

SUMMARY
Although DNA modifications play an important role in gene regulation, the underlying mechanisms remain elusive. We developed EpiSELEX-seq to probe the sensitivity of transcription factor binding to DNA modification in vitro using massively parallel sequencing. Feature-based modeling quantifies the effect of cytosine methylation (5mC) on binding free energy in a position-specific manner. Application to the human bZIP proteins ATF4 and C/EBPβ, and three different Pbx-Hox complexes shows that 5mCpG can both increase and decrease affinity, depending on where the modification occurs within the protein-DNA interface. The TF paralogs tested vary in their methylation sensitivity, for which we provide a structural rationale. We show that 5mCpG can also enhance in vitro p53 binding, and provide evidence for increased in vivo p53 occupancy at methylated binding sites, correlating with primed-enhancer histone marks. Our results establish a powerful strategy for dissecting epigenomic modulation of protein-DNA interactions and their role in gene regulation.
Graphical Abstract

INTRODUCTION
High-throughput profiling of in vitro transcription factor (TF) binding specificities is a powerful approach for obtaining sequence motifs for a variety of TF families, and in several different organisms (Badis et al., 2009; Jolma et al., 2013; Weirauch et al., 2014). However, despite the growing number of known TF motifs, accurate prediction of in vivo TF binding and its effect on target gene expression has remained surprisingly difficult. One of the complications is that protein-protein interactions can modify the DNA binding specificities of transcription factors (Jolma et al., 2015; Miller, 2009; Slattery et al., 2011). Another potential complication is the existence of covalent modifications of DNA, particularly cytosine methylation (5mCpG), which is widespread in vertebrates. Because of their potential to alter chromatin state (Hashimshony et al., 2003) or DNA shape (Lazarovici et al., 2013), an important and hotly debated question is to what extent DNA modifications can influence TF binding and thereby contribute to changes in the epigenetic landscape and gene regulation. Such a regulatory mechanism is conceptually compelling, as DNA modifications could provide an additional layer of temporal and spatial control to fine-tune gene expression.
5mCpG has been shown to be important in gene silencing in normal and cancer cells (Jones and Baylin, 2007; Stein et al., 1982), gene imprinting (Razin and Cedar, 1994), and X chromosome inactivation (Hellman and Chess, 2007; Tribioli et al., 1992). In spite of this progress, there is no general mechanism explaining the impact of DNA methylation on gene expression (Dantas Machado et al., 2015). Several studies have found that despite the overall association between promoter methylation and transcriptional silencing, some promoters can simultaneously be methylated and transcriptionally active (Gutierrez-Arcelus et al., 2013). In addition, systematic studies with cancer cell lines have found that aberrant methylation, such as hypermethylation of specific CpG islands, is a hallmark of cancer progression (Baylin and Jones, 2011; Paz et al., 2003). Recent studies have identified additional modifications such as 5hmC and 6mA in mammalian genomes, raising the possibility that these also influence gene regulation (Fu et al., 2015; Greer et al., 2015; Zhang et al., 2015). To identify the causal determinants of in vivo TF binding among all these correlated variables, detailed quantitative characterization of the effect of DNA modification on in vitro transcription factor binding is a prerequisite.
On a limited scale, the in vitro platform of protein binding microarrays (PBM) has been used to probe TF binding to methylated DNA probes (Hu et al., 2013; Mann et al., 2013). These studies demonstrated that 5mCpGs can have both positive and negative effects on affinity. However, they were limited by the fact that the DNA arrays contained either fully methylated or fully un-methylated sequences (Mann et al., 2013), but not both in competition, or they only considered a select subset of sequences (Hu et al., 2013). In addition, the data analysis in these studies was restricted to oligomer-based methods, which makes it difficult to identify position-specific effects, especially for lower-affinity binding sites that deviate from the consensus motif. To study the effect of cytosine methylation on TF binding at high resolution, a quantitative assay is required that allows for simultaneously probing of methylated and unmethylated DNA probes across all possible sequence contexts.
To address these issues, we developed EpiSELEX-seq, a method that uses a single round of gel electrophoresis to simultaneously assess binding to methylated and unmethylated DNA fragments, thus allowing methylation sensitivity to be analyzed for any TF or TF complex. We apply EpiSELEX-seq to human bZIP and Hox complexes, as well as tetramers of the tumor suppressor protein p53. Using a feature-based Poisson regression model, we quantify position-specific methylation effects on in vitro binding in the low affinity range. For p53, by jointly analyzing whole genome bisulfite sequencing and in vivo binding (ChIP-seq) data, we provide evidence that the increased in vitro affinity for specific DNA sequences due to methylation leads to enhanced occupancy in vivo. These sites of increased binding have a histone modification pattern associated with primed enhancers, supporting a role for p53 as a pioneer factor that can access methylated DNA sites.
RESULTS
Affinity-based selection from mixed pools of methylated and unmethylated DNA ligands
To quantitatively assess the effects of DNA methylation on TF binding, we developed a method in which methylated (Lib-M) and unmethylated (Lib-U) libraries containing a randomized region of a desired length (16 bp or 26 bp) were first separately synthesized, each distinguished by a unique 4 bp barcode located near the variable region (Figure 1A). After treatment of Lib-M with a DNA methyltransferase, both libraries were mixed in equal proportions, incubated with a TF of interest, and subjected to a single round of EMSA selection. Sequencing libraries were prepared from the library mix both before (R0) and after (R1) affinity-based selection (Figure 1B and Figure S1A–B). For each sequenced DNA ligand, the barcode allows us to reconstruct the methylation status at the time of TF binding.
Figure 1. Overview and validation of the EpiSELEX-seq design.

(A) Library design. 4 bp barcodes distinguish unmodified (Lib-U) and modified (Lib-M) DNA ligands. All libraries share a random region, reverse-complement-symmetric flanks and a pair of 5′ and 3′ primer sites. (B) EpiSELEX-seq workflow. Lib-M is methylated and mixed with Lib-U. The mixed pool is incubated with a TF of interest and the bound fraction is separated by an EMSA, purified, split, and amplified using two sets of primers. Unique Illumina barcodes are added for multiplexing. (C) Validation of methylation protocol. Shown are dinucleotide frequencies in Lib-M after various combinations of optional methylation (M+/M−) and bisulfite treatment (BsT+/BsT−), determined by Illumina sequencing. The four CpN dinucleotides, for which the methylation status of the cytosine is unambiguous, are highlighted, as is TpG, which serves as a reference for CpN dinucleotides. (D) TpG-normalized recovery of the four CpN dinucletides. Only the CpGs protected by methylation are retained after bisulfite conversion.
For accurate affinity estimation, it is important that the two cytosines in each CpG base-pair step in Lib-M be fully methylated, as incomplete methylation would lead to underestimation of the impact of 5mCpG on TF binding. We employed two separate tests to confirm full methylation: (i) methylation, bisulfite treatment, and sub-cloning of a test sequence containing four CpGs and (ii) high-throughput sequencing followed by dinucleotide analysis of a methylated library that was either treated or not treated with bisulfite. In the first test, we determined that optimal methylation efficiency is achieved after two successive rounds of methylation with ≤250 ng of input DNA per reaction (Table S1). Using larger amounts of DNA (e.g., the recommended 1 ug) resulted in incomplete methylation of the test probes. The short size of our probes (~50 bp) compared to typical genomic fragments (>1 kb) might be the source of this discrepancy because suboptimal conditions typically resulted in the methylation of either all four CpGs or none, arguing for a processive nature of the DNA methyltransferase. In the second test, bisulfite treatment of an unmethylated library of random 16-mers showed depletion of all CpN dinucleotides, as expected (Figure 1C). In contrast, under optimal methylation conditions, bisulfite treatment of a methylated library showed depletion of all CpN dinucleotides except CpG, which was recovered at levels identical to those observed in non-bisulfite treated, methylated libraries (Figure 1D).
EpiSELEX-seq identifies differences in methylation sensitivity within the bZIP family
To benchmark our method, we considered the basic leucine zipper (bZIP) transcription factors ATF4 and C/EBPβ, previously reported to be sensitive to DNA methylation (Mann et al., 2013). Many bZIP homo- or heterodimers preferentially bind to the cAMP response element (CRE) TGACGTCA and/or the C/EBP consensus TTGCGCAA (Figure 2A–B). These palindromic sequences both contain a central CpG dinucleotide, creating the potential for methylation-sensitive DNA binding. For ATF4 homodimers, as expected, the relative enrichment of 10 bp sequences (encompassing the suspected TF footprint) that do not contain any CpG dinucleotides is similar between Lib-U and Lib-M (Figure 2C). However, sequences that contain at least one CpG fall into distinct groups, each with a different ratio between Lib-M and Lib-U, indicative of a sensitivity to cytosine methylation that depends on the position of the CpG dinucleotide within the binding site (Figure 2C and Figure S2A). When a CpG base pair step is present at the center of the ATF4 binding site, methylation of both cytosines leads to a decrease in affinity. By contrast, sequences that contain a CpG in the flank of the motif (at positions −3/−4 or +3/+4) are bound much more strongly when methylated, leading to an alternative optimal left half-site, (5mC)GAT. Interestingly, these methylation sensitivities are not observed for C/EBPβ (Figure 2D), consistent with a previous observation that in vivo binding by this factor tolerates CpG methylation (Zhu et al., 2016). The methylation sensitivity for ATF4 is also reflected in the energy logos (Foat et al., 2006) that can be derived from the oligomer enrichment tables by considering all possible point mutations away from the optimal sequence (see Experimental Procedures). The logo derived from Lib-M, when compared to its equivalent for the unmethylated library (Lib-U), no longer has a central CpG as the most preferred sequence, and shows an increased preference for a CpG at position −4/−3 (Figure 2E–F). Together, these findings demonstrate that sensitivity to DNA methylation can differ between paralogs from the same structural family.
Figure 2. Probing methylation sensitivity for ATF4 and C/EBPβ.

(A, B) Crystal structure (PDBID: 1GTW) for the human bZIP homodimer C/EBPβ along with the symmetric consensus motifs for ATF4 (A) or for C/EBPβ (B) and the definition of ‘flank’ (green) and ‘center’ (pink) positions in the binding sites. (C) Enlargement of low affinity range comparing the relative enrichment of 10 bp oligonucleotides between Lib-M versus Lib-U for ATF4. Non-CpG sequences (blue) show similar enrichment in both libraries, while distinct subsets of the CpG-containing sequences (red) are either preferred in Lib-U (“center”) or in Lib-M (“flank”). (D) As in C but for C/EBPβ homodimers. Non-CpG and CpG-containing sequences show similar enrichments in both libraries across entire sequence range. Insets in C and D show the marginal distributions and the distribution of methylated/unmethylated ratio for all oligomers with a relative enrichment above 10−3. (E, F) Energy-Logo for ATF4 derived from Lib-U (E) and Lib-M (F). The central CpG is no longer the top choice in the methylated library. 5mCpGs at the equivalent positions −4/−3 and +3/+4 appear as a new sequence feature in Lib-M. (G) Relative affinities (each point represents a 10 bp oligomer) containing either an A (reference base) or a point mutation (C, T, or G) at position −5. The slope of the lines represents the value of ΔΔG associated with each point mutation as estimated from the Lib-U read counts using a feature-based model. (H) Lib-M versus Lib-U 10-mer relative affinity plots in logarithmic scale. Lines represent the ΔΔG coefficients for the position-dependent methylation effects derived from the feature-based model.
Feature-based modeling quantifies position-specific methylation effects
To systematically analyze the quantitative effect of cytosine methylation on binding affinity, we developed a feature-based generalized linear model to estimate the change in binding free energy associated with cytosine modification at any particular offset within the binding site. The frequency of DNA ligand S after one round (R1) of affinity-based selection, F1, is proportional to the frequency of the same probe in the initial (R0) pool, F0, as well as to the relative affinity of the interaction:
We model ΔΔG(S), the difference in binding free energy between ligand S and the optimal ligand Sopt as a sum of contributions due to the specific (binary) features ϕ associated with S:
Some features indicate the presence (X=1) or absence (X=0) of a specific base at a given position within the binding site, while others indicate the methylation status of a particular CpG dinucleotide. We estimate the corresponding coefficients βϕ from the data by fitting a generalized linear model based on counting statistics to the read counts in R1 while accounting for biases in R0 (see Experimental Procedures for details). To validate this modeling approach, we first inferred free energy effects for the three possible substitutions of the optimal base A−5 using the ATF4 homodimer data. Good agreement is observed with the results obtained using oligomer enrichment (Figure 2G). Next, we used an extended model that included features indicating methylation status. The coefficients from this fit indicate that methylation of C−1G+1 represses binding (ΔΔG/RT = 1.5, corresponding to 0.9 kcal/mol, or equivalently, a 4.5-fold reduction in affinity), consistent with the changes in oligomer enrichment between Lib-U and Lib-M (Figure 2H). The coefficients for the equivalent flanking positions C−4G−3 and C+3G+4 are almost identical, as expected based on symmetry, and indicate a strong increase in binding due to methylation (ΔΔG/RT = −2.6). Our model also predicts the combined effect of methylating both C−1G+1 and C+3G+4 (or C−3G−4) by simply adding up the respective free energy coefficients (Figure 2H).
Explaining the effect of cytosine methylation by “thymine mimicry”
Although it has distinct base pairing preferences, 5mC is chemically similar to thymine in that both have a methyl group at the carbon 5 position of the pyrimidine ring (Figure 3A). Therefore, the total impact of a C to T transition on protein-DNA binding free energy, ΔΔG[C → T], can be separated into (i) the effect of the methyl group alone, ΔΔG[C → 5mC], and (ii) changes in charge and base pair interactions, ΔΔG[5mC → T] (Figure 3A). Following this logic, the value of ΔΔG[C → T] and ΔΔG[5mC → T], as estimated using the unmethylated (Lib-U) and methylated (Lib-M) library, respectively, can be subtracted from each other to obtain an estimate of the effect due to methylation ΔΔG[C → 5mC]. This approach was successful when applied to ATF4 to predict the effect of methylating the CpG dinucleotide, both at the central (−1/+1) and the flanking (−4/−3) positions (Figure 3B–D). In agreement with these observations, many bZIP proteins contain two conserved hydrophobic amino acids that in crystal structures make van der Waals (VdW) contacts with the carbon 5 methyl group of thymidine at position −4 in the binding site (Figure S2B). ATF4, but not C/EBPβ, has a valine instead of an alanine at one of these positions, providing a possible mechanistic explanation for the increased preference of ATF4 for 5mC over C, where the gain of a methyl group on the base may compensate for the loss of a methyl group in alanine compared to valine.
Figure 3. Deconvolving the methylation sensitivity for ATF4.

(A) Decomposition of the position-specific DNA-protein binding free energy change associated with a C→T transition. The C→T change is the sum of C→5mC and 5mC→T, allowing an interpretation of methylation sensitivity in terms of “thymine mimicry.” (B) Change in binding free energy associated with C→T transition in each library as derived from an oligomer based PSAM. (C) Position-specific methylation effect on binding free energy, as estimated based on either the oligomer-enrichment-based approach (as in B; grey) or the feature-based-modeling approach (red). (D) The methylation effect as estimated using the feature-based model (red arrows) explains the differences in the C→T transition effect observed for Lib-U and Lib-M.
Deciphering the DNA binding specificity of human Pbx-Hox complexes
An important aspect of gene regulation is the capacity of TFs to form complexes with cofactors. A prominent example of such cooperative binding is that of Hox proteins and their three amino acid loop extension (TALE) cofactors, which play a crucial role in animal development (Merabet and Mann, 2016). As monomers, Hox family members bind to similar DNA sequences in vitro, but have distinct functions in vivo. Previously, we used SELEX-seq to capture the latent binding specificity of all eight Drosophila Hox proteins with their TALE cofactors Extradenticle (Exd) and the HM-isoform (HM) of Homothorax (Hth), which is required for optimal Exd-Hox interaction (Slattery et al., 2011). In mammals, where the Hox cluster has been duplicated several times in the genome, multiple cofactors from the PBC and MEIS class of TALE factors, as well as epigenetic DNA modifications, all have the potential to modulate DNA binding.
Here, we used EpiSELEX-seq to characterize the binding of human heterodimeric Pbx-Hox complexes to DNA (Figure 4A). To cover the three Hox subclasses defined in Slattery et al. (Slattery et al., 2011), we performed these experiments using HoxA1, HoxA5 and HoxA9, each in complex with the cofactor PBX1, which was purified together with the HM domain of MEIS1. Comparing the pattern of 12 bp oligomer enrichment from R0 to R1 for each complex, we found similar cofactor-dependent differences in binding specificity between these Hox proteins as previously observed for their D. melanogaster orthologs (Slattery et al., 2011) (Figure 4B and Figure S4A): the preferred central dinucleotide spacer (underlined) in the binding site consensus NTGAYNNAYNNN (where Y denotes C or T) is TG for anterior (Class I) factor HoxA1, TA for central (Class II) factor HoxA5, and TT for posterior (Class III) factor HoxA9 (Figure 4D).
Figure 4. Methylation-sensitivity of human Pbx-Hox complexes.
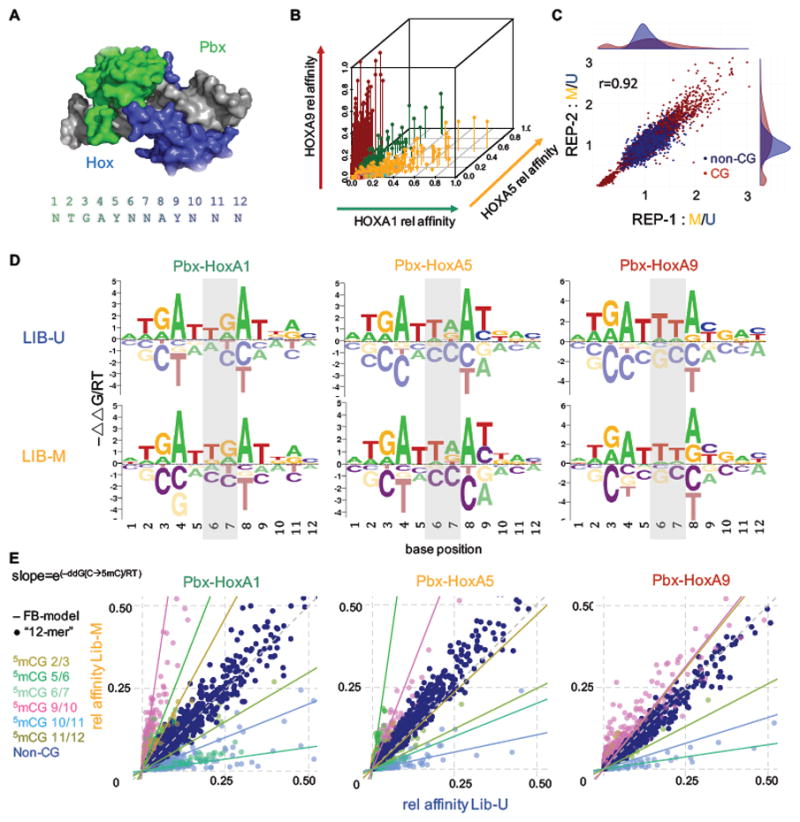
(A) Crystal structure (PDBID: 1PUF) of human Pbx-HoxA9 with Hox shown in blue and Pbx in green. The consensus sequence with position labels is shown as a reference. (B) Relative affinity comparison of Pbx1 plus HoxA1, HoxA5 or HoxA9 (green, orange, red). Each Hox prefers distinct sets of 12-mers. Preferred central spacers (position 6 and 7) are TG, TA and TT for HoxA1, HoxA5 and HoxA9, respectively. (C) Replicate agreement for EpiSELEX-seq of Pbx1-HoxA9. Methylated/unmethylated (M/U) ratios for 12-mers are shown for one replicate versus the other. Sequences with or without CpGs are red or dark blue respectively. Pearson correlation of 0.92. Staggered density plots show a narrow distribution of non-CpG 12-mers around 1, but a much broader and bimodal distribution for CpG 12-mers. (D) Oligomer-based energy logos for all three Pbx-Hox complexes for Lib-U and Lib-M. No obvious differences between the methylated and unmethylated libraries are observed. Central spacer is shaded in grey. (E) Lib-M versus Lib-U relative affinity plots for all three complexes. Points are colored based on the position of the CpG dinucleotide (dark blue for non-CpG sequences). The slopes of the lines represent the exponentiated free energy coefficient for the methylation effect in the feature-based (FB) model.
Human Pbx-Hox dimers show position-specific methylation sensitivity
The EpiSELEX-seq protocol allows us to assess the three human Pbx-Hox complexes for sensitivity to cytosine methylation. We first constructed separate energy logos for Lib-U and Lib-M by considering all possible point mutations from the most enriched 12 bp sequence (Figure 4D). While paralog-dependent differences in the central spacer (shaded area) are readily apparent, the logos for the unmethylated (Lib-U) and methylated (Lib-M) human libraries are otherwise highly similar to each other and to those of their fly orthologs. However, this oligomer enrichment based approach is unable to detect methylation sensitivity for any cytosine that does not occur in a CpG context in the optimal sequence (Figure S3). For ATF4, both cytosine positions at which methylation sensitivity was observed (−4 and −1) were fortuitously followed by a guanine (cf. Figure 2A), but this is not the case for Pbx-Hox. Indeed, when we used our feature-based Poisson regression model to jointly analyze the Lib-U and Lib-M libraries in order to quantify the effect of 5mCpG on binding, all three Hox proteins and Pbx showed significant methylation sensitivity at various positions throughout the binding interface (Figure 4C, E and Figure S4B). The direction and amplitude of the methylation effect are highly position-dependent: methylation of CpG dinucleotides that start at positions 5 or 9 (underlined in the consensus sequence NTGAYNNAYNNN) enhance binding by several fold. In contrast, methylation of CpGs shifted by one position (positions 6 or 10, underlined in NTGAYNNAYNNN) decreases binding by up to 7-fold (Figure 4E). This is reflected in both the energy coefficients (lines in Figure 4E) and in the relative enrichment of 12-mers (points in Figure 4E and Figure S4C). We tested these predictions using competition DNA binding experiments. Consistent with our EpiSELEX-seq analysis, using binding sites that contain a CpG at position 9/10 revealed that a higher concentration was required for unmethylated (IC50 = 45.5 ± 14.7) than for methylated (IC50 = 20.3 ± 2.6) binding sites to compete with a radioactively labeled consensus probe for Pbx- HoxA1 binding (Figure S5).
Thymine mimicry explains variation in methylation sensitivity among Hox paralogs
The effect of methylation on binding not only depends on the position of the CpG dinucleotide within the protein-DNA interface but also differs between Hox paralogs (Figure 5A). At dinucleotide positions 5/6 and 9/10 the strength of methylation sensitivity is collinear with the Hox expression domain along the anterior-posterior axis (HoxA1-HoxA5-HoxA9), similar to other aspects of Hox function (Slattery et al., 2011). To gain more insight into the structural mechanisms underlying these differences in binding, we compared HoxA1 and HoxA9, which show distinct differences in methylation preference at position 9: Pbx-HoxA1 strongly prefers T over C (ΔΔG[C → T]/RT ≈ −2.0), while Pbx-HoxA9 shows no such preference (Figure 5B). Close examination of a Pbx- HoxB1 (a proxy for HoxA1) crystal structure reveals that isoleucine at position 47 (Ile47) within the homeodomain has a VdW interaction with the carbon 5 methyl group on base T9 of the forward DNA strand (Figure 5B). In contrast, in a Pbx-HoxA9 crystal structure Ile47 is closer to and interacts with the C9 base, even without this methyl group. Accordingly, we would predict that HoxB1/A1 should benefit from the methylation of a C9, whereas HoxA9 should be indifferent to methylation. Indeed, ΔΔG[C9 → T9]/RT is similar to (ΔΔG[C9 → 5mC9]/RT) for HoxA1 whereas for HoxA9 ΔΔG[C9 → 5mC9]/RT is close to zero (Figure 5B). As the crystal structures show no further base-specific interactions at position 9, these differences can be fully accounted for by the relative benefit of gaining a methyl group for each paralog.
Figure 5. Collinearity of methylation sensitivity explained by structural differences.

(A) Comparison of the methylation effect for all three Pbx-Hox complexes. The two A9 replicates are shown in different shades of red and have good agreement (blue asterisks indicate that coefficients were fit at sub-optimal affinity thresholds due to low counts). Position 9/10 shows large paralog-dependent differences, with HoxA1 having high, HoxA5 medium, and HoxA9 almost no methylation sensitivity; position 5/6 shows the opposite trend. (B) Comparing Hox-specific C or T read-out for position 9. HoxA1 prefers a T over a C, whereas HoxA9 has equal preference. The observed difference in binding free energy associated with a C→T transition should equal the methylation sensitivity difference between HoxA1 and HoxA9. Alignment of helix3 of several Hox TFs (B1, A1, A5, A9) reveals conservation of Ile47 for the Hox family, but polymorphism at residue 43. Ile47 interacts with the pyrimidine at position 9 in both the HoxB1 and the HoxA9 structures. The distance to the aromatic carbon 5 is 5.4 Å for HoxB1, but only 3.9Å for HoxA9. Addition of a methyl group in HoxB1 reduces the distance to 4.0 Å, allowing for the same VdW interaction as seen in HoxA9. Arg43 (A9) aids in bringing Ile47 closer to the DNA by interacting with the phosphate backbone at nucleotide C9, whereas Thr43 (B1/A1) does not interact with the backbone, but rather pulls Ile47 away from T9. The C→T energy difference between HoxA1 and HoxA9 is most likely driven by the methyl read-out. The table shows that the C→T free energy difference is comparable to the difference in methylation sensitivity (feature-based model) between the two paralogs.
EpiSELEX-seq identifies non-consensus p53 binding sequences whose affinity is increased upon methylation
Because altered methylation patterns are observed in many cancers, we tested if binding by the human tumor suppressor protein p53 might be methylation sensitive. In vivo, p53 is thought to bind as a tetramer to two dimer sites RRRCWWGYYY (which we will refer to as CWWG) separated by a spacer of 0–13 bp (el-Deiry et al., 1992; Funk et al., 1992) (Figure 6A). Consistently, the palindromic sequence GGACATGTCC site independently emerged from our data as the most enriched 10-mer in both Lib-M and Lib-U (Figure S6A). Comparing Lib-M and Lib-U directly reveals that there are three different classes of CpG-containing sequences that show altered p53 binding upon methylation (Figure 6A). Methylation of a CpG occurring at the 3′ end of the half site (RRRCATGYCG, which we will refer to as C+4G+5, relative to the motif center) decreases binding by ~20%, while methylation at a CpG shifted one bp to the left (RRRCATGCGY or C+3G+4) increases binding by ~50%. The largest effect, a ~250% increase in binding affinity, was observed when the CpG is in the core of the binding site (RRRCACGYYY or C+1G+2). Analysis of a p53 crystal structure [3Q06; (Petty et al., 2011)] reveals that the methyl group at carbon 5 of the T+1 base pyrimidine ring in the CATG core is stacked above the polar guanidinium plane of p53 amino acid R280. The latter is crucial for p53 binding as it forms a hydrogen bond with the G+2 base (Figure 6B). The thymine methyl group might thus direct and constrain R280 towards G+2, which has been proposed to serve as a methylation readout mechanism of zinc finger proteins (Liu et al., 2013). T+1→C+1 replacement would thus eliminate the guiding methyl group, providing an explanation for the stabilizing effect of methylation at position C+1.
Figure 6. p53 differentially binds methylated motifs in vivo in distinct chromatin modification states.

A) EpiSELEX-seq 10-mer relative affinity plot showing the consensus motif (RRRCWWGYYY; blue) and 3 classes of CpG-containing motifs. CpG motifs are differentially bound upon methylation, with methylation of a) C4+G5+ (green) halfsites reducing binding about 20%, whereas methylation of b) C3+G4+ (cyan) and c) C1+G2+ (pink) sites increases binding ~1.5 and ~2–3 fold respectively. Non-CpG consensus sites, as expected, show no difference between Lib-U and Lib-M. The slope of the lines represents the value of ΔΔG associated with methylation at each of the identified CpG positions using the feature-based model; methylation effects related by reverse-complement symmetry, estimated independently, are shown as separate lines. (B) p53 structure (PDB-ID 3Q06) showing the DNA interface of a p53 dimer with the RRRCA|TGYYY core (labeled +/− relative to the motif center). The two arginines (R280) form hydrogen bonds with the respective G+2 bases of each pentamer half sites (2.5 and 3 Å; red) guided by the methyl groups of the pyrimidine carbon 5 of the T+1 base, which stack on top of the polar guanidinium plane (3.9 and 4 Å; blue) thus constraining the possible orientations of the positive charge in favor of forming hydrogen bonds with G+2. Methylation of a T+1→C+1 substitution would therefore result in stabilization due to regaining a methyl group at position +1. (C) Comparison of motif-centric analysis and MACS2 peak calling. Left panel: Distribution of induction levels (defined as the logarithm of the ratio of drug-induced and uninduced IP coverage) for all covered CATG or C1+G2+ sites (μ = mean and σ = standard deviation). Right panel: Fraction of decamer sites overlapping with MACS2 peak regions split by their log-transformed induction. For all three drugs and both the consensus CATG and the C1+G2+ motifs there is a highly significant trend between motif-centric induction levels and MACS2 peak calling (D) Feature model fits of drug-induced (5FU, Nutlin, RITA), in vivo P53 ChIP-seq data for MCF7 using Lib-U relative affinities, average methylation levels and CpG density within a 500 bp region as context-dependent predictors and three position-specific binary methylation indicator features. Datasets were sub-sampled to 50 sites for each possible methylation-motif combination (see Experimental Procedure for details). Upper panel shows the significance of the the methylation features with red signifying positive and blue negative effects on binding. Z-scores for C1+G2+ ranges from 3.0 (5Fu) to 6.3 (Nutlin). Lower panel shows the scores for the context dependent, confounding model predictors (highly significant across all drugs). (E) Methylation coefficients for the C1+G2+ sites were computed on the entire dataset using the feature-based model from (D) with increasing cutoffs on the sum of uninduced and drug-induced p53 IP coverage. Pink area shows the expected difference in binding free energy from EpiSELEX-seq results. (F) Overlap with peaks of histone modifications (<1 kb) for methylated and unmethylated C1+G2+ motifs (>2 sd above mean induction, dark shade). Equally sized, methylation-matched random control sets (light shade) show the expected overlap. Primed-enhancer (H3K4me1) and heterochromatin (H3K9me3) modifications but not marks of active transcription are significantly enriched in methylated C1+G2+ sites whereas unmethylated C1+G2+ sites show patterns of active transcription (H3K4me1; H3K4me3; H3K27ac), perhaps reflecting increased accessibility at active promoters. (G) Potential mechanism how aberrant methylation patterns might contribute to altered p53 binding and thus potentially contribute to changes in chromatin landscape and gene regulation.
Evidence for enhanced p53 binding to methylated sites in vivo
When unmethylated, sequences of type C+1G+2 are bound by p53 at a relative affinity of <10%. However, our analysis shows that binding to these sites is strongly enhanced by cytosine methylation. To test whether this effect on in vitro binding is also observable in vivo we jointly analyzed whole-genome bisulfite sequencing (Consortium) and p53 genomic occupancy data – generated by ChIP-seq both before and after induction of p53 – for the cell line MCF7 (Nikulenkov et al., 2012). Using standard peak calling (Zhang et al.) at a false discovery rate of 5%, we detected 40 sites that were both occupied by p53 and had an underlying DNA sequence containing a match to RRRCACGYYY, a sample too small to allow for statistical analysis of the effect of methylation status (Figure S6B). Moreover, the negative effect of methylation on chromatin accessibility in vivo may obscure the positive effect on binding suggested by our SELEX analysis. To address this issue, we developed a motif-centric analysis strategy that avoids peak calling. We started by identifying all individual matches to the most strongly bound RRRCATGYYY sites in the genome, and classifying each of these p53 half-sites in terms of the change in the number of ChIPed DNA fragments covering it before and after p53 induction. We observed a strong and statistically significant trend between motif-centric fold-induction and the probability of falling within a peak region based on MACS2 (Figure 6C), indicating this approach captures the underlying p53 binding signature. In addition, this trend was robust for three different inducers of p53 activity, and was also observed for the CpG containing C+1G+2 motif (Figure 6C).
Encouraged by this observation, we used a generalized linear model that explains how the number of sequenced IP fragments covering an individual genomic match to any of the four decamer half-site motif classes (CATG, C+1G+2, C+3G+4, C+4G+5) is distributed between the uninduced and induced conditions. The CATG motif, which does not match any CpG-containing decamers, serves to estimate the effect of local chromatin context, which is represented by the average methylation level and CpG content of the flanking regions as predictors in the model. To account for variation in binding affinity unrelated to methylation, we also included as a covariate the relative affinity of the 10bp half-site as derived from the DNA sequence using a scoring matrix derived from our Lib-U data (see Experimental Procedures and Supplemental Experimental Procedures for details). Finally, and most importantly, the coefficients associated with three binary indicators for the presence of a methylated CpG dinucleotide at each offset quantify the effect of cytosine methylation on the responsiveness of in vivo p53 binding.
When the model is fit to ChIP-seq data, the position-dependent effects of cytosine methylation within the binding site identified by our EpiSELEX-seq assay are recapitulated in MCF7 cells, with methylation of C+1G+2 having a significant stabilizing effect (Figure 6D). The coefficients for the confounding contributions in the model also behave as expected, with positive effects for CpG density and sequence-derived p53 affinity, and a negative effect for regional methylation (Figure 6D). Considering that the in vivo methylation effects should more closely reflect the in vitro effect at higher levels of ChIP enrichment, where the local chromatin context presumably is more permissive, we repeated our model fit using increasing cutoffs on the sum of induced and uninduced read counts for all consensus matches in the genome (Figure 6E). The coefficient for C+1G+2 behaves as expected, and saturates at ΔΔG/RT = +1.5, corresponding to a ~4.5-fold increase in binding affinity upon full methylation of the CpG dinucleotide (Figure 6E). Thus, the in vivo methylation effect appears to be even higher than in vitro, which could reflect contributions from additional methylated CpG dinucleotides within the full p53 tetramer binding site or cooperativity with other factors. For the other two motif classes (C+3G+4 and C+4G+5), the coverage by IP fragments is too sparse to allow quantification, consistent with the weaker in vitro methylation sensitivity observed for these CpG offsets with our EpiSELEX-seq assay.
It has been suggested that p53 can bind to high-nucleosome-occupancy regions and act as a pioneer factor to alter chromatin accessibility (Laptenko et al., 2011; Sammons et al., 2015). We therefore analyzed five histone modifications that in combination can be used to classify enhancers or promoters as active, closed, or primed (Calo and Wysocka, 2013). Methylated C+1G+2 sites are significantly enriched for H3K9me3 and H3K4me1 but not H3K27ac (associated with active enhancers) or H3K4me3 (associated with active transcription), when compared to a matched control set (see Experimental Procedures for details) (Figure 6F). These histone modifications have been suggested to mark either heterochromatin (H3K9me3) (Grewal and Jia, 2007) or enhancers that are primed to become active (H3K4me1) (Calo and Wysocka, 2013). We observed the same pattern for CATG sites within methylated regions (Figure S6C). By contrast, unmethylated C+1G+2 sites tend to have a strong signature of H3K4me1 and H3K4me3 or H3K27ac (Figure 6F), arguing that ChIP enrichment at those loci may be due to transcriptional activity rather than specific p53 targeting. This again underscores the need to account for confounding effects when analyzing in vivo binding data.
Interestingly, 67 out of 90 (74%, with 44% expected, p-value=3*10−10) of the methylated C+1G+2 sites occur within 3 kb of a protein-coding gene (60 genes total) or a lincRNAs (20 total) (Figure S6D) annotated in GENCODE (Derrien et al., 2012). The enrichment for sites occurring near lincRNAs (21/90 sites, or 23%, with 8% expected, p-value=5*10−7) (Figure S6D) is consistent with previous findings about p53 regulation of lincRNA expression (Leveille et al., 2015).
DISCUSSION
With EpiSELEX-seq we have developed a method that can accurately quantify the change in binding free energy associated with the presence of a methylated cytosine at any position within the protein DNA interface. A key aspect of our approach, which allows us to robustly identify methylation sensitivity, is that modified and unmodified DNA ligands are probed simultaneously in a single reaction, ensuring a direct comparison of TF occupancy. One round of selection is sufficient to accurately capture methylation effects, even for lower-affinity sites that deviate from the consensus and thus readily escape detection when binding to methylated and unmethylated ligands is assayed separately or over multiple rounds of selection. The context-sensitive nature of our analysis is essential, because opposing methylation effects can occur within a single binding site, making it difficult or impossible to detect the impact of methylation using less precise approaches such as oligomer enrichment only. This point is illustrated by our analysis of human Pbx-Hox heterodimers, whose DNA binding specificity we studied here for the first time at high resolution. The net effect of methylation on binding is close to neutral, but methylation of different CpGs in the binding site can modulate the binding affinity by up to 7-fold in either direction. This also illustrates why it may be difficult to detect methylation sensitivity by looking at motif enrichment in differentially methylated regions (DMR). Pbx-Hox sequence logos constructed separately for the unmethylated and methylated libraries were nearly indistinguishable and did not reveal significant methylation sensitivity of Pbx-Hox complexes (Figure 4D). Only when we examined the consequences of methylation at specific positions were we able to identify clear effects.
Despite an ongoing debate to what extent CpG methylation is a driver of gene silencing or the consequence thereof (Ambrosi et al., 2017), the general view is that methylation has a repressive effect on TF binding. For example, in a study that compared binding of TFs between wild-type and Dnmt1-knockout ESC cells (Domcke et al., 2015), the authors showed that removal of methylation marks at specific nuclear respiratory factor 1 (NRF1) binding sites led to increased binding and expression of nearby genes. In addition, experimentally induced methylation reduced NRF1 binding to those sites. Here, and in agreement with recently published data (Yin et al., 2017), we demonstrate that endogenous methylated motifs containing a CpG at specific sites within the protein-DNA interface can also increase binding and that the mechanisms underlying the epigenetic control of TF binding and thus gene expression are more nuanced then previously thought.
For p53, despite a general negative effect of regional methylation on genomic occupancy, the increased binding to methylated RRRCACGYYY sites that our analysis revealed implies that methylated binding sites are functional and might direct p53 to alter previously inaccessible loci in the genome. This conclusion is supported by our finding that these occupied and methylated binding sites are associated with a histone modification pattern that indicates either compacted chromatin (Grewal and Jia, 2007) or transcriptionally poised enhancers. Additional evidence that p53 can access nucleosomal DNA in vitro and in vivo, and thus might be a pioneer factor, also supports this notion (Laptenko et al., 2011; Sammons et al., 2015). Many diseases, in particular many forms of cancer, are accompanied by aberrant methylation patterns (Kulis and Esteller, 2010) raising the question whether even subtle changes in the methylome could trigger differential TF binding and thus contribute to the onset of disease. Interestingly, H3K4me1 has also been shown to be significantly associated with loss of methylation during aging in multiple human cell types (Fernandez et al., 2015), providing yet additional support for the functionality and importance of such sites.
EXPERIMENTAL PROCEDURES
Protein expression and purification
Human proteins, either full-length or nearly full length, were affinity purified using a HIS-tag. p53 protein was purified as described in (Laptenko et al., 2015) containing a deletion in the C-terminal basic region to prevent non-specific DNA binding contributions outside the core DNA-binding domain.
Library design and methylation
Full library sequences were as follows: 5′-GGTAGTGGAGG-TGGG-CCTGG-16(26)xN-CCAGG-GAGGTGGAGTAGG-3′ for Lib-U and 5′-GGTAGTGGAGG-GCAC-CCTGG-16(26)xN-CCAGG-GAGGTGGAGTAGG-3′ for Lib-M. Libraries were double-stranded by annealing und extension using Klenow polymerase (NEB). Lib-M was methylated with M.SssI (NEB) using only ~250 ng per 1x reaction and two subsequent incubation cycles of 2h at 37°C each. Up to 400ng of 1x methylated DNA can be combined in the second step using Oligo-Clean-up columns (Zymo) for purification.
EpiSELEX-seq protocol
ElectroMobility Shift Assays (EMSAs) and extraction of bound DNA were performed as described previously (Slattery et al., 2011) with an equal mix of Lib-U and Lib-M. Purified, bound DNA was split in two and amplified using a 15 cycle PCR protocol with high-fidelity enzymes (Phusion or Q5; NEB) with overhang primers adding TruSeq Illumina adapter sites in two orientations respectively to allow sequencing from both 5′- and 3′-ends. Efficient splitting was analyzed by comparing the number of reads resulting from each set of primers (Figure S1). Specific Illumina barcodes were added by a five cycle PCR using NEBNext Multiplex Oligos for Illumina sequencing and Phusion/Q5 polymerase. Indexed libraries were gel-purified as described previously (Slattery et al., 2011), pooled, and sequenced using a v2 75 cycle high-output kit on an Illumina NEXTSeq Series desktop sequencer. For initial (R0) and enriched (R1) libraries, 5–35 million single-end reads were obtained.
Testing for methylation efficiency
A DNA probe containing 4 CpG sites was methylated, bisulfite treated, and cloned into pBlueScript vector; 4–8 colonies were picked for sequencing. We assessed methylation efficiency by counting retained CpG dinucleotides. Alternatively, Lib- M was split in three parts (methylated and not treated; methylated and treated; unmethylated and treated with bisulfite) and prepared for sequencing as described above. Dinucleotide frequencies were computed and CpN ratios were compared across all treatments to assess methylation efficiency, using “TpG” as a reference.
EpiSELEX-seq data processing
FASTQ files were pre-processed using the FASTX toolkit (Hannon lab). Files were reverse-complemented, merged, and trimmed to have unidirectional reads starting from the 4 bp barcode site. Each data set was assigned to either Lib-U or Lib-M. A 5th-order Markov Model was generated from the R0 libraries for Lib-U and Lib-M, respectively, using the R package bioconductor.org/packages/SELEX (Riley et al., 2014).
Oligomer enrichment analysis
Relative affinities for oligomers of length k were estimated as described previously (Slattery et al., 2011). Fold-enrichments were normalized based on the most enriched oligomer for each library. Position-specific affinity matrices (PSAMs) (Foat et al., 2006) were generated by considering all 3k point mutations away from the most enriched oligomer. Binding free energy differences ΔΔG/RT between the mutated and optimal sequence were computed as the negative logarithm of the relative fold-enrichment. The binding free energy change ΔΔG/RT for a C→T transition was calculated separately for Lib-U and Lib-M, and used to estimate the effect of methylation on binding as follows:
Feature-based modeling
A feature-based Poisson regression model was fit to the R1 read counts. First, PSAMs were constructed from oligomer enrichment tables for each sample and used to estimate relative binding affinity, in either orientation, and at each offset relative to the random region, allowing for up to 2 bp overlap with the constant flanks. Only probes for which a single offset/orientation contributing at least 95% of the total were kept. To avoid bias, R1 reads were randomly split; one half was used to define the set of oligomers that correspond to the rows in the design matrix, and the other to obtain R1 counts. Regression models were fit in two ways: (i) using the Lib-U R1 count for a particular motif of length k, the markov model prediction from the corresponding R0 as an offset, and 4k base indicator features at each position in the motif as independent variables; (ii) same as before, but including Lib-M and using both base and 5mCpG features. As expected, adding 5mCpG features and jointly fitting to Lib-U and Lib-M did not affect the base feature estimates (Figure S1C). For p53, a separate intercept was fit for each library.
Competition assay for Pbx-HoxA1
Two 12-mer competitor probes with identical sequence – ATGATTGACGAC – but different methylation status at position 9 were tested for their capacity to compete with a labeled probe for Pbx-HoxA1 binding in an EMSA. Pbx-HoxA1 and labeled-probe concentrations were held constant while increasing the concentrations of the unlabeled competitor DNA over a 1000-fold range. Experiments were performed in duplicate. IC50 values were calculated using ImageJ for quantification and R-package drc to fit a dose-response curve.
Data processing for in vivo p53 binding
We downloaded ChIP-seq data (FASTQ files; Sequence Read Archive Accession number: SRP007261) for p53 in MCF7 cells (no-drug control, and drugs Nutlin, RITA and 5FU), and MCF7 whole-genome bisulfite sequencing data (BED file; GSM1328112). FASTQ files were aligned to hg19 (Bowtie2) and converted to coverage tracks (deeptools/bamCoverage) after extending reads by 200 nt. MACS2 was run with options –g hs and –q 0.05 using the uninduced p53 IP as control. BED peak files (hg19) for five histone modifications in MCF7 were downloaded from ENCODE (ENCSR000EWP, ENCSR000EWQ, ENCSR000EWR, ENCSR493NBY, ENCSR985MIB). GTF files for the current releases (v25) of human whole genome annotation and lincRNA specific annotation data were downloaded from GENCODE (mapped to GRCh37/hg19).
In vivo, motif-centric p53 binding analysis
Analysis was done in R. The hg19 genome was scanned for sites mapping to the consensus RRRCATGYYY or the three CpG-containing motif classes (RRRCATGYCG, RRRCATGCGY, RRRCACGYYY). CpG sites were intersected with WGBS BED files and methylation status was assigned based on the percentage methylated (“1” for >80% and “0” for < 10% methylated), keeping only sites with ≥ 10x coverage. Average methylation levels and CpG density of the 500 bp centered around the motif were computed and the per-motif p53 coverage for uninduced (control) and drug-induced (IP) were obtained, keeping only motifs with ≥ 1x coverage in both control and IP. Individual genomic motif occurrences correspond to the rows of the design matrix (X); columns are the in vitro 10-mer affinities (unmethylated EpiSELEX-seq), the three position-specific binary 5mCpG indicators, average methylation level, and CpG density. The glm function with family=”binomial” was used in R to fit the following model of the probability of a specific motif being bound:
Where
Regression coefficients and Z-scores quantifying their statistical significance were obtained for each model fit. To avoid bias in the size of motif classes in the training data, sub-sampling (~200 times) was applied in a way that guarantees an equal number of occurrences of methylation status and motif class (for CATG motif, a 50% regional methylation level was used as a threshold). Models for enriched p53 occupancy were fit by sequentially removing rows for which the sum of drug-induced and uninduced IP fragment counts fell below a certain threshold. Fishers’s exact test was used to compute statistical associations between MACS2 peaks (1 kb around peak summit) and grouped motif-centered log-transformed enrichment values.
Overlap with GENCODE annotation and histone marks
Enrichment for either GENCODE annotations or histone marks was scored by computing the overlap between the motif sets (log-induction > 2 SD above the mean) and either the gene annotations (within 3kb) or the histone peaks (within 1kb). P-values were computed by generating >100 random, methylation-matched sets from the WGBS data and calculating the probability of observing the actual overlap based on the sampling of random overlaps.
Supplementary Material
Acknowledgments
We thank Remo Rohs, Timothy Bestor, Barry Honig, as well as the members of the Bussemaker, Mann, and Rohs labs, for valuable discussions. This work was supported by an HHMI International Student Research Fellowship (J.F.K.) and NIH grants R01HG003008 to H.J.B. and R35GM118336 to R.S.M and grants P01CA087497 and R01CA196234 to C.P. Columbia University’s Shared Research Computing Facility is supported by NIH grant G20RR030893 and NYSTAR contract C090171.
Footnotes
AUTHOR CONTRIBUTIONS
J.F.K., R.S.M. and H.J.B. conceived of the experiments; J.F.K. carried out the experiments with help from O.L. and S.C.; J.F.K., G.D.M., and H.J.B. constructed the feature-based model; J.F.K. analyzed the ChIP-seq and WGBS data; O.L., W.A.F.-P. and C.P. helped interpret the p53 data; J.F.K., R.S.M., and H.J.B. interpreted the data and wrote the paper. All authors discussed the findings and contributed to the manuscript.
ACCESSION NUMBERS
The raw sequencing data and tables including the PSAM-filtered Lib-M and Lib-U relative affinities and respective oligomer sequences for all protein complexes have been deposited with the NCBI Gene Expression Omnibus under accession number GSE98652.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambrosi C, Manzo M, Baubec T. Dynamics and Context-Dependent Roles of DNA Methylation. J Mol Biol. 2017 doi: 10.1016/j.jmb.2017.02.008. [DOI] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas Machado AC, Zhou T, Rao S, Goel P, Rastogi C, Lazarovici A, Bussemaker HJ, Rohs R. Evolving insights on how cytosine methylation affects protein-DNA binding. Brief Funct Genomics. 2015;14:61–73. doi: 10.1093/bfgp/elu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–579. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Fernandez AF, Bayon GF, Urdinguio RG, Torano EG, Garcia MG, Carella A, Petrus-Reurer S, Ferrero C, Martinez-Camblor P, Cubillo I, et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res. 2015;25:27–40. doi: 10.1101/gr.169011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foat BC, Morozov AV, Bussemaker HJ. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22:e141–149. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Dore LC, et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161:879–892. doi: 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y. DNA Methylation on N6-Adenine in C. elegans. Cell. 2015;161:868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T, Romano L, Planchon A, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen HN, Shin J, Cox E, Rho HS, Woodard C, et al. DNA methylation presents distinct binding sites for human transcription factors. Elife. 2013;2:e00726. doi: 10.7554/eLife.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015;527:384–388. doi: 10.1038/nature15518. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- Laptenko O, Beckerman R, Freulich E, Prives C. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc Natl Acad Sci U S A. 2011;108:10385–10390. doi: 10.1073/pnas.1105680108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Shiff I, Freed-Pastor W, Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T, Manfredi J, et al. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell. 2015;57:1034–1046. doi: 10.1016/j.molcel.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, Sabo PJ, Lu Y, Rohs R, Stamatoyannopoulos JA, et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci U S A. 2013;110:6376–6381. doi: 10.1073/pnas.1216822110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveille N, Melo CA, Rooijers K, Diaz-Lagares A, Melo SA, Korkmaz G, Lopes R, Akbari Moqadam F, Maia AR, Wijchers PJ, et al. Genome-wide profiling of p53- regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun. 2015;6:6520. doi: 10.1038/ncomms7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Blumenthal RM, Cheng X. A common mode of recognition for methylated CpG. Trends Biochem Sci. 2013;38:177–183. doi: 10.1016/j.tibs.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, Vinson C. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res. 2013;23:988–997. doi: 10.1101/gr.146654.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet S, Mann RS. To Be Specific or Not: The Critical Relationship Between Hox And TALE Proteins. Trends Genet. 2016;32:334–347. doi: 10.1016/j.tig.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. The importance of being flexible: the case of basic region leucine zipper transcriptional regulators. Curr Protein Pept Sci. 2009;10:244–269. doi: 10.2174/138920309788452164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kivioja T, Ignatiev I, Kel A, Taipale J, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, Esteller M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- Petty TJ, Emamzadah S, Costantino L, Petkova I, Stavridi ES, Saven JG, Vauthey E, Halazonetis TD. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011;30:2167–2176. doi: 10.1038/emboj.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A, Cedar H. DNA methylation and genomic imprinting. Cell. 1994;77:473–476. doi: 10.1016/0092-8674(94)90208-9. [DOI] [PubMed] [Google Scholar]
- Riley TR, Slattery M, Abe N, Rastogi C, Liu D, Mann RS, Bussemaker HJ. SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods Mol Biol. 2014;1196:255–278. doi: 10.1007/978-1-4939-1242-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammons MA, Zhu J, Drake AM, Berger SL. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015;25:179–188. doi: 10.1101/gr.181883.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein R, Razin A, Cedar H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci U S A. 1982;79:3418–3422. doi: 10.1073/pnas.79.11.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribioli C, Tamanini F, Patrosso C, Milanesi L, Villa A, Pergolizzi R, Maestrini E, Rivella S, Bione S, Mancini M, et al. Methylation and sequence analysis around EagI sites: identification of 28 new CpG islands in XQ24-XQ28. Nucleic Acids Res. 1992;20:727–733. doi: 10.1093/nar/20.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017:356. doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565. doi: 10.1038/nrg.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.