

Abstract
Proton transfer is crucial for many enzyme reactions. Here, we show that in addition to protonatable amino acid side chains, water networks could constitute proton-binding sites in proteins. A broad IR continuum absorbance change during the proton pumping photocycle of bacteriorhodopsin (bR) indicates most likely deprotonation of a protonated water cluster at the proton release site close to the surface. We investigate the influence of several mutations on the proton release network and the continuum change, to gain information about the location and extent of the protonated water network and to reveal the participating residues necessary for its stabilization. We identify a protonated water cluster consisting in total of one proton and about five water molecules surrounded by six side chains and three backbone groups (Tyr-57, Arg-82, Tyr-83, Glu-204, Glu-194, Ser-193, Pro-77, Tyr-79, and Thr-205). The observed perturbation of proton release by many single-residue mutations is now explained by the influence of numerous side chains on the protonated H bonded network. In situ hydrogen/deuterium exchange Fourier transform IR measurements of the bR ground state, show that the proton of the release group becomes localized on Glu-204 and Asp-204 in the ground state of the mutants E194D and E204D, respectively, even though it is delocalized in the ground state of wild-type bR. Thus, the release mechanism switches between the wild-type and mutated proteins from a delocalized to a localized proton-binding site.
Keywords: bacteriorhodopsin, hydrogen bonded network, proton release
Proton transfer reactions are key steps in protein catalysis. An excellent system to study the basic principles of proton transfer is the light-driven proton pump bacteriorhodopsin (bR) (1–3), a seven α-helical membrane protein in the archaebacterium Halobacterium salinarum. The retinal chromophore is bound to the protein by a protonated Schiff base to K216, separating the cytoplasmic and the extracellular halves of bR. Absorption of light by the chromophore induces vectorial proton transfer from the cytoplasmic to the extracellular medium, building up the electrochemical gradient across the membrane, which is used for ATP synthesis.
Structural models of the bR ground state (BR) and different photointermediates with resolutions to 1.43 Å have been reported from cryoelectron microscopy and x-ray diffraction (4, 5). After light-excitation bR undergoes a proton pumping photocycle with the intermediates J, K, L, M, N, and O in order of their appearance (6). The intermediates are characterized by UV–visible (7), resonance Raman (8, 9), and IR spectroscopy (10, 11), as well as by their structural changes. Crucial events in the pumping mechanism are the light-induced all-trans- to 13-cis-retinal isomerization in the K intermediate (12), the deprotonation of the central proton-binding site, the protonated Schiff base, and the corresponding protonation of the nearby counterion D85 in the L-to-M transition (13–15), the reprotonation of the Schiff base by D96 in the M-to-N transition (14, 15), the 13-cis to all-trans reisomerization in the N-to-O transition (16), and deprotonation of D85 in the O-to-BR reaction (10).
Despite extensive investigation of bR, essential parts of the proton transport mechanism have remained unclear, especially the proton release mechanism. It is generally agreed that during the photocycle a proton is released at the extracellular surface with a time constant of ≈80 μs at pH 7 (17), roughly concomitant with the deprotonation of the protonated Schiff base and the protonation of D85 (15, 18), but the exact nature of the proton release group is still disputed. Early studies assumed deprotonation of a single amino acid residue. Three different candidates were proposed, R82, E194, and E204, because mutation of these residues blocks proton release during the rise of the M intermediate, as shown by measurements with pH-dependent indicator dyes (19–21). First, E204 was suggested to be the proton release group (20). This model was expanded to a more complex mechanism: the proton, which is bound to the so called “E204 site” in BR (21, 22), is released during the L-to-M transition via the transiently protonated E194. These models were obtained by investigations of the E204Q, E204D, and E194D mutants. But time-resolved IR data for the wild-type (WT) bR show no evidence for deprotonation of a carboxylic acid in the L-to-M transition (23, 24). The role of R82 is also not yet clear; it may deprotonate (25) or undergo H bond changes, but it might not be essential for the proton release (19). In a more recent model it was proposed that the proton resides within an H-bonded network consisting of water molecules, stabilized by E204, E194, and R82 (23, 26). In some respects, such a proton would behave similarly to excess protons in water (27–30), which can either form an  complex, in which an H3O+ core is strongly H bonded to three H2O molecules [Eigen cation (31)], or it can be more delocalized by being shared between two H2O molecules in an
complex, in which an H3O+ core is strongly H bonded to three H2O molecules [Eigen cation (31)], or it can be more delocalized by being shared between two H2O molecules in an 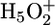 complex [Zundel cation (32)]. The H bonds in the Zundel cation are characterized by low to nonexistent barriers for proton transfer and short equilibrium distances. The spectral signature of excess protons in water is a broad IR absorption extended up to 2,500 cm-1 with a maximum between 1,500 and 2,000 cm-1 (29, 32–34). This so-called continuum absorbance is observed in many liquid systems and proteins, in which H bonds with a double-minimum proton potential or H-bonded networks with a multiminimum proton potential are present. The continuum is caused by the Zundel polarization, where strong electrostatic coupling of the environment gives rise to deformations of the proton transfer potential and fluctuations of the equilibrium H-bond lengths. This situation leads to a continuity of energy level differences (32). Such continuum absorbance changes are actually observed during the photocycle of bR, and they indicate proton transfer via protonated water networks (23, 32, 35–37).
complex [Zundel cation (32)]. The H bonds in the Zundel cation are characterized by low to nonexistent barriers for proton transfer and short equilibrium distances. The spectral signature of excess protons in water is a broad IR absorption extended up to 2,500 cm-1 with a maximum between 1,500 and 2,000 cm-1 (29, 32–34). This so-called continuum absorbance is observed in many liquid systems and proteins, in which H bonds with a double-minimum proton potential or H-bonded networks with a multiminimum proton potential are present. The continuum is caused by the Zundel polarization, where strong electrostatic coupling of the environment gives rise to deformations of the proton transfer potential and fluctuations of the equilibrium H-bond lengths. This situation leads to a continuity of energy level differences (32). Such continuum absorbance changes are actually observed during the photocycle of bR, and they indicate proton transfer via protonated water networks (23, 32, 35–37).
The structural models from x-ray crystallography support the existence of a protonated water network near E194 and E204. Although excess protons and the protonation states of glutamic acids could not be observed directly, the structural models show at least three water molecules in the vicinity of E194 and E204, sufficient for containing an excess proton (4). pKa calculations verified that the protonated water network near these two charged carboxylic acids is energetically possible, and that this network is more likely to be protonated than one of the nearby glutamic acids (26). A long-time molecular dynamics (MD) simulation was used to reveal participation of amino acids in the stabilization of the protonated water network by H bonding (38). The simulation shows two extensive water densities located at the extracellular side of bR, separated from each other by the side chain of R82 (Fig. 1) and not in direct contact with the surrounding bulk water. The water densities correspond to the space occupied by the fluctuating water molecules, rather than to the fixed positions of water molecules provided by the x-ray structure analysis, and are defined in Kandt et al. (38). The largest water cluster, density V, is close to the extracellular surface (Fig. 1). Kandt et al. predict, furthermore, amino acids that are H bonded to density V. Mutations of these stabilizing residues should disturb the protonated water cluster and affect the continuum absorbance change. The candidates for such interaction were identified as Y57, S193, E194, E204, Y83, P77, and R82, whereas replacing T205 and Y79 should have no influence (38).
Fig. 1.

View of the extracellular side of bR, showing water density V and its H-bonded amino acids (38). The water density characterizes the space occupied by the fluctuating water molecules, an analogue to electron densities (38). These are Y57, R82, Y83, Y79, T205, E204, E194, S193, and P77. The red balls demonstrate the discrete positions of water molecules taken from x-ray structure (4). Y57 and R82 are located between water clusters IV and V, and R82 provides the separation of the two clusters. Water cluster V is located in the region that Rammelsberg et al. (23) suggested for the protonated water cluster.
In this work we probed the delocalization of the proton in water cluster V by mutating all of the surrounding amino acids, except P77. The WT and the mutants were investigated by time-resolved step-scan Fourier transform IR spectroscopy with 30-ns time resolution in the spectral region between 2,500 and 1,000 cm-1. We compare our findings to earlier results from other groups, and we resolve some of the controversies around the source of the released proton. This work provides insight into the proton release mechanism for the WT system, and we derive alternative proton release mechanisms for the mutants E204D and E194D.
Materials and Methods
Mutagenesis and Mutant Expression. The site-specific mutants of bR were prepared as described in ref. 39, mutagenesis is described in ref. 40, and isolation as purple membrane sheets was according to ref. 41.
Step-Scan Measurements. Pure membrane sheets were suspended (200 μg in 1 M KCl/100 mM Tris·HCl buffer at pH 7, R82Q at pH 8), centrifuged for 2 h at 200,000 × g, and squeezed between two CaF2 windows. The time-resolved step-scan Fourier transform IR measurements were performed at room temperature (20–23°C) with a Bruker IFS66v spectrometer as described in ref. 42. Spectra were recorded between 3,200 and 900 cm-1 with up to 30-ns time-resolution and a spectral resolution of 4 cm-1. The bR samples were excited by an neodymium:yttrium/aluminum garnet laser at 532 nm (Spectra-Physics) with a laser energy density of 2 mJ/cm2 per pulse.
A linear photovoltaic HgCdTe detector (KV100-1-B-7/190 from Kolmar Technologies, Newburyport, MA) was used, to prevent nonlinear response of the detector-preamplifier system causing an artificial time course of the base line in the difference spectra, which would interfere with the very small broad continuum absorbance (42).
Visible-Light Measurements. Transient pH changes were measured by using pyranine (8-hydroxyl-1,3,6-pyrenetrisulfonate from Molecular Probes) and covalently bound fluorescein (FITC or 5,6-carboxyfluorescein succinimidyl ester, Molecular Probes) in a UV–visible photolysis apparatus (42) as described in ref. 17.
In Situ Hydrogen/Deuterium (H/D) Exchange Measurements. The experiments were performed in a homemade apparatus. Pure membrane sheets were suspended at a concentration of 5 mg/ml in 0.5 mM KCl/0.5 mM Tris·HCl buffer at pH 8. Two 10-μl aliquots of this solution were dried on two CaF2 windows. The windows were placed in a homemade airtight holder connected to a homemade mixing device with three gas valves, connected to pure N2, N2 enriched with H2O vapor, and N2 enriched with D2O vapor. With this equipment, all protonatable groups were either protonated or deuterated without removing the sample from the spectrometer. Thus, an in situ H/D-exchange difference spectrum is obtained from the same sample. The bands of all protonatable groups become visible through their isotopic H/D shifts. A H/D exchange difference spectrum can be obtained for the ground state as well as for steady-state accumulated intermediates such as the M intermediate. The relative humidity of the N2/H2O(D2O) vapor mix was ≈85%, the level at which the proton release is not perturbed (43). The exchange is controlled by the disappearance of the O–H stretch and the appearance of the O–D stretch of water. The detailed strategy for measuring such exchange spectrum will be described elsewhere. An H/D-exchange difference spectrum of bR is calculated between the spectra of BR in H2O and BR in D2O after repeating the measuring procedure at least 20 times. To reduce the influence of the H2O and D2O absorbance the second derivatives of the H/D-exchange difference spectra were calculated (with 25-point smoothing). Whereas the H/D exchange was made at 18°C, all of the spectra were taken at -35°C. This procedure reduces the amount of H2O and D2O vapor within the sample caused by condensation. For fast and precise temperature regulation, a Peltier element was integrated in the sample holder and connected to a thermostat capable of maintaining the low temperature.
Results and Discussion
Fig. 2 shows the time-resolved Fourier transform IR difference spectrum between 2,500 and 1,720 cm-1 taken 300–400 μs after laser excitation, corresponding mainly to M intermediates. This M - BR difference spectrum reveals a broad negative band in the spectral region between 2,400 and 1,800 cm-1, representing the continuum absorbance change discussed above. No other bands overlap with the continuum absorbance in this spectral region. Below 1,800 cm-1 the continuum absorbance overlaps with other bands of the protein, for example at 1,762 cm-1, where protonation of D85 is seen, and which is used to calibrate the amplitude of the continuum band. The experimentally observed spectral characteristics agree with the spectrum proposed for an 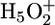 complex within bR, which is based on recent quantum-mechanical/molecular-mechanical (QM/MM) calculations (44). In these calculations a protonated water complex is considered in the protein environment and not only in water. The continuum spectrum of both a Zundel-like and an Eigen-like complex are calculated. The Eigen complex shows an increase of the continuum band at higher wavenumbers with a maximum at 2,500 cm-1, whereas the Zundel complex shows a decrease from 1,800 cm-1 to higher wavenumbers with zero amplitude at ≈2,500 cm-1. The agreement of the spectral dependence of the calculated Zundel complex shown in figure 2 of ref. 44 and the measured continuum absorbance shown here is astonishing. Therefore, the protonated water cluster resembles mostly a Zundel-like complex. To probe the delocalization of the proton within the water cluster V (Fig. 1), the continuum absorbance was measured in mutated proteins. When side chains that are H bonded to the protonated water cluster are replaced the continuum absorbance change should be disturbed. Fig. 3 shows the time courses of the continuum absorbance change integrated between 1,900 and 1,800 cm-1 in the WT and in mutants changed at residues proposed to be H bonded to the protonated water cluster (38): E204Q, E204D, E194Q, E194D, R82Q, Y57F, Y83F, S193A, Y79F, and T205V. The initial, larger, decrease of the continuum absorbance change of WT in the L-to-M transition (line) is caused by deprotonation of the water cluster, releasing a proton to the extracellular surface (23). The second, smaller, decrease in the M-to-N transition (dashed) is assigned to the proton transfer via an H-bonded network providing a Grotthuss-like proton path between D96 and the Schiff base (36). The presence of a linear chain of four H-bonded water molecules that links these groups is evident in the x-ray diffraction structure of the N state (45). Of the two, only the L-to-M continuum absorbance change of WT will be considered here and compared with that of the mutants.
complex within bR, which is based on recent quantum-mechanical/molecular-mechanical (QM/MM) calculations (44). In these calculations a protonated water complex is considered in the protein environment and not only in water. The continuum spectrum of both a Zundel-like and an Eigen-like complex are calculated. The Eigen complex shows an increase of the continuum band at higher wavenumbers with a maximum at 2,500 cm-1, whereas the Zundel complex shows a decrease from 1,800 cm-1 to higher wavenumbers with zero amplitude at ≈2,500 cm-1. The agreement of the spectral dependence of the calculated Zundel complex shown in figure 2 of ref. 44 and the measured continuum absorbance shown here is astonishing. Therefore, the protonated water cluster resembles mostly a Zundel-like complex. To probe the delocalization of the proton within the water cluster V (Fig. 1), the continuum absorbance was measured in mutated proteins. When side chains that are H bonded to the protonated water cluster are replaced the continuum absorbance change should be disturbed. Fig. 3 shows the time courses of the continuum absorbance change integrated between 1,900 and 1,800 cm-1 in the WT and in mutants changed at residues proposed to be H bonded to the protonated water cluster (38): E204Q, E204D, E194Q, E194D, R82Q, Y57F, Y83F, S193A, Y79F, and T205V. The initial, larger, decrease of the continuum absorbance change of WT in the L-to-M transition (line) is caused by deprotonation of the water cluster, releasing a proton to the extracellular surface (23). The second, smaller, decrease in the M-to-N transition (dashed) is assigned to the proton transfer via an H-bonded network providing a Grotthuss-like proton path between D96 and the Schiff base (36). The presence of a linear chain of four H-bonded water molecules that links these groups is evident in the x-ray diffraction structure of the N state (45). Of the two, only the L-to-M continuum absorbance change of WT will be considered here and compared with that of the mutants.
Fig. 2.

Time-resolved Fourier transform IR difference spectrum in the 2,500- to 1,720-cm-1 region, integrated over the time range 300–400 μs, representing the changes in the M intermediate.
Fig. 3.

Time courses of the continuum absorbance change between 1,900 and 1,800 cm-1 for WT (blue circles) and various bR mutants (red squares) at pH 7 (R82Q at pH 8). The absorbance changes together with the fit-curves are shown. The L-to-M fit component of the WT (blue lines) and the corresponding fit component of the mutants (red lines) are shown in addition above the absorbance changes. All mutated residues are H-bonded to the lower water cluster (Fig. 1). Y79 and T205 are H-bonded only by their backbone. The kinetics of the continuum absorbance of Y57F, Y83F, and S193A are superimposed with their proton release kinetics, which was measured by using fluorescein (flu-, black triangle), and, additionally, pyranine (pyr-, green) for Y57F. The black lines show the fits. Again, the first component of the fit for the fluorescein measurement is shown in addition above the absorbance changes.
Some of these mutants show different photocycle and proton release kinetics as well.
E204Q and E194Q. The glutamates E194 and E204 on the extracellular side of the protein were suggested to be part of the proton release group, because mutations to the nonprotonatable glutamines show the delayed proton release characteristic of an inactive proton release system (20, 21). In this case the proton is released during the recovery of the BR state, most probably by D85. In these mutants the continuum absorbance change is not observed, indicating that the proton release mechanism is altered as compared with WT, and no protonated water network is formed.
E204D. It is an open question whether D204 is protonated when E204 is exchanged for an aspartate (20, 23). This question will be addressed by in situ H/D-exchange measurements (see below). E204D has a normal proton release in the L-to-M transition, and it shows a continuum absorbance change like WT (20, 23). In contrast to WT, however, the amplitude of the continuum absorbance change is much smaller. This observation is consistent with the earlier finding that at this pH the early proton release is partially blocked because of the lower pKa of the proton release complex in the ground state of this mutant (21).
E194D. In this mutant D194 is the proton release group (21). D194 is deprotonated in BR, becomes transiently protonated in M, and deprotonates later as a pH indicator dye detects a proton at the surface. The source of the proton was expected to be E204, but the corresponding negative band, which should appear in the M - BR spectra due to deprotonated E204, could not be observed by Dioumaev et al. (21). So it is an open question whether E204 is protonated in the BR state of E194D. As in the case of E204D, this question will be addressed below by H/D-exchange measurements. E194D shows no continuum absorbance change similarly to the E194Q mutant, therefore no protonated water network is established. Thus, the donor in the protonation of D194 cannot be a protonated water cluster.
R82Q. In R82Q the L-to-M transition is much faster than in WT, but proton release is delayed to 1 ms at pH > 7.5 (19), and it is delayed until the end of the photocycle at pH < 7.5 (20). The absence of rapid proton release is consistent with the absence of the first, larger, component of the continuum change in R82Q. But in comparison with the WT, there is a significant continuum absorbance decrease in R82Q well before the proton release (<100 ns). Different structural arrangements in WT and R82Q might explain the different continuum change. In WT there is a separation between the upper and lower water cluster (Fig. 1). No water exchange takes place between these two clusters (38), because the side chain of R82 acts as a barrier. However, mutation of this residue could result in merging of the two water clusters (38). As a result of this merging, the additional fast continuum absorbance change may indicate the rearrangement of water molecules near the Schiff base after photoisomerization. Because R82 stabilizes the protonated water cluster, changes of the arginine H bonding by deprotonation of the water cluster should cause changes of the R82 bands. Actually, changes of R82 are observed in the L-to-M transition (25) at 1,660 cm-1 and 1,556 cm-1. However, in contrast to our hypothesis, the authors propose that these bands indicate a deprotonation of R82 rather than H bonding change and therefore propose R82 to be the proton release group. However, R82 might also share a proton with Y57 (Fig. 1), and the conformational change of R82 in M might shift the proton equilibria and cause the two IR bands even if R82 is not the proton release group (19). Further quantum-mechanical/molecular-mechanical calculations might clarify the origin of the 1,660/1,556 cm-1 bands and the involvement of R82.
Y57F. A delayed proton release, with a time constant of 50 ms, was reported for Y57F (46), but our own measurements show more complex kinetics. Besides the early proton release in the L-to-M transition detected by fluorescein, there is an additional pyranine signal at the end of the photocycle (ref. 47; Fig. 3). This signal indicates changed pKa of the proton release complex, resulting in the existence of two Y57F species, one with an early and one with a late proton release. The former species should demonstrate WT-like continuum changes, whereas the latter may be expected to behave similarly to R82Q, as proposed above. Indeed, Y57F shows a continuum absorbance change that looks like a superposition of those from WT and R82Q (Fig. 3), with a significant contribution at the beginning of the photocycle.
Y83F and S193A. These mutations have smaller influence on the bR photocycle and on the continuum absorbance change. The proton release kinetics (Fig. 3, black lines) were measured by using fluorescein covalently bound to K129 at the extracellular surface (17). There is tight kinetic coupling between the proton release and the continuum absorbance change in Y83F, as in WT (23): the proton release is delayed as much as the continuum absorbance change.
In contrast, there is a small delay between the continuum absorbance change and the proton release in S193A. S193 prevents water exchange between the lower water cluster at E204/E194 and bulk water and is located at the proton release pathway. Upon replacement of the hydrophilic serine with the more hydrophobic alanine the exit might be even more blocked, which could lead to transient protonation of E204 or E194 after deprotonation of the protonated water network but before the proton release.
Y79F and T205V. These mutations have no influence on the continuum absorbance change. [Measurements of Y79F and T205V were performed at room temperature (≈23°C), whereas the WT measurement was made at 20°C, therefore small differences are seen in the kinetics.] The amino acids Y79 and T205 are connected by H bonds to the lower water cluster only by their backbones. Therefore mutations of these residues should not disturb water cluster V (Fig. 1), as indeed was observed here.
Summing up, these mutation studies probe the extent of proton delocalization within the water network. The influences of the studied mutations on the continuum absorbance change show that water cluster V in Fig. 1 describes adequately the space in which the proton fluctuates and the amino acids stabilizing the protonated water complex
Protonation States of E194, D194, E204, and D204. Knowing the protonation states of E204 and E194 in BR is crucial for defining the release mechanism. The question arises whether the proton is shared between the water network and the carboxylic acids or is confined entirely within the water network. Because the light-induced difference spectra can probe the protonation state of carboxylic acids only if they protonate during the photocycle (similarly to D85 in M), deprotonate (similarly to D96 in N), or undergo H-bonding changes (similarly to D96 in L), an alternative experimental approach was developed.
In Situ H/D-Exchange Measurements. H/D exchange induced by changing the solvent from H2O to D2O shifts the C=O stretching vibration frequency of a protonated carboxylic acid down by ≈10 cm-1. Usually, difference spectra are taken between BR and the intermediate state of interest in H2O and in D2O. Afterward the two spectra are compared to reveal the shifted carbonyl stretching vibrations. Here we report difference spectra taken directly (in situ) between BR in H2O and BR in D2O. This approach does not need two different protein states.
Fig. 4 a and c shows the enlarged 1,770- to 1,700-cm-1 region of the H/D-exchange spectra of WT and D96N. (The double mutant D96N/D115N and the complete spectrum as well as the H/D-exchange spectra of D115N are shown, respectively, in Figs. 7 and 8, which are published as supporting information on the PNAS web site.) The carbonyl region of WT can be deconvoluted into four Gaussian functions, which originate from the protonated and deuterated carboxylic amino acids of D96 and D115. The spectrum of D96N shows only the H-D-shift of D115. This measurement demonstrates that spectral deconvolution of in situ H/D-exchange spectra is capable of revealing the protonation states of D96 and D115 without measuring them in the photocycle and shows that there are no protonated carboxylic acids other than D96 and D115 in bR WT. In the deconvolution procedure, three additional functions (Fig. 4, gray lines) are necessary to account for the increase due to the strong amide I and water absorbance bands, which might mask the bands of protonated E204 or E194, which are expected to lie below 1,720 cm-1. By using the second derivative of the H/D-exchange spectrum the broad bands are eliminated and more details are revealed (Figs. 4 b and d, Fig. 7 f and h, and 8). In such a spectrum, a maximum appears as a minimum with two side maxima and vice versa. The isotopic downshift of the protonated carboxylic acid band of D115 in D96N is reflected by a minimum at 1,736 cm-1 and a maximum at 1,726 cm-1 (Fig. 4d, solid line). The second derivative of the H/D spectrum of the double mutant D96N/D115N (Fig. 4d, dotted line) contains, most convincingly, no carbonyl band shifts at all.
Fig. 4.

In situ H/D-exchange measurements and its second derivatives of bR WT and D96N. (a) The carbonyl region of WT can be deconvoluted into four Gaussian functions. The positive bands (green 1,742 cm-1, blue 1,736 cm-1) originate from the carbonyl stretch of the protonated carboxylic amino acids D96 and D115; negative bands (magenta 1,731 cm-1, red 1,725 cm-1), from the corresponding deuterated residues. (b) Second derivative of the H/D-exchange spectrum of WT. (c) The D96N spectrum is deconvoluted into two Gaussian functions showing protonated and deuterated D115 at 1,736 and 1,725 cm-1, respectively. (d) Second derivative of the H/D-exchange spectrum of D96N (solid line) and the double mutant D96N/D115N (dotted line). In D96N/D115N unprotonated carboxylic amino acids are detectable.
To verify that protonation changes can be monitored by this technique as well the protonation state, the second derivatives of the BR and M H/D-exchange spectra are compared for WT and D96N in Fig. 5 a and b. Here, both the protonation of D85 at 1,762 cm-1 and the downshift of this band to 1,749 cm-1 due to deuteration can be resolved. The mutant is used because M can be accumulated in larger amounts (compare Fig. 5 a and b). There is no protonation change of another carboxylic acid observed. If the released proton were to originate from a protonated carboxylic acid, an additional C=O stretch difference band should appear in this spectral region. E204Q shows the same spectrum (data not shown).
Fig. 5.

Second derivatives of the in situ H/D-exchange spectra of WT, D96N, E204D, and E194D for BR and M. The M spectra reveal an additional difference band due to protonated D85 (highlighted in green) (a and b). (c and d) The comparison of WT and E204D (c) reveals an additional difference band due to a protonated carbonyl band in the ground state of E204D. The protonated form is highlighted in blue and the deuterated in red. The same is observed for E194D (d). (e and f) In e the BR spectrum of E204D is compared with its M spectrum. The additional difference carbonyl band disappears in M. This observation shows that a protonated carbonyl group deprotonates in M. This is different for E194D (f). In this mutant the proton is transferred from E204 to D194 in the L-to-M transition and released later (21).
The same procedure is used to reveal the bands of carboxylic acids in the mutants E204D and E194D, where they are expected to absorb between 1,720 and 1,700 cm-1 if they are protonated. In Fig. 5c BR states of WT (black) and E204D (red) are compared. This comparison reveals an additional difference band for E204D, indicating a protonated carboxylic acid in the ground state of E204D. The protonated form is highlighted in blue, and the deuterated in red. A very similar band of a protonated carboxylic acid is observed in E194D (Fig. 5d). Because D194 is unprotonated in the BR state of this mutant (21), the band should also represent the protonated E204.
In the next step, it had to be resolved whether these groups undergo a protonation change in M. Therefore in Fig. 5e the BR spectrum of E204D (red dashed line) is compared with the M spectrum (green line). The additional difference band disappears in M. This observation shows that D204 deprotonates in M. In the E204D mutant, an equilibrium exists between the 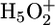 complex and protonated D204 because of their similar pKa values. The bands are different for E194D (Fig. 5d). In its M intermediate the E204 band disappears also (compare with the dashed line) as in E204D, and in addition the protonated band of D194 appears at 1,718/1,706 cm-1. The carbonyl band of D194 is assigned at 1,720 cm-1, as by Dioumaev et al. (21). This observation confirms that in the E194D mutant D194 becomes protonated in M by deprotonation of E204.
complex and protonated D204 because of their similar pKa values. The bands are different for E194D (Fig. 5d). In its M intermediate the E204 band disappears also (compare with the dashed line) as in E204D, and in addition the protonated band of D194 appears at 1,718/1,706 cm-1. The carbonyl band of D194 is assigned at 1,720 cm-1, as by Dioumaev et al. (21). This observation confirms that in the E194D mutant D194 becomes protonated in M by deprotonation of E204.
The results of the in situ H/D-exchange measurements are summarized in Table 1.
Table 1. Positions of vibrational bands (wavenumbers in cm-1) of the protonated carboxylic groups revealed by in situ H/D-exchange measurements.
| WT
|
E194D
|
E204D
|
|||||
|---|---|---|---|---|---|---|---|
| Intermediate | Residue | H2O | D2O | H2O | D2O | H2O | D2O |
| BR | D96 | 1,742 | 1,731 | 1,742 | 1,731 | 1,742 | 1,731 |
| D115 | 1,736 | 1,725 | 1,736 | 1,725 | 1,736 | 1,725 | |
| E/D204 | Deprotonated | 1,712 | 1,703 | 1,711 | 1,703 | ||
| M | D96 | 1,741 | 1,730 | 1,741 | 1,730 | 1,741 | 1,730 |
| D115 | 1,737 | 1,726 | 1,737 | 1,726 | 1,737 | 1,726 | |
| D85 | 1,761 | 1,749 | 1,763 | 1,750 | 1,761 | 1,749 | |
| E/D194 | Deprotonated | 1,718 | 1,706 | Deprotonated | |||
The wavenumbers in italics are results obtained from D96N or D115N mutants. The measurements of the D96 and D115 bands in M are not shown in this article.
Conclusions
The continuum absorbance change during the bR photocycle between 2,500 and 1,800 cm-1 indicates most likely that there is an excess proton within a network of internal bound water molecules, which is released to the extracellular medium during the L-to-M transition. The experimental results shown here in combination with earlier molecular dynamics simulations (38) reveal that the proton is delocalized within a network of five water molecules surrounded by Y57, R82, Y83, E204, E194, S193, P77, Y79, and T205 (Fig. 1). Replacement of six of nine residues on this list abolishes or strongly affects the continuum change, as expected. The Y79F and T205V mutations have no influence because Y79 and T205 are H bonded to the protonated water cluster by their backbone carbonyl groups (38). The fluctuation of the proton within the water cluster explains why a large variety of different mutations disturb or inhibit the proton release. Any time a group that stabilizes the protonated water complex is mutated the fine-tuned equilibrium is disturbed and the protonated water complex is destroyed. In contrast to earlier conclusions, none of the groups in this region represent a single proton-binding site. Rather, all of them contribute to the stabilization of the protonated water cluster. Thereby, the earlier controversy around the source of the released proton is resolved.
On the basis of the agreement between the experimentally observed continuum absorbance change and the quantum-mechanical/molecular-mechanical calculated continuum spectrum of a Zundel-like cation in density V (44), we conclude that an 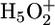 complex mostly stabilized by E194 and E204 is responsible for the proton release, as also proposed by Spassov et al. (26). This mechanism is illustrated in Fig. 6.
complex mostly stabilized by E194 and E204 is responsible for the proton release, as also proposed by Spassov et al. (26). This mechanism is illustrated in Fig. 6.
Fig. 6.

Schematic drawing of the proton release mechanism for bR WT and E194D. In WT a protonated water cluster (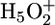 complex) is located at the extracellular side near the unprotonated E204 (Left). In the E194D mutant, the two water molecules do not form a
complex) is located at the extracellular side near the unprotonated E204 (Left). In the E194D mutant, the two water molecules do not form a 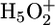 complex, but E204 is protonated (Right). In the E204D mutant, an equilibrium exists between the
complex, but E204 is protonated (Right). In the E204D mutant, an equilibrium exists between the 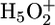 complex and protonated D204 because of their similar pKa values.
complex and protonated D204 because of their similar pKa values.
It is now established by the in situ H/D-exchange experimental approach described here that neither E204 nor E194 is protonated in the BR state of the WT. This situation is different in E204D and E194D, where a protonated carboxylic acid is observed in the ground state. In the E194D mutant, E204 is the protonated one (Fig. 6) and it is the proton donor to D194 in the L-to-M transition. The release, as measured with a dye, is somewhat delayed relative to WT, but coincident with deprotonation of D194 (21). Thus, the proton released in this mutant is a localized one. These conclusions are now in full agreement with earlier proposals on the protonation states and mechanisms of E204D and E194D (20, 21). In these mutants the release mechanism is changed from a delocalized proton fluctuating within an aqueous network in the WT protein to a localized proton-binding site. It appears that if the geometric relationship of the two carboxyl groups is perturbed by a glutamate- to-aspartate mutation, the proton will be located not in the cluster but on either E204 (in E194D) or D204 (in E204D).
The following proton release mechanism is proposed: The proton released in the L-to-M transition is mostly shared between two water molecules in a Zundel complex stabilized by E204 and E194. The protonated water cluster is stabilized by nine amino acids. Any mutation disturbs the complex and therefore the release mechanism. R82 plays a key role in the proton release mechanism: together with Y57 it separates the water cluster close to the Schiff base from the protonated water cluster V. Protonation of D85 induces a downward movement of the side chain of R82 to the extracellular side as proposed in ref. 48 and as the crystallographic structure of the M state had suggested (49). Upon movement of R82 the two water clusters merge (38, 50), thus providing a Grotthuss-like proton pathway from D85 to the proton release group (4). Movement of the charged arginine side chain toward 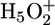 Zundel cation should disturb its symmetry and shift the equilibrium toward an H3O+ cation. This complex is energetically unfavorable and deprotonates (26). Thereby, protonation of D85 and the proton release is coupled.
Zundel cation should disturb its symmetry and shift the equilibrium toward an H3O+ cation. This complex is energetically unfavorable and deprotonates (26). Thereby, protonation of D85 and the proton release is coupled.
It is furthermore shown that in situ H/D-exchange measurements are able to reveal the protonation states of carboxylic acids in membrane proteins without having to induce changes in their protonation or H-bonding state. Therefore other proteins can be investigated that do not have the advantage of having at least two different states like the intermediates in the photocycle of bR.
Supplementary Material
Acknowledgments
We are grateful to J. Tittor for providing us with the D115N mutant and C. Kandt for producing parts of Fig. 1. F.G. and K.G. thank N. Bourdos for a critical review of the manuscript and the Deutsche Forschungsgemeinschaft (GE 599/12-1) and Sonderforschungsbereich 642 for financial support. This work was also supported in part by grants to J.K.L. from the National Institutes of Health (R01-GM29498) and the Department of Energy (DEFG03-86ER13525).
Author contributions: F.G., K.G., and J.K.L. designed research; F.G. and L.S.B. performed research; F.G. and K.G. analyzed data; and F.G., L.S.B., J.K.L., and K.G. wrote the paper.
Abbreviations: bR, bacteriorhodopsin; BR, bR ground state; H/D, hydrogen/deuterium.
References
- 1.Oesterhelt, D. & Stoeckenius, W. (1971) Nat. New Biol. 233, 149-152. [DOI] [PubMed] [Google Scholar]
- 2.Haupts, U., Tittor, J. & Oesterhelt, D. (1999) Annu. Rev. Biophys. Biomol. Struct. 28, 367-399. [DOI] [PubMed] [Google Scholar]
- 3.Lanyi, J. K. (2000) J. Phys. Chem. B 104, 11441-11448. [Google Scholar]
- 4.Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. (1999) J. Mol. Biol. 291, 899-911. [DOI] [PubMed] [Google Scholar]
- 5.Lanyi, J. K. & Schobert, B. (2003) J. Mol. Biol. 328, 439-450. [DOI] [PubMed] [Google Scholar]
- 6.Lozier, R. H., Bogomolni, R. A. & Stoeckenius, W. (1975) Biophys. J. 15, 955-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie, A. H., Nagle, J. F. & Lozier, R. H. (1987) Biophys. J. 51, 627-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames, J. B., Bolton, S. R., Netto, M. M. & Mathies, R. A. (1990) J. Am. Chem. Soc. 112, 9007-9009. [Google Scholar]
- 9.Stockburger, M., Klusmann, W., Gattermann, H., Massig, G. & Peters, R. (1979) Biochemistry 18, 4886-4900. [DOI] [PubMed] [Google Scholar]
- 10.Hessling, B., Souvignier, G. & Gerwert, K. (1993) Biophys. J. 65, 1929-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothschild, K. J. (1992) J. Bioenerg. Biomembr. 24, 147-167. [DOI] [PubMed] [Google Scholar]
- 12.Braiman, M. & Mathies, R. (1982) Proc. Natl. Acad. Sci. USA 79, 403-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelhard, M., Gerwert, K., Hess, B., Kreutz, W. & Siebert, F. (1985) Biochemistry 24, 400-407. [DOI] [PubMed] [Google Scholar]
- 14.Gerwert, K., Hess, B., Soppa, J. & Oesterhelt, D. (1989) Proc. Natl. Acad. Sci. USA 86, 4943-4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerwert, K., Souvignier, G. & Hess, B. (1990) Proc. Natl. Acad. Sci. USA 87, 9774-9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith, S. O., Pardoen, J. A., Mulder, P. P., Curry, B., Lugtenburg, J. & Mathies, R. (1983) Biochemistry 22, 6141-6148. [Google Scholar]
- 17.Heberle, J. & Dencher, N. A. (1992) Proc. Natl. Acad. Sci. USA 89, 5996-6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao, Y., Brown, L. S., Needleman, R. & Lanyi, J. K. (1993) Biochemistry 32, 10239-10248. [DOI] [PubMed] [Google Scholar]
- 19.Govindjee, R., Misra, S., Balashov, S. P., Ebrey, T. G., Crouch, R. K. & Menick, D. R. (1996) Biophys. J. 71, 1011-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown, L. S., Sasaki, J., Kandori, H., Maeda, A., Needleman, R. & Lanyi, J. K. (1995) J. Biol. Chem. 270, 27122-27126. [DOI] [PubMed] [Google Scholar]
- 21.Dioumaev, A. K., Richter, H. T., Brown, L. S., Tanio, M., Tuzi, S., Saito, H., Kimura, Y., Needleman, R. & Lanyi, J. K. (1998) Biochemistry 37, 2496-2506. [DOI] [PubMed] [Google Scholar]
- 22.Koyama, K. & Miyasaka, T. (2004) Electrochemistry 72, 2-4. [Google Scholar]
- 23.Rammelsberg, R., Huhn, G., Lubben, M. & Gerwert, K. (1998) Biochemistry 37, 5001-5009. [DOI] [PubMed] [Google Scholar]
- 24.Zscherp, C., Schlesinger, R., Tittor, J., Oesterhelt, D. & Heberle, J. (1999) Proc. Natl. Acad. Sci. USA 96, 5498-5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao, Y., Hutson, M. S., Belenky, M., Herzfeld, J. & Braiman, M. S. (2004) Biochemistry 43, 12809-12818. [DOI] [PubMed] [Google Scholar]
- 26.Spassov, V. Z., Luecke, H., Gerwert, K. & Bashford, D. (2001) J. Mol. Biol. 312, 203-219. [DOI] [PubMed] [Google Scholar]
- 27.Agmon, N. (1995) Chem. Phys. Lett. 244, 456-462. [Google Scholar]
- 28.Marx, D., Tuckerman, M. E., Hutter, J. & Parrinello, M. (1999) Nature 397, 601-604. [Google Scholar]
- 29.Vuilleumier, R. & Borgis, D. (1999) J. Chem. Phys. 111, 4251-4266. [Google Scholar]
- 30.Day, T. J. F., Schmitt, U. W. & Voth, G. A. (2000) J. Am. Chem. Soc. 122, 12027-12028. [Google Scholar]
- 31.Eigen, M. (1964) Angew. Chem. Int. Ed. 3, 1-19. [Google Scholar]
- 32.Zundel, G. (2000) Adv. Chem. Phys. 111, 1-218. [Google Scholar]
- 33.Eigen, M., Wicke, E. & Ackermann, T. (1954) Z. Phys. Chem. (Munich) 1, 340-364. [Google Scholar]
- 34.Kim, J., Schmitt, U. W., Gruetzmacher, J. A., Voth, G. A. & Scherer, N. E. (2002) J. Chem. Phys. 116, 737-746. [Google Scholar]
- 35.Garczarek, F., Wang, J., El-Sayed, M. A. & Gerwert, K. (2004) Biophys. J. 87, 2676-2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Coutre, J., Tittor, J., Oesterhelt, D. & Gerwert, K. (1995) Proc. Natl. Acad. Sci. USA 92, 4962-4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, J. P. & El-Sayed, M. A. (2000) J. Phys. Chem. 104, 4333-4337. [Google Scholar]
- 38.Kandt, C., Schlitter, J. & Gerwert, K. (2004) Biophys. J. 86, 705-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrando, E., Schweiger, U. & Oesterhelt, D. (1993) Gene 125, 41-47. [DOI] [PubMed] [Google Scholar]
- 40.Needleman, R., Chang, M., Ni, B., Varo, G., Fornes, J., White, S. H. & Lanyi, J. K. (1991) J. Biol. Chem. 266, 11478-11484. [PubMed] [Google Scholar]
- 41.Oesterhelt, D. & Stoeckenius, W. (1974) Methods Enzymol. 31, 667-678. [DOI] [PubMed] [Google Scholar]
- 42.Rammelsberg, R., Hessling, B. & Chorongiewski, H. (1997) Appl. Spectrosc. 51, 558-562. [Google Scholar]
- 43.Dencher, N. A., Sass, H. J. & Buldt, G. (2000) Biochim. Biophys. Acta 1460, 192-203. [DOI] [PubMed] [Google Scholar]
- 44.Rousseau, R., Kleinschmidt, V., Schmitt, U. W. & Marx, D. (2004) Angew. Chem. Int. Ed. 43, 4804-4807. [DOI] [PubMed] [Google Scholar]
- 45.Schobert, B., Brown, L. S. & Lanyi, J. K. (2003) J. Mol. Biol. 330, 553-570. [DOI] [PubMed] [Google Scholar]
- 46.Govindjee, R., Kono, M., Balashov, S. P., Imasheva, E., Sheves, M. & Ebrey, T. G. (1995) Biochemistry 34, 4828-4838. [DOI] [PubMed] [Google Scholar]
- 47.Brown, L. S. (2000) Biochim. Biophys. Acta 1460, 49-59. [DOI] [PubMed] [Google Scholar]
- 48.Bashford, D. & Gerwert, K. (1992) J. Mol. Biol. 224, 473-486. [DOI] [PubMed] [Google Scholar]
- 49.Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. (1999) Science 286, 255-260. [DOI] [PubMed] [Google Scholar]
- 50.Kandt, C., Gerwert, K. & Schlitter, J. (2004) Proteins 58, 528-537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}