macrophage infiltration (MOMA-2+ cells) as evaluated by immunohistochemical staining, and the effects could be markedly suppressed by overexpression of CYP2J2 (Fig. 2B and D). Moreover, CD68+ cells detected by immunocytofluorescence staining were also used to confirm the macrophages infiltration, and the results were consistent with that from immunohistochemical staining of MOMA-2 (Supplemental Figure 1E). The infiltration of neutrophils (Gr-1+) showed no differences in all groups (Supplemental Figure 1G and H).
Fig. 2.

CYP2J2 suppressed Iso or Ang II-induced inflammatory response in hearts of mice. (A) and (C) NF-κB p65 nuclear translocation activity was increased in WT mice with Iso(A) or Ang II (C) infusion, and were reduced in Tr mice. Proteins were normalized to β-actin or LaminB. (B) and (D) Immunohistochemical analyses of mice hearts and number of MOMA-2-positive cells per mm2. Scale bar, 40 μm. (E) and (F) Gene expression and protein levels of IL-1β, IL-6, IL-10 and MCP-1 in LV analyzed by real-time polymerase chain reaction and ELISA. LV, left ventricle; WT, wild type mice; Tr, CYP2J2 transgenic mice; Iso, isoprenaline; Ang II, angiotensin II. n = 5 each group. *P < 0.05 vs. WT + saline, #P < 0.05 vs. WT + Iso or WT + Ang II.
We further assayed the effect of CYP2J2 on expression of inflammatory cytokines induced by Iso or Ang II. As shown in Fig. 2E, Iso and Ang II significantly increased the mRNA and protein levels of IL-1β, IL-6 and MCP-1 in left ventricle, and overexpression of CYP2J2 markedly suppressed these inflammatory cytokines. On the contrary, IL-10, an anti-inflammatory cytokine, was markedly increased in the hearts of Iso or Ang II-infused CYP 2J2 Tr mice (Fig. 2E and F). Similar results were also observed when we examined serum IL-1β and IL-10 levels by ELISA (Supplemental Figure 1F). All data suggest that cardiac-specific CYP2J2 transgene attenuates inflammatory response induced by Ang II and Iso-infusion.
3.3. 14,15-EETs inhibited inflammatory response induced by Iso or Ang II in cardiomyocytes
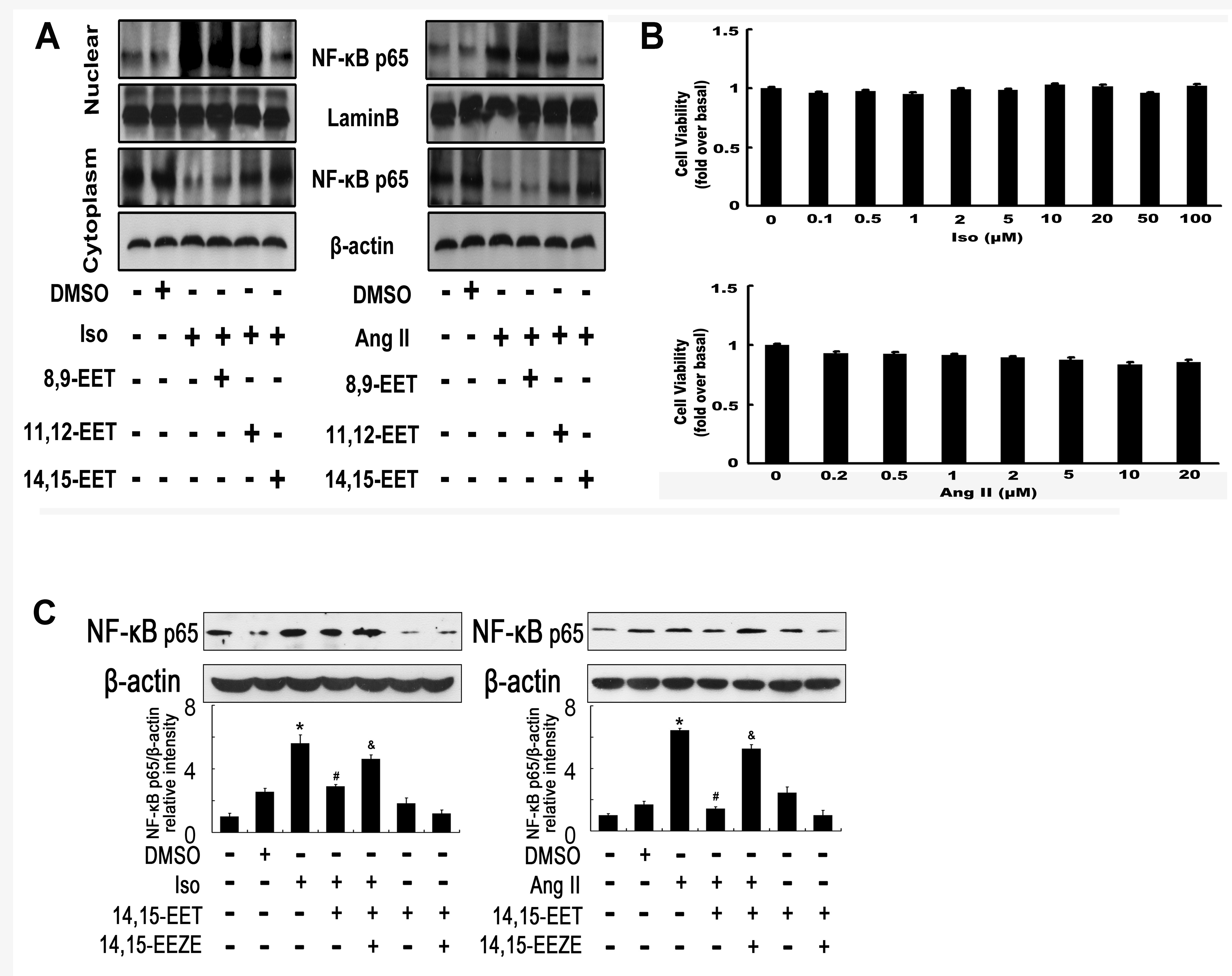
To investigate the effects of CYP2J2 on myocardial fibrosis in vitro, the cultured rat H9c2 cells were pretreated with three kinds of EETs (8,9-, 11,12- and 14,15-EETs; 1 μM) before exposing to Iso (10 μM) or Ang II (1 μM). And the results showed that 14,15-EET had the most powerful effect on inhibiting NF-κB p65 activation (Supplemental Figure 2A), so we selected 14,15-EET as the equivalent of CYP2J2 in the following experiments, After that, the cell viability in increasing concentrations of Iso (0–100 μM) or Ang II (0–20 μM) for 12 h was assessed using a CellTiter 96® Non-Radioactive Cell Proliferation (MTT) Assay. And the concentration-response analysis demonstrated that under these conditions 10 μM Iso and 1 μM Ang II treatment did not affect cellular viability (Supplemental Figure 2B). Then, we found the nuclear translocation activity and total expression of NF-κB p65 was induced by Iso or Ang II. However, 14,15-EET pretreatment effectively inhibited its activation and expression induced by Iso and Ang II, (Fig. 3A and B; Supplemental Figure 2C). This effect was further confirmed by immunocytofluorescence staining for cytosolic and nuclear NF-κB p65 (Fig. 3C and D). In addition, the NF-κB p65 activity of DNA binding was detected by EMSA, and the results showed that Iso or Ang II increased the DNA binding activity of NF-κB p65, while 14,15-EET pretreatment suppressed the effect (Fig. 3E and F). Consistent with the real-time PCR and ELISA results, 14,15-EET also down-regulated the expression of pro-inflammatory cytokine genes and proteins such as IL-1β and IL-6, and up-regulated the anti-inflammatory factor IL-10, (Fig. 3G and H). However, 14,15-EEZE (1 μM), the special inhibitor of 14,15-EET, reversed the anti-inflammatory effect of 14,15-EET.
Fig. 3.

14,15-EETs inhibited inflammatory response induced by Iso or Ang II in Cardiomyocytes. (A) and (B) H9c2 cells were administered with Iso (10 μM) or Ang II (1 μM) with or without pretreatment with the exogenous 14,15-EET, and cell lysates were analyzed by Western blotting. NF-κB p65 were activated after Iso (A) or Ang II (B) stimulation, and 14,15-EET inhibited NF-κB p65 activation induced by Iso or Ang II. And 14,15-EEZE could abolish the above effect of 14,15-EET. Proteins were normalized to β-actin or LaminB. (C) and (D) Representative immunocytofluorescence staining for NF-κB p65 in H9c2 cells. 14,15-EET pretreatment markedly attenuated NF-κB p65 nuclear translocation stimulated by Iso (C) or Ang II (D), 14,15-EEZE could abolish the above effect of 14,15-EET. (E) and (F) NF-κB p65 activity of DNA binding detected by EMSA. Iso (E) or Ang II (F) increased the DNA binding activity, 14,15-EET inhibited it and 14,15-EEZE abolished the effect of 14,15-EEZE. (G) and (H) 14,15-EET suppressed Iso (G) or Ang II (H)-induced IL-1β, IL-6 and MCP-1 in mRNA and protein levels, and increased IL-10 expression, 14,15-EEZE reversed the above effect of 14,15-EET. (I) and (J) 14,15-EETs inhibited nuclear translocation of NF-κB p65 induced by Iso (G) or Ang II (I) in neonatal cardiomyocytes. Proteins were normalized to β-actin or LaminB. (I) and (J) Inflammatory factors IL-1β, IL-6 and IL-10 in the supernate of neonatal cardiomyocytes were detected by ELISA. Iso, isoprenaline; Ang II, angiotensin II. n = 3–5 for each experiment; *P < 0.05 vs. control, #P < 0.05 vs. Iso or Ang II, &P < 0.05 vs. Iso or Ang II + 14,15-EET.
Supplementary Figure 2 related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.prostaglandins.2015.01.004.
We also verified the effects of 14,15-EET exposure to Iso and Ang II in primary rat cardiomyocytes, and the results demonstrated that 14,15-EET prevented cardiomyocytes from inflammation. As shown in Fig. 3I and J, the Iso- or Ang II-induced NF-κB p65 nuclear translocation activity was suppressed in the presence of 14,15-EET, and the expression of inflammatory factors (IL-1β, IL-6 and IL-10) in the supernatant also confirmed the results from H9c2 cells. These data suggest that 14,15-EET attenuates Iso or Ang II-stimulated inflammatory response in cardiomyocytes.
3.4. Pro-inflammation transfers from cardiomyocytes to macrophages
To investigate whether inflammation in cardiomyocytes promotes inflammation in macrophages, we treated H9c2 cells or neonatal rat cardiomyocytes with Iso or Ang II. We used 12-h cardiomyocytes inflammation-conditioned medium to explore possible effects on the murine macrophage cell line (RAW 264.7) or the primary macrophages in a model in which cell-free inflammation-conditioned medium was transferred to macrophages (Fig. 4A). After cultivation for 12 h, we noted that the pro-inflammatory NF-κB p65 signaling was activated in macrophages (Fig. 4B, K and L). However, pretreatment of 14,15-EET in cardiomyocytes inhibited inflammation response-conditioned medium transferring pro-inflammation to macrophages (Fig. 4B, K and L). Moreover, the NF-κB p65 activity of DNA binding was detected by EMSA, and the results showed that Iso or Ang II increased the DNA binding activity of NF-κB p65, while 14,15-EET pretreatment suppressed the effect (Fig. 4C and D). Furthermore, the ELISA results showed that the supernatant concentration of pro-inflammatory cytokines such as IL-1β, IL-6 and MCP-1 were up-regulated in macrophages, while the anti-inflammatory factor IL-10 was decreased (Fig. 4E, F, M and N). We also assessed gene expression and supernatant concentration of the fibrosis marker TGF-β1, and found that TGF-β1 increased in RAW 264.7 macrophages (Fig. 4G, H, I and J). However, pretreatment of 14,15-EET in cardiomyocytes also down-regulated the pro-inflammatory cytokine and TGF-β1, and increased the expression of IL-10 (Fig. 4E–J, M and N). Taken together, these data indicate that H9c2 cells under inflammatory response secrete mediators that cause macrophages to initiate inflammatory response and secretion of pro-inflammatory cytokines and TGF-β1.
Fig. 4.

Pro-inflammation transfers from cardiomyocytes to macrophages. (A) Scheme of macrophage culture in inflammation responded cardiomyocytes-conditioned medium. Briefly, cardiomyocytes were treated as indicated, conditioned medium harvested and transferred to macrophages for the indicated times, and inflammatory cytokine production in macrophages assayed by RT-qPCR and ELISA. (B) The nuclear translocation of NF-κB p65 was induced in RAW 264.7 culturing in Iso or Ang II stimulated H9c2 cell-conditioned medium, respectively. 14,15-EET pretreatment suppressed the activation of NF-κB p65 in RAW 264.7. Proteins were normalized to β-actin or LaminB. *P < 0.05 vs. H9c2 control c.m., #P < 0.05 vs. H9c2 Iso.c.m. or H9c2 Ang II c.m. (C) and (D) NF-κB p65 activity of DNA binding detected by EMSA. (E)–(J) Inflammation and profibrotic response were induced in RAW 264.7 culture in Iso or Ang II stimulated H9c2 cell-conditioned medium, and 14,15-EET pretreatment inhibited the effects above. Inflammatory factors IL-1β, IL-6, IL-10 and MCP-1 were detected by ELISA, profibrotic factor TGF-β1 was detected by RT-qPCR and ELISA. n = 3 to 5 for each experiment. *P < 0.05 vs. H9c2 Control c.m., #P < 0.05 vs. H9c2 Iso or Ang II c.m., &P < 0.05 vs. H9c2 Iso or Ang II + 14,15-EET c.m. (K) and (L) The nuclear translocation of NF-κB p65 was induced in primary macrophages culturing in Iso or Ang II stimulated primary cardiomyocytes-conditioned medium, respectively. 14,15-EET pretreatment suppressed the activation of NF-κB p65 in primary macrophages. Proteins were normalized to β-actin or LaminB. (M) and (N) Expression of inflammatory factors (IL-1β, IL-6, IL-10 and MCP-1) in the conditioned supernate from primary macrophages. n = 3–5 for each experiment. CM: primary cardiomyocytes; *P < 0.05 vs. CM Control c.m., #P < 0.05 vs. CM Iso or Ang II c.m., &P < 0.05 vs. CM Iso or Ang II + 14,15-EET c.m. Differences between data groups were evaluated for significance using ANOVAs.
Compared to the conditioned medium, normal media were added as an additional control. Iso or Ang II was directly used to stimulate RAW 264.7 cells with or without 14,15-EET pretreatment. As shown in supplemental Figure 3A and B, Iso and Ang II increased the nuclear translocation of NF-κB p65, but 14,15-EET weakened this effect. With regard to the results in normal and conditioned media, 14,15-EET may regulate the inflammation in macrophages through two pathways.
Supplementary Figure 3 related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.prostaglandins.2015.01.004.
3.5. Macrophages conditioned medium promotes fibrosis in fibroblasts
To confirm the observed effects, we performed the following experiments. The conditioned medium of macrophages cultured in cardiomyocytes-conditioned medium after 12 h was harvested and transferred to fibroblasts for another 12 h. Collagen I, collagen III and α-smooth muscle actin (α-SMA; a marker of myofibroblast differentiation) were used to detect the collagen expression and fibroblasts activity. As shown in Fig. 5A and B, collagen I, III, and α-SMA were induced in NIH/3T3 cells following the treatment of supernatant from macrophages after incubation with Iso-or Ang II-stimulated cardiomyocytes-conditioned medium, while 14,15-EET pretreatment suppressed their expression. These results were further confirmed in primary fibroblasts (Fig. 5C and D), which showed that the expression of collagen I, III, and α-SMA were up-regulated following incubation of conditioned medium from primary macrophages with pretreatment of Iso- or Ang II-induced conditioned medium from primary cardiomyocytes, while 14,15-EET pretreatment inhibited collagen deposition and fibroblasts activity.
Fig. 5.

Macrophages conditioned medium promotes fibrosis in fibroblasts. The conditioned medium of macrophages culturing in inflammation-responded cardiomyocytes-conditioned medium after 12 h, harvested and transferred to fibroblasts for another 12 h. Collagen I, collagen III and α-SMA were induced in NIH 3T3 cells in culture of RAW 264.7 after incubation in Ang II (A) or Iso (B) stimulated H9c2 cell-conditioned medium, while 14,15-EET pretreatment suppressed the expression of them in NIH/3T3 cells. Proteins were normalized to β-actin. *P < 0.05 vs. RAW control c.m., #P < 0.05 vs. RAW Ang II or Iso c.m. &P < 0.05 vs. RAW Ang II or Iso + 14,15-EET c.m. (C) and (D) The conditioned medium of primary macrophages (Mac) culturing in inflammation-responded CM-conditioned medium after 12 h, harvested and transferred to primary fibroblasts for another 12 h. Collagen I, collagen III and α-SMA were induced in primary fibroblasts, while 14,15-EET pretreatment suppressed the expression of them in primary fibroblasts. Proteins were normalized to β-actin. *P < 0.05 vs. Mac control c.m., #P < 0.05 vs. Mac Ang II or Iso c.m. &P < 0.05 vs. Mac Ang II or Iso + 14,15-EET c.m.
Compared to the conditioned medium, normal media were added as an additional control. Iso or Ang II was directly used to stimulate NIH/3T3 cells with or without 14,15-EET pretreatment. As shown in supplemental Figure 3C and D, Iso and Ang II increased expression of collagen I, III and α-SMA, but 14,15-EET weakened this effect. With regard to the results in normal and conditioned media, 14,15-EET may regulate the collagen deposition in fibroblasts through two pathways.
To further detect what in the supernatant of macrophages may induce the collagens deposition, exogenous cytokines (IL-1β, IL-6, IL-10, TGF-β1 and MCP-1) were directly added to the NIH/3T3 cells, and the results demonstrated the cytokines (IL-1β, IL-6, IL-10, TGF-β1 and MCP-1) secreted by conditioned-macrophages maybe the reason of collagens deposition (Supplemental Figure 3E). These results pointed out different kinds of cytokines in conditioned medium from macrophages induced collagen deposition in fibroblasts.
4. Discussion
Cardiac fibrosis plays an important role in maladaptive hypertrophy and heart failure. Our findings in this study demonstrate that CYP2J2 and their metabolites, EETs, exert marked cardiovascular protective effects. First, we found cardiac-specific overexpression of CYP2J2 markedly inhibited Iso- or Ang II-infusion induced inflammatory response, and alleviated cardiac hypertrophy and fibrosis and attenuated heart failure in mice. Moreover, our observations showed that EETs, especially 14,15-EET, suppressed Iso or Ang II-induced inflammatory response in H9c2 cells, and prevented macrophages activation. We further showed that macrophages cultured in conditioned medium from inflammation-stressed H9c2 cells underwent inflammatory response while pretreatment of EETs in H9c2 cells inhibited the expression of pro-inflammatory cytokine and fibrosis marker TGF-β1 in macrophages. Moreover, we proved RAW 264.7 conditioned medium promoted the differentiation of fibroblasts to myofibroblast and secretion of collagens in NIH/3T3 cells. Thus, our study demonstrates that CYP2J2-derived EETs play a critical role in regulating Iso- or Ang II-induced cardiac fibrosis.
Cardiac fibrosis is an important pathological condition that causes ventricular stiffness, diastolic dysfunction and arrhythmia [33,34]. To date, no therapeutic strategy has been developed to specifically target fibrosis in the heart. There is growing evidence that anti-inflammatory therapy may be a promising strategy against cardiac fibrosis [35]. NF-κB is a pleiotropic transcription factor that, in addition to playing fundamental roles in immunity, also regulates the expression of genes that contribute to various cardiac diseases including cardiac fibrosis [36]. Cardiac tissue consists mainly of cardiomyoctyes, which are surrounded by extracellular matrix [37]. Herein, we were interested in the mechanisms of cardiomyocytes on regulating cardiac fibrosis. In present study, we found that inflammatory response in cardiomyocytes induced by insults, such as Iso or Ang II, could be transmitted to macrophages, and as a result, the macrophages get activated and secrete TGF-β1 which promotes collagen deposition.
Earlier studies have identified CYP epoxygenases and EETs played important roles in cardiovascular protection through multiple biological signaling pathways. However, the cellular effects of CYP-derived EETs and their regulation of various inflammatory processes have become increasingly appreciated in recent years, suggesting that the role of this pathway in the cardiovascular system extends beyond the maintenance of vascular tone. Collectively, previous studies have demonstrated that CYP-derived EETs significantly attenuate pathologically relevant inflammatory responses in the cardiovascular system, including endothelial activation and leukocyte adhesion, which is mediated at least in part through inhibition NF-κB activation [38–40]. However, whether CYP2J2-derived EETs could suppress transmission of pro-inflammation from cardiomyocytes to macrophages in cardiac fibrosis is incompletely understood. Interestingly, we provided the evidence that CYP2J2-derived EETs have the potential to attenuate inflammatory response and blunt Iso or Ang II-induced cardiac hypertrophy and fibrosis. Herein, a new possible mechanism is that CYP2J2 or EETs may play an important role in cardiovascular disease through mediating inflammation in cardiomyocytes. In this study, we found the activation of NF-κB was reduced in CYP2J2 transgenetic mouse models with heart failure. Then we observed that CYP2J2 transgene decreased macrophage filtration into myocardium induced by Iso-or Ang II-infusion, and also 14,15-EETs suppressed the transfer of inflammatory response from cardiomyocytes to macrophages.
Pathological stimuli can stimulate tissue fibroblasts differentiation into myofibroblasts, which has been considered as a major mechanism responsible for fibrosis [41]. TGF-β1 signaling has an essential role in the development of fibrosis. Macrophages are known to trigger the differentiation of fibroblasts into myofibroblasts mainly through TGF-β1-dependent signaling in Ang II-infused heart [41,42]. Our data showed that active TGF-β1 was increased in WT mice infused with Iso or Ang II and CYP2J2 transgene inhibited the expression of TGF-β1 induced by Iso and Ang II infusion in mice, as assessed by reduced protein and mRNA levels in hearts and serum concentration of TGF-β. In vitro, we also assessed gene expression and serum concentration of the fibrosis marker TGF-β1, and found that TGF-β1 increased in RAW 264.7 macrophages simulated by medium from Iso- or Ang II-treated H9c2 cells. However, pretreatment of 14,15-EET in H9c2 cells also down-regulated the TGF-β1 at the level of protein and mRNA. Then, we found Collagen I, Collagen III and α-SMA were increased in NIH/3T3 fibroblast cells after administration with the conditioned medium from macrophages which were simulated by medium from Iso- or Ang II-treated H9c2 cells, while the expression of the above three markers were inhibited after pretreatment with supernatant from the 14,15-EET conditioned-H9c2 cells. Therefore, it is likely that myocardial fibrosis is triggered by macrophages mediated by inflammatory process in cardiomyocytes.
In conclusion, CYP-2J2 or EETs inhibits NF-κB signaling pathway initially induced by injury, such as Iso and Ang II, in cardiomyocytes. Then pro-inflammatory response transferred from cardiomyocytes to macrophages is suppressed, and TGF-β1 and other cytokines secreted by macrophages are decreased in the process. As we know, TGF-β1 promotes the fibrotic deterioration. Therefore, CYP2J2 or EETs attenuates the cardiac fibrosis and heart failure (Fig. 6).
Fig. 6.

Proposed mechanism of CYP2J2 or EETs in cardiac fibrosis. Iso or AngII activates NF-κB signal pathway in cardiomyocyte and promotes the transmission of pro-inflammation from cardiomyocytes to macrophages, which stimulates the secretion of TGF-β1 in macrophages. TGF-β1 augments the collagen secretion in myofibroblasts. Inhibition the inflammation in cardiomyocytes will reduce the profi-brotic process. CYP2J2 or EETs can suppress the inflammation in cardiomyocytes, which plays an important role in preventing cardiac fibrosis.
Our findings herein raise a number of interesting mechanistic questions for future study. First, when exposed to conditioned medium, macrophages underwent inflammatory response. What are the transmitters in the supernatant of cardiomyocytes and the sensory receptors in macrophages transferring the pro-inflammatory response from the cardiomyocytes to macrophages. Second, what is the target receptor of EETs. Some studies reported the nuclear receptors peroxisome-proliferator activated receptors (PPAR) were the possible receptors of CYP2J2 [43]. However, whether there are other receptors as the binding site of EETs still remains to be explored. Third, crosstalk in the three types of cells is the main pathway we interested in this manuscript. However, the independent roles of Iso or Ang II in each type of cells are also worth researching, because we found Iso or Ang II could directly induce inflammation in macrophages and increase collagens generation in fibroblasts. So which pathway dominates in the process of cardiac fibrosis.
In summary, we demonstrated that CYP2J2 and 14,15-EET could suppress cardiac fibrosis and heart failure through suppressing transmission of pro-inflammation from cardiomyocytes to macrophages, which suggests that increasing endogenous or exogenous EETs level especially in cardiomyocytes could be considered as a potential strategy to prevent cardiac fibrosis and heart failure.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by a National Basic Research Program (973) of China (2012CB518004) and National Natural Science Foundation of China (Nos. 31130031 and 81000097). The authors would like to thank Dr. Darryl C. Zeldin for giving cardiac-specific human CYP2J2 transgenic mice as gift.
Abbreviations
- CYP2J2
cytochrome P450 2J2 epoxygenase
- EET
epoxye-icosatrienoic acids
- Iso
isoprenaline
- Ang II
angiotensin II
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- TGF-β1
transforming growth factor-beta 1
- β-SMA
alpha smooth muscle actin
- ECM
extracellular matrix
- PPAR
nuclear receptors peroxisome-proliferator activated receptor
- WT
wild type
- Tr
transgene
Footnotes
Conflicts of interest
All authors have reported that they have no relationships with industry and financial associations relevant to the contents of this paper to disclose and no conflict of interests to declare.
References
- 1.Dobaczewski M, Frangogiannis NG. Chemokines and cardiac fibrosis. Front Biosci (Sch Ed) 2009;1:391–405. doi: 10.2741/s33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H, Tian A, Wu J, et al. Danshensu inhibits beta-adrenergic receptors-mediated cardiac fibrosis by ROS/p38 MAPK axis. Biol Pharm Bull. 2014;37(6):961–7. doi: 10.1248/bpb.b13-00921. [DOI] [PubMed] [Google Scholar]
- 3.Zhang YG, Li YG, Liu BG, et al. Urotensin II accelerates cardiac fibrosis and hypertrophy of rats induced by isoproterenol. Acta Pharmacol Sin. 2007;28(1):36–43. doi: 10.1111/j.1745-7254.2007.00485.x. [DOI] [PubMed] [Google Scholar]
- 4.Qi GM, Jia LX, Li YL, Li HH, Du J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology. 2014;155(6):2254–65. doi: 10.1210/en.2013-2011. [DOI] [PubMed] [Google Scholar]
- 5.Ferrario CM, Strawn WB. Role of the renin–angiotensin–aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98(1):121–8. doi: 10.1016/j.amjcard.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 6.Jia L, Li Y, Xiao C, Du J. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci (Landmark Ed) 2012;17:221–31. doi: 10.2741/3923. [DOI] [PubMed] [Google Scholar]
- 7.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170(3):818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma F, Li Y, Jia L, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE. 2012;7(5):e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63(8):597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Edin ML, Wang Z, Bradbury JA, et al. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011;25(10):3436–47. doi: 10.1096/fj.11-188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung O, Brandes RP, Kim IH, et al. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45(4):759–65. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 12.Lee CR, Imig JD, Edin ML, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24(10):3770–81. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merabet N, Bellien J, Glevarec E, et al. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol. 2012;52(3):660–6. doi: 10.1016/j.yjmcc.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Ulu A, Davis BB, Tsai HJ, et al. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52(4):314–23. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao B, Li X, Yan J, et al. Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J Pharmacol Exp Ther. 2010;334(3):784–94. doi: 10.1124/jpet.110.167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao G, Wang J, Xu X, et al. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-alpha-induced injury. J Lipid Res. 2012;53(3):456–66. doi: 10.1194/jlr.M017319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng C, Wang L, Li R, et al. Gene delivery of cytochrome p450 epoxygenase ameliorates monocrotaline-induced pulmonary artery hypertension in rats. Am J Respir Cell Mol Biol. 2010;43(6):740–9. doi: 10.1165/rcmb.2009-0161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tse MM, Aboutabl ME, Althurwi HN, Elshenawy OH, Abdelhamid G, El-Kadi AO. Cytochrome P450 epoxygenase metabolite, 14,15-EET, protects against isoproterenol-induced cellular hypertrophy in H9c2 rat cell line. Vasc Pharmacol. 2013;58(5–6):363–73. doi: 10.1016/j.vph.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Samokhvalov V, Alsaleh N, El-Sikhry HE, et al. Epoxyeicosatrienoic acids protect cardiac cells during starvation by modulating an autophagic response. Cell Death Dis. 2013;4:e885. doi: 10.1038/cddis.2013.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ai D, Pang W, Li N, et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106(2):564–9. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Ni L, Yang L, et al. CYP2J2-derived epoxyeicosatrienoic acids suppress endoplasmic reticulum stress in heart failure. Mol Pharmacol. 2014;85(1):105–15. doi: 10.1124/mol.113.087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seubert J, Yang B, Bradbury JA, et al. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95(5):506–14. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 23.Nienaber JJ, Tachibana H, Naga Prasad SV, et al. Inhibition of receptor-localized PI3K preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112(7):1067–79. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christe M, Jin N, Wang X, et al. Transgenic mice with cardiac-specific over-expression of MLK7 have increased mortality when exposed to chronic beta-adrenergic stimulation. J Mol Cell Cardiol. 2004;37(3):705–15. doi: 10.1016/j.yjmcc.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Georgakopoulos D, Mitzner WA, Chen CH, et al. In vivo murine left ventricular pressure–volume relations by miniaturized conductance micromanometry. Am J Physiol. 1998;274(4 Pt 2):H1416–22. doi: 10.1152/ajpheart.1998.274.4.H1416. [DOI] [PubMed] [Google Scholar]
- 26.Cingolani OH, Yang XP, Cavasin MA, Carretero OA. Increased systolic performance with diastolic dysfunction in adult spontaneously hypertensive rats. Hypertension. 2003;41(2):249–54. doi: 10.1161/01.hyp.0000052832.96564.0b. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P, Liaudet L, Bai P, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107(6):896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 28.Ni L, Zhou C, Duan Q, et al. beta-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PLoS ONE. 2011;6(11):e27294. doi: 10.1371/journal.pone.0027294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 30.Jiang JG, Ning YG, Chen C, et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 2007;67(14):6665–74. doi: 10.1158/0008-5472.CAN-06-3643. [DOI] [PubMed] [Google Scholar]
- 31.Cokkinos DV, Pantos C. Myocardial remodeling, an overview. Heart Fail Rev. 2011;16(1):1–4. doi: 10.1007/s10741-010-9192-4. [DOI] [PubMed] [Google Scholar]
- 32.Wiesner P, Choi SH, Almazan F, et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107(1):56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thakur S, Li L, Gupta S. NF-kappaB-mediated integrin-linked kinase regulation in angiotensin II-induced pro-fibrotic process in cardiac fibroblasts. Life Sci. 2014;107(1–2):68–75. doi: 10.1016/j.lfs.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 34.Weber KT. From inflammation to fibrosis: a stiff stretch of highway. Hypertension. 2004;43(4):716–9. doi: 10.1161/01.HYP.0000118586.38323.5b. [DOI] [PubMed] [Google Scholar]
- 35.Taqueti VR, Mitchell RN, Lichtman AH. Protecting the pump: controlling myocardial inflammatory responses. Annu Rev Physiol. 2006;68:67–95. doi: 10.1146/annurev.physiol.68.040104.124611. [DOI] [PubMed] [Google Scholar]
- 36.Gaspar-Pereira S, Fullard N, Townsend PA, et al. The NF-kappaB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol. 2012;180(3):929–39. doi: 10.1016/j.ajpath.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Li L, Chu H, Sun X, Ge Z. Oral sophocarpine protects rat heart against pressure overload-induced cardiac fibrosis. Pharm Biol. 2014;52:1045–51. doi: 10.3109/13880209.2013.877038. [DOI] [PubMed] [Google Scholar]
- 38.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fleming I, Michaelis UR, Bredenkotter D, et al. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88(1):44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Zhang Y, Schmelzer K, et al. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102(46):16747–52. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibro-proliferative diseases. J Clin Invest. 2007;117(3):524–9. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren J, Yang M, Qi G, et al. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by angiotensin II infusion. Am J Hypertens. 2011;24(6):701–7. doi: 10.1038/ajh.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wray JA, Sugden MC, Zeldin DC, et al. The epoxygenases CYP2J2 activates the nuclear receptor PPARalpha in vitro and in vivo. PLoS ONE. 2009;4(10):e7421. doi: 10.1371/journal.pone.0007421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.