

Abstract
We propose that the forward and reverse halves of a flash-induced protein-protein electron transfer (ET) photocycle should exhibit differential responses to dynamic interconversion of configurations when the most stable configuration is not the most reactive, because the reactants exist in different initial configurations: the flash-photoinitiated forward ET process begins with the protein partners in an equilibrium ensemble of configurations, many of which have little or no reactivity, whereas the reactant of the thermal back ET (the charge-separated intermediate) is formed in a nonequilibrium, “activated” protein configuration. We report evidence for this proposal in measurements on (i) mixed-metal hemoglobin hybrids, (ii) the complex between cytochrome c peroxidase and cytochrome c, and (iii and iv) the complexes of myoglobin and isolated hemoglobin α-chains with cytochrome b5. For all three systems, forward and reverse ET does respond differently to modulation of dynamic processes; further, the response to changes in viscosity is different for each system.
Keywords: cytochrome c, dynamics, hemoglobin, myoglobin, cytochrome c peroxidase
The long-range transfer of a single electron from donor to acceptor in a condensed phase is a fascinating and widely studied process (1, 2). Much of this work seeks to understand the electron transfer (ET) process itself. However, when the ET event involves a dynamic protein-protein interface, the observed kinetics frequently are controlled not by the ET process itself, but by the dynamics of recognition and binding and/or conversion within an ensemble of bound configurations (3, 4).
Studies of interprotein ET (3, 5-10) began with the implicit assumption of a protein-protein binding-energy landscape with a single reactive complex (Fig. 1A Left), implying a direct correlation between binding and reactivity. When the landscape for complex formation has several discrete minima (Fig. 1A Center) the reactive conformation may differ from the most stable one, in which case the observed ET kinetics are controlled by the rates and/or energetics of conformational conversion within a complex (11-13). Recent studies of ET between myoglobin (Mb) and cytochrome b5 (Fe3+b5), which bind to each other by weak electrostatic interactions, disclosed a new dynamic docking paradigm in protein-protein reaction dynamics: the landscape involves numerous configurations of similar affinity, only a subset of which is active in ET (Fig. 1A Right) (4, 14).
Fig. 1.

Protein-protein binding-energy landscapes. (A) Tier-1 energy landscapes for ET complexes. (B) Proposed tier-2 energy landscapes. • represent the system point for the initial ensemble of configurations for the photoinitiated ET process (Left) and the return of the ET intermediate to the ground state (Right). The rainbows represent ET-active configurations.
A majority of these studies have used a photocycle in which laser-flash excitation of the metallo-porphyrin in a metal-substituted (M = Zn or Mg) hemoprotein to its triplet excited state (3D) triggers ET from the triplet to the metal center of an acceptor protein (A) across a protein-protein interface, with rate constant, kf, Eq. 1:
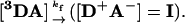 |
[1] |
The acceptor metal center of A typically is a ferri-heme center, (9, 15-17) but also can be a Cu(II) center (18). The product of the photoinitiated 3D → A ET process is the charge-separated intermediate with a metalloporphyrin π-cation radical in the donor protein and a reduced acceptor protein, I = [D+A-]; it returns to the ground state by a thermal ET process, rate constant kb (Eq. 2), completing the photocycle.
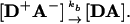 |
[2] |
Given the exponentially steep fall-off in the matrix element for ET between the two redox centers (19, 20), there will be only a subset of conformations in which the ET matrix element is maximized, and these will dominate both the forward reaction, 1, and back reaction, 2. However, the forward ET reaction is initiated by excitation of the protein partners in an equilibrium ensemble of mostly nonreactive configurations (Fig. 1B Left), which in general must involve conformational conversion to a reactive configuration. In contrast, the reactant complex of 2 is the ET intermediate I generated by 1 and formed in a highly reactive configuration (Fig. 1B Right). As a result, the back ET reaction is initiated within a nonequilibrium ensemble of ET-optimized configurations and would be expected to return to the ground state with the fastest rate constant possible unless conformational interconversion first “breaks the connection” and the complex converts to a nonreactive substate. According to this analysis, the two halves of the photocycle (Eqs. 1 and 2) might be expected to respond differently to changes in the dynamics of configurational conversion: in the simplest picture, the forward reaction should be slowed as dynamic processes are slowed, say by an increase in viscosity; the back reaction could show an increased rate under conditions that suppress interconversion, whether by viscosity increase or strengthened binding.
Here, we present some preliminary results with four ET complexes that indicate that the above ideas will indeed provide a fruitful approach to the study of the dynamics of protein-protein complexes.
Mixed-metal hemoglobin (Hb) hybrids provide an ideal system with which to study ET between redox partners at a fixed and crystallographically known distance and orientation (16). For the purposes of ET studies, the tetramer can be treated as two independent, predocked [α1β2] protein-protein ET complexes, each of which has been represented by a simple docking energy landscape (Fig. 1 A Left). In this most “static” of complexes we nonetheless find evidence for differential influence of dynamic processes on Eqs. 1 and 2 by studying the effect of viscosity on the ET photocycle in the 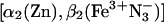 hybrid.
hybrid.
The physiological ET partners cytochrome c peroxidase (CcP) and cytochrome c (Cc) often are viewed as the paradigmatic interprotein ET pair, but in fact they comprise a quite complex ET system. First, CcP contains two redox centers, the heme and an adjacent tryptophan, each of which can be oxidized under appropriate circumstances (21). To eliminate this aspect of the problem, we discuss here only the CcP(W191F) mutant, for which the photocycle of Eqs. 1 and 2 involves only the hemes of the partners (22, 23). Second, although interprotein ET between CcP and Cc was long assumed to occur solely through a tight-binding 1:1 complex, as visualized by x-ray diffraction (24), our work has shown that CcP can bind two Cc at distinct domains (3). Thus, one can view CcP as providing an energy landscape of the gating type (Fig. 1 A Center) but with reaction possible when Cc is bound in either “well.”
In this article we take advantage of the fact that the association constant for formation of the tightly bound 1:1 complex is sufficiently high (K = 107 M-1) that at small ratios of Fe3+Cc/ZnCcP one can treat the system as stoichiometrically forming this complex; in effect, the complex functions as though it were represented by the simple-docking landscape. However, because the stability of this complex is far less than that of the Hb tetramer, the [ZnCcP(W191F),Fe3+Cc] complex offers the possibility that intracomplex dynamics within the “tight-site energy well” may play a larger role than in the Hb hybrids. Indeed, it should be recalled that the beautiful 1:1 structure for this complex was derived from a partly dehydrated crystal; in the as-formed crystals, the Cc adopted multiple positions (either statically or dynamically) and was not apparent in the x-ray structure (24). We present evidence for the differential influence of dynamic processes on Eqs. 1 and 2 when measuring the effects of increasing viscosity on the progress curves for I in the ET photocycle in Fig. 2.
Fig. 2.

Intracomplex ET photocycle, simplified by ignoring association/dissociation of the ground and photo-excited states.
Our recent studies of ET between Mb and Fe3+b5 showed that they were represented by the dynamic docking landscape of Fig. 1 A Right (4, 14). This paradigm in protein-protein ET was revealed through the decoupling of binding and reactivity: increasing the surface charge on Mb caused the second-order rate for ET from 3ZnMb to Fe3+b5 to increase by nearly three orders of magnitude without significantly varying Mb's binding affinity for Fe3+b5. We find that further modulation of the electrostatic interaction with three other Mb surface mutants provides dramatic evidence for the analysis presented above, with further evidence provided by the effects of changing viscosity in the analogous system of Hb α-chains with Fe3+b5.
Materials and Methods
Protein Purification. Hb was isolated from human erythrocytes and purified by ion exchange chromatography (25). The [α(Zn),β(FeCO)] Hb hybrid was prepared by the chain method (26) with the modifications described by Naito et al. (27). The [α(Zn),β(FeCO] hybrid was oxidized with potassium ferricyanide at pH 6 and converted to the azidomet form by copiously washing the sample on a size-exclusion resin equilibrated with a buffered solution containing 50 μM sodium azide.
The plasmids for expressing the trypsin-solubilized fragment of bovine Fe3+b5 (28), human Fe3+b5 (29), and horse Fe3+Mb (30) were obtained from Grant Mauk (University of British Columbia, Vancouver). Two additional single mutants of Mb [Mb(D44K) and Mb(D60K)] and the Mb(D44K/D60K) double mutant were constructed by using the method of overlap extension. Cultures of Escherichia coli containing a plasmid for recombinant Mb, the Mb mutants, or either of the two Fe3+b5s were grown, cells were isolated, and the Fe3+Mb and Fe3+b5 proteins were extracted and purified as described (28, 30). The Mb mutants were characterized by electrospray MS (Quattro II mass spectrometer, Micromass, Manchester, U.K.): D44K and D60K [observed mass = 17,095.4 D for Mb(D44K) and 17,095.5 D for Mb(D60K); calculated mass = 17,095 D]. All of the Mbs were reconstituted with Zn-deuteroporphyrin IX (Frontier Scientific, Logan, UT) following established methods (9).
The plasmid for CcP(W191F) was obtained from Dave Goodin (The Scripps Research Institute, La Jolla, CA) (31, 32). The protein was overexpressed in E. coli containing this plasmid, cells were isolated, and apo-CcP was extracted, purified, reconstituted with hemin, and crystallized following the procedure published for the recombinant CcP(MKT) (32). Zn-substituted-CcP was prepared by heme extraction and reconstitution with Zn-protoporphyrin IX (Frontier Scientific) following the procedure of Asakura and Yonetani (33). The plasmid for yeast iso-1 Fe3+Cc(C102T) was obtained from Marcellus Ubbink (Leiden University, Leiden, The Netherlands), and Fe3+Cc was prepared as described (34).
Flash Photolysis Measurements. Samples for ET studies were prepared in the dark under a N2 atmosphere as described (4, 14). Briefly, protein stock solutions for flash photolysis were exchanged into working buffers: 10 mM potassium phosphate (KPi) at pH 7 for experiments with the [ZnDMb,Fe3+b5] complex; 5 mM KPi, 5 mM sodium azide, 50 μM phytic acid, pH 8 for the experiments with the Hb hybrids; 10 mM KPi, pH 7.0 for measurements with the [ZnCcP(W191F),Fe3+Cc] complex; and 10 mM KPi, pH 6.5 for experiments with the [α(Zn),Fe3+b5] complex.
For the experiments with the Hb hybrid and the [ZnCcP(W191F),Fe3+Cc] complex, cryosolvents of varying viscosity were made at a selected (vol/vol) composition of glycerol and deionized water. Stock solutions of monobasic and dibasic phosphate were prepared either by dissolving the potassium salts in the cryosolvent at the desired composition or by adding an appropriate volume of a 1 M aqueous stock. The cryobuffer was adjusted to the desired pH by titrating the stock monobasic with the stock dibasic cryosolution. For the experiments with the [ZnDMb,Fe3+b5(bovine)] and the [α(ZnD),Fe3+b5(human)] complexes, the cryosolvents were prepared by weight. The absolute viscosity of water and glycerol cryosolutions were taken from tables (35). Small volumes of aqueous stock solutions of the proteins were added to the cryosolution.
Flash photolysis measurements were performed with an LKS60 photolysis unit (Applied Photophysics, Surrey, U.K.) equipped with a pulsed Xe arc lamp and a frequency-doubled Nd:YAG pulsed laser (YG660A, Continuum, Santa Clara, CA) as the excitation source. Triplet decays were recorded at 475 nm, and signals for the ET intermediates were acquired at triplet isosbestic points. Measurements were performed at 20°C.
For the Hb hybrids and the complexes with Fe3+b5, the time courses for the ET intermediates were fit to the scheme of Fig. 2 in the limit koff = 0. For the [ZnCcP(W191F),Fe3+Cc] complex kinetic constants, including, koff, were determined from fits with the kinetics simulation software package scop (Simulations Resources, Redlands, CA).
Results and Discussion
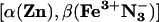 Mixed-Metal, Hb Hybrids. In the absence of ET, the 3Zn porphyrin in an α-chain of the [α(Zn),β(Fe2+)] hybrid decays exponentially, with an intrinsic decay rate constant, kD = 50 s-1 (36). In the
Mixed-Metal, Hb Hybrids. In the absence of ET, the 3Zn porphyrin in an α-chain of the [α(Zn),β(Fe2+)] hybrid decays exponentially, with an intrinsic decay rate constant, kD = 50 s-1 (36). In the 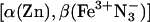 hybrid, the 3Zn-α decay remains exponential (rate constant kobs) but is enhanced through ET quenching to form the charge-transfer intermediate, I = D+A- (Eq. 1), with an ET rate constant of kf = kobs - kD. The rate constant for the return of I to ground (Eq. 2, kb) is greater than kobs, and as a result, I appears with a rate constant that corresponds to kb and decays with one that corresponds to kobs (Fig. 3 Inset).
hybrid, the 3Zn-α decay remains exponential (rate constant kobs) but is enhanced through ET quenching to form the charge-transfer intermediate, I = D+A- (Eq. 1), with an ET rate constant of kf = kobs - kD. The rate constant for the return of I to ground (Eq. 2, kb) is greater than kobs, and as a result, I appears with a rate constant that corresponds to kb and decays with one that corresponds to kobs (Fig. 3 Inset).
Fig. 3.

Viscosity dependence of the relative rate constants (krel) for the 3ZnP → Fe3+P forward (kf, ▪) and ZnP+ → Fe2+P back (kb, •) ET reactions within the 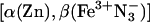 mixed-metal Hb hybrid. The relative rate constant is reported as the ratio of the rate constant at the viscosity of the buffered cryosolution to that in aqueous buffer. (Inset) Progress curve for the ET intermediate in 40% glycerol cryobuffer (η = 3.3 cP); time is in ms. Conditions were: ≈5 μM
mixed-metal Hb hybrid. The relative rate constant is reported as the ratio of the rate constant at the viscosity of the buffered cryosolution to that in aqueous buffer. (Inset) Progress curve for the ET intermediate in 40% glycerol cryobuffer (η = 3.3 cP); time is in ms. Conditions were: ≈5 μM 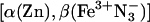 ; 5 mMNaN3; 5 mM KPi; 50 μM inositol hexaphosphate, pH 8; ≈436 nm wavelength; 20°C.
; 5 mMNaN3; 5 mM KPi; 50 μM inositol hexaphosphate, pH 8; ≈436 nm wavelength; 20°C.
The two rate constants for the ET photocycle were determined for solutions of the hybrid whose viscosity (η) ranged from η = 1 centipoise (cP) (buffer) to η = 13 cP (75% cryobuffer; 20°C). To best compare the effects of viscosity on the two rate constants (kf and kb), we normalized each to its respective value in aqueous buffer (Fig. 3). The forward ET rate constant in aqueous buffer is small (kf = 25 s-1 at η = 1 cP) and shows a distinct decrease with the initial increase in viscosity, and then a continued slow fall, k ∝ η-0.3, qualitatively as expected for a reaction that depends on dynamical processes that are progressively being slowed. The back rate constant is much larger (kb = 234 s-1 at η = 1 cP) and behaves quite differently, being virtually independent of η in the range studied. This differential effect of η on kf and kb suggests a complex where forward ET is facilitated by conformational fluctuations; if back ET depends on viscosity, the low solution viscosities used are less than the “internal viscosity” (18) associated with the relevant fluctuations. We note that both forward and reverse ET processes must persist as the viscosity increases “without bound,” as the ET photocycle is observed down to helium temperatures (37).
The [ZnCcP(W191F);Fe3+Cc] Complex. The decay of photo-excited 3ZnCcP(W191F) is exponential, with decay constant, kD ≈ 80 s-1, which decreases mildly with increasing viscosity. When a substoichiometric quantity of Fe3+Cc is added, it quantitatively binds to the tight-binding CcP site. The [ZnCcP,Fe3+Cc] complex is in slow exchange with its components, and as a result, the 3ZnCcP(W191F) decay becomes biexponential. The fraction of CcP involved in the complex decays with a rate constant increased by photo-initiated ET, kobs = kD + kf; the unbound fraction decays with kD. During a titration of ZnCcP with Fe3+Cc, the decay constants remain unchanged, and the fraction of ZnCcP bound in a complex increases according to a hyperbolic binding isotherm. The intermediate, I, appears exponentially, with a rate constant that corresponds to the triplet decay of the complex, and returns to the ground state through back ET with a smaller rate constant, kb < kobs (Fig. 4 Inset). A second phase in the decay at long time shows that I in part dissociates (rate constant koff), with the separated components subsequently undergoing second-order back ET to the ground state (Fig. 2). For the ET intermediate formed in aqueous solution, the fit of the progress curve with the mechanism in Fig. 2 gives rate constants: kf = 214 s-1; kb = 64 s-1; koff = 6 s-1; and k2 = 1.2 × 107 M-1·s-1.
Fig. 4.

Viscosity dependence of the relative ET rate constants (krel) for the 3ZnP → Fe3+P forward (kf, ▪) and ZnP+ → Fe2+P back (kb, •) ET reactions within the [ZnCcP(W191F),Fe3+Cc] complex. The values of krel are determined as the ratio of the rate constant in the cryobuffer to that in aqueous buffer. (Inset) Normalized kinetic progress curves for the ET intermediates; x axis is time in ms. Conditions were: 20°C; 10 mM KPi, pH 7.0 with η = 1.0 cP (red), η = 2.6 cP (blue), and η = 6.0 cP (green). Samples contained ≈5 μM ZnCcP(W191F) and 0.8 equivalents of Fe3+Cc. Kinetic data were collected at 548 nm.
We have measured the viscosity dependence of kf, kb, and koff. To best compare the effects of viscosity on the two ET rate constants (kf and kb), we again normalized them to the respective values given above for aqueous buffer (η = 1 cP at 20°C) (Fig. 4). The rate constant for photo-initiated ET, kf, doesn't change with increasing η until η ≈3 cP, then gently falls with further increase in η, behavior that appears quite different in detail from that of the hybrid. The value of kb for the [ZnCcP,Fe3+Cc] complex also changes weakly with the initial increase in η, but actually appears to show an initial small increase with increasing viscosity. As the viscosity for the complex is raised beyond η ≈3 cP, kb falls more steeply than does kf. Thus, the forward and back ET processes for the complex, like those of the hybrid, show differential dynamics, as reflected in the different dependencies on η. Unlike the hybrids, results to date suggest that intracomplex ET may not persist as the viscosity increases without bound (38).
[ZnDMb(X),Fe3+b5] Complexes, X = WT, D44K, D60K, and D44K/D60K, and the [Zn-α,Fe3+b5] Complex. Mb surface charge variants. Earlier work showed that when Fe3+b5 is titrated into a solution of ZnDMb or one of a suite of surface-charge variants ET to Fe3+b5 quenches the triplet; the decay remains exponential and the decay constant varies linearly with quencher concentration (kobs = kD + kf2[Fe3+b5]), indicating that the affinities of all of the variants are low and that the complex and components are in rapid exchange (4, 14). These studies showed that the bimolecular, 3ZnDMb → Fe3+b5 forward ET rate constant, kf2, increases dramatically as the positive charge on Mb is changed by the nominal amounts, -1 ≤ Δqnom ≤ +3, but without a substantial increase in the binding affinity (4). To extend these observations, we used the macrodox simulation program (7) to predict the relative reactivities of D/E-to-K charge-reversal mutants of Mb. Three of these Δqnom = +2 variants (D44K, D60K, and E85K) were predicted to show a substantial enhancement of the ET rate constant, and we have prepared Zn-deuteroporphyrin-substituted versions of two of them (D44K and D60K), as well as the double mutant (D44K/D60K), Δqnom = +4.
In the absence of Fe3+b5, all of the variants exhibit exponential triplet decays, kD = 52 s-1, as reported for ZnDMb (9). When Fe3+b5 is titrated into a solution of ZnDMb or a Δqnom = +2 variant, ET to Fe3+b5 quenches the triplet; as with previous variants, the decay remains exponential and the decay constant varies linearly with quencher concentration (kobs = kD + kf2 [Fe3+b5]). The bimolecular forward ET rate constants (kf2) for Mb(D44K) and Mb(D60K) are an order of magnitude greater than for Mb(WT) (Table 1), but an order of magnitude less than that for ZnD-(dme)Mb (37), also with Δqnom =+2; this finding likely reflects the different locations of the mutations on the Mb surface, but also may involve increased hydrophobicity of the interaction domain. The quenching constant for the Mb(D44K/D60K) double mutant, Δqnom =+4, is double that for Mb(dme), Δqnom =+2. The titration curve for the double mutant shows slight curvature, suggesting that the increased net charge of this ZnDMb finally has increased the binding constant enough for us to see hints of saturation.
Table 1. Bimolecular rate constants (k2) for the quenching of the triplets of ZnMb variants by Fe3+b5 at low ionic strength (pH 7.0, μ = 18 mM, 20°C).
| ZnMb variants | Δqnom* | k2 [M-1·s-1] | k2/k2 (ZnDMb) |
|---|---|---|---|
| ZnDMb | 0 | 4.3 × 106 | 1 |
| ZnDMb(D44K) | 2 | 6.4 × 107 | 15 |
| ZnDMb(D60K) | 2 | 9.5 × 107 | 22 |
| ZnD(dme)Mb | 2 | 5.6 × 108 | 130 |
| ZnDMb(D44K/D60K) | 4 | 9.8 × 108 | 228 |
Nominal change in charge relative to native Fe2+Mb or ZnMb
Fig. 5 shows progress curves of the [ZnD+Mb,Fe2+b5] ET intermediates for Mb(WT) and the three additional Mb variants. The intermediate I formed by the forward ET in the complexes of the horse ZnDMb variants with Fe3+b5 forms with a rate constant, kf, that is faster than the decay of the photo-excited triplet state, and it disappears slowly, with a concentration-dependent rate, indicating that association/dissociation of all states must be considered; similar results were reported for the reaction of whale ZnDMb with Fe3+b5 (9). Detailed analyses of these traces will be forthcoming; for present purposes it is instructive to consider the apparent first-order rate constants obtained by fitting the time courses of I to a simple exponential rise and fall. With such a fit, the rate constant for the appearance of the intermediate corresponds roughly to the rate constant for triplet decay; with an appreciable excess of Fe3+b5 as used here, this rate constant is essentially the pseudo first-order forward ET rate constant: kf = kf2[b5]. The time, t1/2 = ln2/kb, corresponds to the half-time for back ET under the experimental conditions.
Fig. 5.

Progress curves for the ET intermediate, I, of the [ZnMb(X),Fe3+b5] complexes, X = WT, D44K, D60K, and D44K/D60K. Conditions were: 10 mM potassium phosphate buffer, pH 7; 20°C; 563 nm wavelength; ≈13 μM ZnDMb; ≈41 μM Fe3+b5.
As Δqnom for the Mb variants increases, the kf increases strongly and the kb less strongly; as a result, the maxima in the progress curves shift to earlier time. In addition, the amount of I that accumulates decreases, dramatically so for the Δqnom = +4 double mutant. This finding is extremely anomalous. In any simple docking (Fig. 1) kinetic ET scheme for I, when the rate of appearance of I (kf) increases more rapidly with Δqnom than the disappearance rate, then the maximum concentration of the intermediate must increase with Δqnom, regardless of whether the product complex is in slow or fast exchange with its components. However, the behavior of I for Mb and its mutants, particularly the double mutant, is precisely the opposite (Fig. 5).
We suggest that this behavior is explained by conformational interconversion of I within the dynamic docking landscape (Fig. 1 A Right and B). The increase in observed kf2 with increasing Δqnom is attributed to increased electrostatic interaction that raises the binding constants for the reactive configuration(s) of the [ZnDMb,Fe3+b5] complex and hence the probability that [3DA] will achieve such a configuration before deactivation. I is necessarily formed in a reactive configuration, and we further propose that the strengthened electrostatic attraction in the reactive configuration(s) of the mutants, and most especially the double mutant, slows conformational conversions of I that break the connection through which back ET can promptly return I to the ground state. Only those complexes that successfully undergo the conversion to a nonreactive configuration accumulate for detection.
Viscosity studies. Triplet quenching titrations and ET time courses also were obtained for the [ZnDMb(WT),Fe3+b5(bovine)] and [α(ZnD),Fe3+b5(human)] complexes as a function of viscosity. The net positive charge of the α(ZnD) chain is greater than that of Mb, and correspondingly, the quenching rate constant for the reaction of Fe3+b5 with α(Zn) in aqueous solution can be fit to a one-site binding curve with kf = 3,600 s-1 and Ka = 1.1 × 104 M-1, in agreement with our previous findings for Hb tetramers (27, 38). Both Ka and kf for reaction of Fe3+b5 with the α(Zn) chain decrease progressively with increasing viscosity.
For [α(ZnD),Fe3+b5], the time course for the ET intermediate again is complex, and we thus discuss apparent rate constants, kf and kb, obtained by fitting the progress curves of I to a simple rise-and-fall function. For both ZnDMb and α(Zn) complexes, kf decreases monotonically with viscosity, according to the function, kf ∝ η-0.6. The deviation of the exponent from unity, the expected value for a diffusive second-order process (39), indicates that intracomplex motions are involved as well. Likewise, in both systems the back rate constant has a steeper dependence on viscosity. Thus, these systems resemble the [ZnCcP(W191F);Fe3+Cc] complex after the crossover of the curves for the two rate constants, whereas the mixed-metal Hb hybrid resembles it before the crossover.
Summary
The preliminary observations described above were assembled to explore the idea that the two halves of the ET photocycle, Eqs. 1 and 2, might be influenced differently by dynamic processes. Forward ET is initiated with the protein partners in an equilibrium ensemble of configurations, most of which have little or no reactivity, whereas the reactant of the back reaction (the ET intermediate I) is formed in a nonequilibrium set of activated protein configurations. The studies on four systems chosen for their different energy landscapes support this idea. They give us the opportunity to obtain detailed information about the differential dynamic properties of the initial ensembles of configurations for the forward and back ET reactions and the time scale of their conversion to inactive ones.
We have taken two approaches with these studies. First, we study the coupling of dynamic processes to interprotein ET through the use of high-viscosity cryosolvent solutions. This approach has long been used, through such studies as our own work with mixed-metal Hb hybrids (40), the [ZnCcP,Fe3+Cc] complex (41, 42), a study of ET in sol-gels (43), the beautiful work by Kostic and coworkers with [ZnCc,CuPc] (18, 44), and [ZnCc6,Fe3+Cf] complexes (45), which presented the first comparisons of the viscosity dependence of kf and kb, numerous studies of the photosynthetic reaction center reacting with Cc (46), and a recent study of a tethered ET pair (47).
For all three systems studied here, forward and reverse ET respond differently to modulation of dynamic processes; further, the response to changes in viscosity is different for each system. The mere observation of viscosity effects on ET within the most rigid of protein complexes, the Hb hybrids, was something of a surprise to us. For the Hb hybrid under these conditions, kb is essentially invariant with viscosity, while kf falls significantly. For the [ZnCcP, Fe3+Cc] complex, with small increases in viscosity, kf again is invariant with increasing η; kb is nearly so, although it appears to exhibit a gentle rise. At higher viscosities, kb falls more rapidly than kf, in contrast to the hybrids but similar to the complexes with Fe3+b5. Clearly, the complicated responses of these several systems to changes in viscosity shows that the analysis of Fig. 1 has disclosed a fruitful area for examination, but falls far short of a complete picture of the influence of intracomplex dynamics on interprotein ET reactions. Among the issues to be addressed are the degree to which the addition of glycerol may be (i) changing the energy landscapes themselves, not merely the rates with which the landscapes are traversed (48), and (ii) influencing water activity (49-51).
The second approach has been the use of the Mb surface mutants. Perhaps the single most dramatic observation in this report is the loss of the ET intermediate in the [ZnDMb(D44K/D60K),Fe3+b5] complex (Fig. 5), which we interpret in terms of the nonequilibrium initial conformation(s) of I.
This article presents exploratory efforts that are being extended, and the ideas presented here could well provide the impetus to rethink earlier viscosity studies (18, 40, 43-47). More broadly, consecutive ET reactions can be considered as prototypes for multistage enzyme-catalyzed chemical reaction, and studying the effects of dynamics and binding energetics on the rich behavior of these ET reactions can further the understanding of the redox processes in these complex biological systems.
Note Added in Proof. Under other conditions, kb for the hybrids distinctly increases with viscosity.
Acknowledgments
This work was supported by National Institutes of Health Grant HL 62303.
Author contributions: B.M.H., J.W., I.V.K., and J.M.N. designed research; L.M.C., D.A.C., A.D.P., J.L.S., K.E.W., J.W., and J.Y. performed research; L.M.C., D.A.C., A.D.P., J.L.S., K.E.W., J.W., J.Y., and J.M.N. analyzed data; and B.M.H. and J.M.N. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ET, electron transfer; Mb, myoglobin; Hb, hemoglobin; Cc, cytochrome c; Ccp, Cc peroxidase; cP, centipoise; Fe3+b5, cytochrome b5.
References
- 1.Balzani, V. (2001) Electron Transfer in Chemistry (Wiley, New York).
- 2.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265-322. [Google Scholar]
- 3.Nocek, J. M., Zhou, J. S., De Forest, S., Priyadarshy, S., Beratan, D. N., Onuchic, J. N. & Hoffman, B. M. (1996) Chem. Rev. 96, 2459-2489. [DOI] [PubMed] [Google Scholar]
- 4.Liang, Z.-X., Kurnikov, I. V., Nocek, J. M., Mauk, A. G., Beratan, D. N. & Hoffman, B. M. (2004) J. Am. Chem. Soc. 126, 2785-2798. [DOI] [PubMed] [Google Scholar]
- 5.Salemme, F. R. (1976) J. Mol. Biol. 102, 563-568. [DOI] [PubMed] [Google Scholar]
- 6.McLendon, G. & Hake, R. (1992) Chem. Rev. 92, 481-490. [Google Scholar]
- 7.Northrup, S. H., Thomasson, K. A., Miller, C. M., Barker, P. D., Eltis, L. D., Guillemette, J. G., Inglis, S. C. & Mauk, A. G. (1993) Biochemistry 32, 6613-6623. [DOI] [PubMed] [Google Scholar]
- 8.Bendall, D. S. (1996) Protein Electron Transfer (BIOS Scientific, Oxford).
- 9.Nocek, J. M., Sishta, B. P., Cameron, J. C., Mauk, A. G. & Hoffman, B. M. (1997) J. Am. Chem. Soc. 119, 2146-2155. [Google Scholar]
- 10.Zhu, Z., Cunane, L. M., Chen, Z.-W., Durley, R. C. E., Mathews, F. S. & Davidson, V. L. (1998) Biochemistry 37, 17128-17136. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman, B. M., Ratner, M. A. & Wallin, S. A. (1990) in Advances in Chemistry, eds. Johnson, M. K., King, R. B., Kurtz, D. M., Jr., Kutal, C., Norton, M. L. & Scott, R. A. (Am. Chem. Soc., Washington, DC), Vol. 226, pp. 125-146. [Google Scholar]
- 12.Pletneva, E. V., Fulton, D. B., Kohzuma, T. & Kostic, N. M. (2000) J. Am. Chem. Soc. 122, 1034-1046. [Google Scholar]
- 13.Davidson, V. L. (1996) Biochemistry 35, 14035-14039. [DOI] [PubMed] [Google Scholar]
- 14.Liang, Z.-X., Nocek, J., Huang, K., Hayes, R. T., Kurnikov, I. V., Beratan, D. N. & Hoffman, B. M. (2002) J. Am. Chem. Soc. 124, 6849-6859. [DOI] [PubMed] [Google Scholar]
- 15.Naito, N., Huang, H., Sturgess, W., Nocek, J. M. & Hoffman, B. M. (1998) J. Am. Chem. Soc. 120, 11256-11262. [Google Scholar]
- 16.Natan, M. J., Kuila, D., Baxter, W. W., King, B. C., Hawkridge, F. M. & Hoffman, B. M. (1990) J. Am. Chem. Soc. 112, 4081-4082. [Google Scholar]
- 17.Qin, L. & Kostic, N. M. (1994) Biochemistry 55, 12392-12399. [Google Scholar]
- 18.Ivkovic-Jensen, M. M., Ullmann, G. M., Crnogorac, M. M., Ejdebaeck, M., Young, S., Hansson, O. & Kostic, N. M. (1999) Biochemistry 38, 1589-1597. [DOI] [PubMed] [Google Scholar]
- 19.Beratan, D. N., Onuchic, J. N., Winkler, J. R. & Gray, H. B. (1992) Science 258, 1740-1741. [DOI] [PubMed] [Google Scholar]
- 20.Winkler, J. R. (2000) Curr. Opin. Chem. Biol. 4, 192-198. [DOI] [PubMed] [Google Scholar]
- 21.Ho, P. S., Hoffman, B. M., Kang, C. H. & Margoliash, E. (1983) J. Biol. Chem. 258, 4356-4363. [PubMed] [Google Scholar]
- 22.Mauro, J. M., Fishel, L. A., Hazzard, J. T., Meyer, T. E., Tollin, G., Cusanovich, M. A. & Kraut, J. (1988) Biochemistry 27, 6243-6256. [DOI] [PubMed] [Google Scholar]
- 23.Seifert, J. L. (2005) Ph.D. thesis (Northwestern University, Evanston, IL).
- 24.Pelletier, H. & Kraut, J. (1992) Science 258, 1748-1755. [DOI] [PubMed] [Google Scholar]
- 25.Williams, R. C. & Tsay, K. (1973) Anal. Biochem. 54, 137-145. [DOI] [PubMed] [Google Scholar]
- 26.Yip, Y. K., Waks, M. & Beychok, S. (1977) Proc. Natl. Acad. Sci. USA 74, 64-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naito, A., Hui, H. L., Noble, R. W. & Hoffman, B. M. (2001) Biochemistry 40, 2060-2065. [DOI] [PubMed] [Google Scholar]
- 28.Funk, W. D., Lo, T. P., Mauk, M. R., Brayer, G. D., MacGillivray, R. T. A. & Mauk, A. G. (1990) Biochemistry 29, 5500-5508. [DOI] [PubMed] [Google Scholar]
- 29.Lloyd, E., Ferrer, J. C., Funk, W. D., Mauk, M. R. & Mauk, A. G. (1994) Biochemistry 33, 11432-11437. [DOI] [PubMed] [Google Scholar]
- 30.Guillemette, J. G., Matsushima-Hibiya, Y., Atkinson, T. & Smith, M. (1991) Protein Eng. 4, 585-592. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald, M. M., Churchill, M. J., McRee, D. E. & Goodin, D. B. (1994) Biochemistry 33, 3807-3818. [PubMed] [Google Scholar]
- 32.Goodin, D. B., Davidson, M. G., Roe, J. A., Mauk, A. G. & Smith, M. (1991) Biochemistry 30, 4953-4962. [DOI] [PubMed] [Google Scholar]
- 33.Asakura, T. & Yonetani, T. (1969) J. Biol. Chem. 244, 537-544. [PubMed] [Google Scholar]
- 34.Pollack, W. B. R., Rosell, F. I., Twitchett, M. B., Dumont, M. E. & Mauk, A. G. (1998) Biochemistry 37, 6124-6131. [DOI] [PubMed] [Google Scholar]
- 35.Miner, C. S. & Dalton, N. N. (1953) Glycerol (Reinhold, New York).
- 36.McGourty, J. L., Peterson-Kennedy, S. E., Ruo, W. Y. & Hoffman, B. M. (1987) Biochemistry 26, 8302-8312. [DOI] [PubMed] [Google Scholar]
- 37.Liang, Z.-X., Nocek, J. M., Kurnikov, I. V., Beratan, D. N. & Hoffman, B. M. (2000) J. Am. Chem. Soc. 122, 3552-3553. [Google Scholar]
- 38.Naito, N. R. (1999) Ph.D. thesis (Northwestern University, Evanston, IL).
- 39.Steinfeld, J. I., Francisco, J. S. & Hase, W. L. (1989) Chemical Kinetics and Dynamics (Prentice-Hall, Englewood Cliffs, NJ).
- 40.Peterson-Kennedy, S. E., McGourty, J. L., Kalweit, J. A. & Hoffman, B. M. (1986) J. Am. Chem. Soc. 108, 1739-1746. [Google Scholar]
- 41.Nocek, J. M., Stemp, E. D. A., Finnegan, M. G., Koshy, T. I., Johnson, M. K., Margoliash, E., Mauk, A. G., Smith, M. & Hoffman, B. M. (1991) J. Am. Chem. Soc. 113, 6822-6831. [Google Scholar]
- 42.Nocek, J. M., Liang, N., Wallin, S. A., Mauk, A. G. & Hoffman, B. M. (1990) J. Am. Chem. Soc. 112, 1623-1625. [Google Scholar]
- 43.Nocek, J., Hatch, S. L., Seifert, J. L., Hunter, G., Thomas, D. D. & Hoffman, B. M. (2002) J. Am. Chem. Soc. 124, 9404-9411. [DOI] [PubMed] [Google Scholar]
- 44.Ivkovic-Jensen, M. M. & Kostic, N. M. (1997) Biochemistry 36, 8135-8144. [DOI] [PubMed] [Google Scholar]
- 45.Grove, T. Z. & Kostic, N. M. (2003) J. Am. Chem. Soc. 125, 10598-10607. [DOI] [PubMed] [Google Scholar]
- 46.Tetreault, M., Rongey, S. H., Feher, G. & Okamura, M. Y. (2001) Biochemistry 40, 8452-8462. [DOI] [PubMed] [Google Scholar]
- 47.Feng, C., Kedia, R. V., Hazzard, J. T., Hurley, J. K., Tollin, G. & Enemark, J. H. (2002) Biochemistry 41, 5816-5821. [DOI] [PubMed] [Google Scholar]
- 48.Balabin, I. A. & Onuchic, J. N. (2000) Science 290, 114-117. [DOI] [PubMed] [Google Scholar]
- 49.Autenrieth, F., Tajkhorshid, E., Schulten, K. & Luthey-Schulten, Z. (2004) J. Phys. Chem. B 108, 20376-20387. [Google Scholar]
- 50.Guo, M., Bhaskar, B., Li, H., Barrows, T. P. & Poulos, T. L. (2004) Proc. Natl. Acad. Sci. USA 101, 5940-5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldbeck, R. A., Paquette, S. J. & Kliger, D. S. (2001) Biophys. J. 81, 2919-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]