

Summary
CD4 T cells are critical for protective immunity against Mycobacterium tuberculosis (Mtb), the cause of tuberculosis (TB). Yet, to-date, TB vaccine candidates that boost antigen-specific CD4 T cells have conferred little or no protection. Here we examined CD4 T cell responses to two leading TB vaccine antigens, ESAT-6 and Ag85B, in Mtb infected-mice and in vaccinated humans with and without underlying Mtb infection. In both species, Mtb infection drove ESAT-6-specific T cells to be more differentiated than Ag85B-specific T cells. The ability of each T cell population to control Mtb in the lungs of mice was restricted for opposite reasons; Ag85B-specific T cells were limited by reduced antigen expression during persistent infection, whereas ESAT-6-specific T cells became functionally exhausted due to chronic antigenic stimulation. Our findings suggest that different vaccination strategies will be required to optimize protection mediated by T cells recognizing antigens expressed at distinct stages of Mtb infection.
In Brief
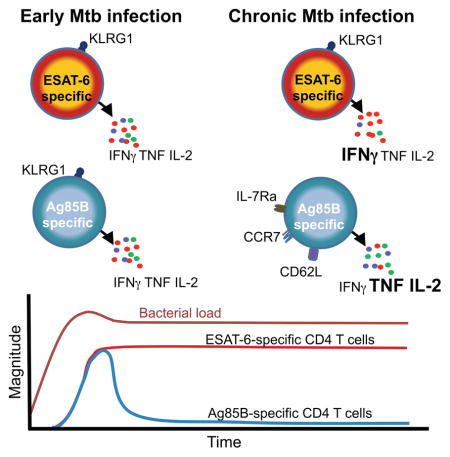
Moguche and Musvosvi et al. show that two leading Mycobacterium tuberculosis vaccine antigens Ag85B and ESAT-6 are differentially expressed during infection. As a result, CD4 T cells recognizing these antigens exhibit distinct patterns of differentiation and their capacities to mediate protective immunity are restricted in different ways.
The central role for CD4 T cells in protective immunity against TB is evidenced by the extreme susceptibility of animals and humans lacking CD4 T cells (Caruso et al., 1999; Lawn et al., 2009; Mogues et al., 2001). Cognate interactions between antigen-specific CD4 T cells and Ag/MHC complexes expressed on Mtb-infected macrophages are required to optimally kill Mtb or restrict its replication (Srivastava and Ernst, 2013). Although the complete arsenal employed by CD4 T cells to mediate this protection is unclear (Gallegos et al., 2011; Sakai et al., 2016; Walzl et al., 2011), production of IFN-γ is critical (Green et al., 2013). Nevertheless, simply boosting IFN-γ-producing CD4 T (Th1) cells is not sufficient to improve immunity against Mtb (Jasenosky et al., 2015; Urdahl, 2014). This was highlighted by the recent failure of a TB vaccine candidate, MVA85A, to enhance protection against TB beyond that conferred by BCG immunization of infants despite significantly boosting circulating Th1 cells specific for the Mtb antigen Ag85A (Tameris et al., 2013).
One factor likely to govern the ability of CD4 T cells to mediate protection is the nature of the Mtb antigen recognized. Numerous Mtb antigens are recognized by CD4 T cells, each with unique expression patterns at different stages of infection. Prior murine studies have suggested that immunodominant Mtb antigens contained in numerous TB vaccine candidates, including Ag85A, Ag85B, and ESAT-6, exhibit distinct expression patterns. Ag85A and Ag85B, mycolyl transferases involved in mycobacterial cell wall synthesis (Kremer et al., 2002), exhibit reduced mRNA expression three weeks post-infection (Rogerson et al., 2006; Shi et al., 2004), which correlates with the onset of adaptive immunity and decreased bacterial replication (Gill et al., 2009). Two reports that examined CD4 T cell responses to Ag85B concluded that Mtb evades immunity by reducing antigenic expression during chronic infection (Bold et al., 2011; Egen et al., 2011). However, reduced Mtb antigen expression during chronic infection is not universal; for example, ESAT-6, a secreted virulence factor (Pagan and Ramakrishnan, 2015), is produced and recognized by T cells throughout Mtb infection (Moguche et al., 2015). How Mtb evades immunity conferred by T cells recognizing persistently expressed antigens is unclear. Regardless, the varied dynamics of Mtb antigen expression is an important consideration for TB vaccine design. Reliably preventing establishment of Mtb infection in all individuals may not be achievable because Mtb has developed numerous strategies to avoid alerting host immunity upon infection (Behar et al., 2014; Cambier et al., 2014; Urdahl, 2014). Thus, in those with established infection, containing bacterial replication and curbing progression to symptomatic and transmissible disease may be more realistic. For such a vaccine to be effective, T cells that recognize Mtb antigens expressed throughout the course of infection would likely need to be primed. The first novel TB vaccine candidate to reach human efficacy trials in many years contained a single Mtb antigen (Ag85A) that was found to be poorly expressed during chronic Mtb infection in mice (Rogerson et al., 2006; Shi et al., 2004). TB stage-specific differences in antigen availability have not been fully considered in the design and prioritization of TB vaccines.
In this study we utilized the highly tractable mouse TB model to determine how differences in TB stage-specific antigen expression impact the differentiation, maintenance, function, and protective capacity of CD4 T cells. Predictions from the mouse were then validated in vaccinated humans with or without Mtb infection, providing evidence that antigen availability shapes T cell differentiation similarly in both species. Our studies focused on Ag85B-specific T cells that recognized antigen during early, but not chronic, infection and ESAT-6-specific T cells that recognized antigen throughout infection. We showed that CD4 T cells recognizing both types of Mtb antigens are restricted in their capacity to control Mtb infection in the mouse lung, but for opposite reasons. Ag85B-specific T cells initially expanded in numbers within the infected lung but then contracted at four weeks post-infection. They developed characteristics of memory T cells and produced multiple cytokines, but their protective capacity was limited by low antigen expression by the pathogen. In contrast, ESAT-6-specific CD4 T cells were maintained in high numbers within the lung parenchyma by continued antigenic stimulation. They were driven towards terminal differentiation, and their protective capacity was limited by functional exhaustion. These findings have important implications for the selection of antigens and delivery systems for vaccines that seek to promote stage-specific protection against TB.
Results
Chronic antigenic stimulation promotes maintenance of CD4 T cells during Mtb infection
Prior murine studies have shown that mRNA expression of Ag85B, but not ESAT-6, is reduced during chronic Mtb infection (Rogerson et al., 2006; Shi et al., 2004). To directly compare antigen availability for T cell recognition, we adoptively transferred carboxyfluorescein succinimidyl ester (CFSE)-labeled transgenic CD4 T cells specific for Ag85B (Tamura et al., 2004) or ESAT-6 (Gallegos et al., 2008) into mice at various stages after aerosolized Mtb infection. Both specificities of T cells proliferated robustly when transferred prior to two weeks post-infection (Figure 1A). However, only ESAT-6-specific cells exhibited substantial proliferation when transferred at 4 weeks or later. Using MHCII tetramers, we compared the kinetics of endogenous Ag85B and ESAT-6-specific CD4 T cell responses in the lungs. Both populations exhibited similar expansion and peaked four weeks post-infection (Figure 1B). Thereafter, however, ESAT-6-specific CD4 T cells were maintained at high numbers, while Ag85B-specific CD4 T cells contracted ~10-fold. Similar patterns of proliferation and cellular kinetics were observed in mice infected with a highly virulent clinical Mtb isolate (W-Beijing strain, SA161) (Figure S1A and B) (Ordway et al., 2011). To assess whether these differences were due to antigen availability or distinct intrinsic properties of Ag85B and ESAT-6-specific cells, we generated a recombinant Mtb strain that constitutively expresses Ag85B. In mice infected with this strain, adoptively transferred transgenic Ag85B-specific CD4 T cells, like ESAT-6 T cells, proliferated throughout chronic infection, verifying the availability of Ag85B for T cell recognition even at late stages of infection (Figure S1C). Importantly, in these mice, Ag85B-specific CD4 T cells were maintained chronically, analogous to ESAT-6-specific cells (Figure S1D). These results indicate that high numbers of ESAT-6-specific CD4 T cells are maintained during chronic infection with WT Mtb due to ongoing antigenic stimulation, whereas Ag85B-specific cells undergo contraction due to diminished antigenic exposure.
Figure 1. Antigen availability promotes maintenance of CD4 T cells during Mtb infection.

(A) CFSE dilution in adoptively transferred transgenic T cells isolated from the lung 5 days after transfer.
(B) Enumeration of Ag85B and ESAT-6-specific tetramer+ CD4 T cells in the lung. Data are representative of 2 independent experiments with 3–5 mice per group and per time point.
(C) Enumeration of IFN-γ-producing Ag85B or ESAT-6-specific T cells in latently infected adolescents. The p-value was calculated using the Wilcoxon signed-rank test. See also Figure S1.
Next, we assessed frequencies of Ag85B and ESAT-6-specific T cells in human Mtb infection. Diagnosis of human Mtb infection relies upon identification of circulating Mtb-specific T cells, which cannot be detected until several weeks after Mtb exposure (Jasenosky et al., 2015). As a consequence, we could not study Ag85B and ESAT-6-specific T cells during early infection. Overlapping peptides spanning ESAT-6 or Ag85B were used to stimulate peripheral blood mononuclear cells (PBMC) of adolescents with latent Mtb infection (QuantiFERON Gold In-Tube positive, QFT+), and frequencies of T cells producing IFN-γ to each antigen were determined by ELISpot. Although Ag85B is much larger than ESAT-6 (325 amino acids vs. 95aa), significantly higher frequencies of T cells responded to ESAT-6 than to Ag85B (Figure 1C). Thus, like mice, humans exhibited higher frequencies of T cells recognizing ESAT-6 than Ag85B during chronic Mtb infection, consistent with another recent report (Lindestam Arlehamn et al., 2016).
Ag85A and Ag85B are regulated similarly and share common epitopes
Murine studies indicate that expression of Ag85A, the sole Mtb antigen contained in the MVA85A vaccine, is reduced during chronic infection similarly to Ag85B (Rogerson et al., 2006; Shi et al., 2004). To assess the global regulation of Ag85A and Ag85B, we examined expression of Ag85A, Ag85B, and ESAT-6 in 2323 publicly available Mtb expression profiles (Peterson et al., 2014) (www.tbdb.org). Collectively, these data assessed Mtb gene expression over a wide variety of environmental conditions (e.g. stresses, drugs) and genetic perturbations. Overall the correlation coefficient between the expression of Ag85A and Ag85B transcripts was approximately 0.4 (Spearman’s rho) (Figure S2A), which is higher than 95% of randomly selected Mtb gene pairs, indicating statistically significant co-expression between Ag85A and Ag85B. In contrast, expression of ESAT-6 transcript was not correlated with either Ag85A or Ag85B.
Because Ag85A and Ag85B are homologous proteins with 79% sequence identity, many Ag85B peptides used in Figure 1C are shared with Ag85A. To investigate commonality between the specific Ag85A and Ag85B epitopes, we compared T cell recognition of overlapping 15mer peptides spanning Ag85A and Ag85B in 24 healthy QFT+ adolescents. We identified three immunodominant peptides within the two proteins that mapped to virtually identical amino acid sequences (Figure S2B and C). Thus, when re-assessed, the data depicted in Figure 1C suggest that T cells recognizing ESAT-6 are represented at higher frequencies during latent Mtb infection than those recognizing Ag85A or Ag85B.
Chronic antigenic stimulation drives CD4 T cell exhaustion during murine Mtb infection
Given the contraction of Ag85B-specific CD4 T cells due to diminished antigen exposure during chronic infection, we predicted that Ag85B-specific cells would assume characteristics of memory T cells. At the peak of Ag85B-driven T cell expansion (d28 post-infection), ~one-third of Ag85B-specific T cells expressed KLRG1 (Figure 2A), a marker of Th1 terminal differentiation in murine TB (Moguche et al., 2015; Reiley et al., 2010; Torrado et al., 2015). Later during infection, however, few Ag85B-specfic CD4 T cells expressed KLRG1, but many expressed CD62L, CCR7, and IL-7Rα (Figure 2B), surface markers associated with less differentiated T cells, such as central memory cells (Pepper and Jenkins, 2011). In contrast, ESAT-6-specific T cells maintained a large population of KLRG1+ cells and did not express these memory markers (Figures 2A and B). Labeling cells in the vasculature by intravenous antibody administration revealed that KLRG1− cells in both populations were almost exclusively located in the lung parenchyma, whereas most KLRG1+ cells resided in the lung vasculature consistent with recent reports (Moguche et al., 2015; Sakai et al., 2014a). Thus, a higher proportion of ESAT-6-specific T cells were located in the lung vasculature during chronic infection (Figure S3A). In mice infected with recombinant Mtb constitutively expressing Ag85B, Ag85B-specific T cells resembled ESAT-6-specific cells throughout infection, showing a substantial proportion of KLRG1+ cells and low expression of CD62L, CCR7, and IL-7Rα (Figure S3B and S3C). These findings validated our hypothesis that diminished Ag85B expression during chronic infection causes Ag85B-specific CD4 T cells to be less terminally differentiated than their ESAT-6-specific counterparts and to resemble central memory T cells.
Figure 2. Chronic antigenic stimulation drives murine Mtb-specific CD4 T cells towards terminal differentiation.

(A) PD1 and KLRG1 expression on Ag85B and ESAT-6-specific CD4 T cells in the lung. The percentage of KLRG1+ cells is shown.
(B) CD62L, CCR7 and IL-7Rα expression on lung naïve, Ag85B-specific, or ESAT-6-specific CD4 T cells at 103 days post-infection. Graphs depict the mean fluorescence intensity (MFI) for individual mice. See also Figure S3.
We predicted that chronic antigenic stimulation may compromise the ability of ESAT-6-specific, but not Ag85B-specific, CD4 T cells to produce cytokines. At the initiation of the Ag85B and ESAT-6 T cell response (d16 post-infection), we identified similar frequencies of antigen-specific CD4 T cells using tetramers as we did by intracellular cytokine staining (ICS) after in vitro peptide stimulation (Figure 3A and B). This ~1:1 ratio of tetramer:cytokine-producing cells was sustained for Ag85B-specific T cells throughout Mtb infection until at least d150 post-infection. In contrast, this ratio steadily increased for ESAT-6-specific T cells and by d150 we detected ~4x more cells using tetramers than ICS (Figure 3A and B).
Figure 3. Murine Ag85B-specific CD4 T cells have a higher cytokine producing capacity than ESAT-6-specific cells.

(A) Enumeration of Ag85B and ESAT-6 T cells in the lung using tetramers (top) or cytokine production following in vitro peptide stimulation (bottom).
(B) Ratio of tetramer binding cells to TNF and IFN-γ double cytokine producing cells as identified in (A).
(C) IFN-γ expression by lung naïve CD44low CD4 T cells, Ag85B-specific, and ESAT-6-specifc CD4 T cells from mice injected with OVA (left), Ag85B, or ESAT-6 peptides (right). The IFN-γ MFI for Ag85B and ESAT-6-specific CD4 T cells in each group are depicted below.
(D) Combinatorial analysis of IFN-γ, TNF and IL-2 production by tetramer+ Ag85B and ESAT-6-specific CD4 T cells isolated from lungs of mice infected with Mtb for 200 days. Data are depicted as mean ± SD, and statistical significance was determined by t-test. All detectable functional subsets are depicted in the figure; no TNF or IL-2 single producers or TNF/IL-2 double-producers were detected. Data are representative of 2–3 independent experiments with 4–5 mice per experiment. See also Figure S4.
To examine functional capacities of Mtb-specific T cells in vivo, antigenic peptides from Ag85B, ESAT-6, or chicken ovalbumin (OVA, as a control) were intravenously infused into mice infected with Mtb 28 days prior. Two hours later, intracellular IFN-γ was assessed directly ex vivo in Ag85B and ESAT-6 tetramer+ CD4 T cells recovered from the lung. In OVA-infused controls, a small percentage of both Ag85B and ESAT-6-specific CD4 T cells expressed low levels of IFN-γ (Figure 3C). Upon in vivo stimulation with cognate peptide, however, the vast majority of Ag85B-specific cells showed high expression of IFN-γ. In contrast, most ESAT-6-specific cells expressed little or no IFN-γ, and only a small percentage expressed high levels. Similar findings were observed with the highly virulent clinical Mtb isolate (Figure S4A). We considered the possibility that the large population of non-responding ESAT-6 tetramer+ cells was simply not stimulated by the infused peptide. However, in vitro peptide restimulation of lung cells isolated from ESAT-6 peptide-infused mice did not reveal an increased number of IFN-γ producing cells, suggesting that the infused peptide reached most ESAT-6 T cells in vivo (Figure S4B). In mice infected with recombinant Mtb constitutively expressing Ag85B, IFN-γ production by Ag85B and ESAT-6-specific T cells was similar (Figure S4C), indicating that differences in IFN-γ production between Ag85B and ESAT-6-specific T cells were not intrinsic to their specificities but reflected differences in antigenic exposure. At 200 days post-infection, ~25% of Ag85B-specific CD4 T cells were polyfunctional and co-produced IFN-γ, TNF, and IL-2 after in vivo peptide infusion, whereas only ~20% failed to produce these cytokines (Figure 3D). In contrast, only ~5% of ESAT-6-specific cells produced all three cytokines, and ~40% produced none. Thus, chronic antigenic stimulation of ESAT-6-specific, but not Ag85B-specific, CD4 T cells results in functional exhaustion.
Human Mtb infection drives ESAT-6-specific T cells towards terminal differentiation
Does human Mtb infection, like murine Mtb infection, drive CD4 T cells recognizing ESAT-6 to be more differentiated than those recognizing Ag85B? Our ability to address this in our study population of adolescents with latent Mtb infection was limited by the very low frequencies of antigen-specific CD4 T cells in peripheral blood (undetectable in most individuals using MHCII tetramers (Figure 4A)). To overcome this problem, we studied individuals enrolled in the THYB-04 phase I clinical trial of H1:IC31, a vaccine comprised of an Ag85B:ESAT-6 fusion protein in a cationic particle based adjuvant (Mearns et al., 2016). We reasoned that vaccination would boost Ag85B and ESAT-6 specific T cells, allowing us to readily assess their phenotypes. If Mtb-infection drove differentiation of ESAT-6-specific T cells to a greater degree we hypothesized we would observe differences between vaccine-expanded Ag85B and ESAT-6 specific T cells in Mtb-infected (QFT+) adolescents, but not in uninfected (QFT−) controls.
Figure 4. Transcriptomic profiles show that human ESAT-6-specific CD4 T cells are more differentiated cells than Ag85B-specific cells during latent Mtb infection.

(A) Ag85B and ESAT-6 HLA class II tetramer staining of CD4 T cells from H1:IC31 vaccinated adolescents (day 14). HLA allele-matched tetramers bearing irrelevant peptide antigens were used as controls. Numbers represent the frequencies of tetramer+ CD4 T cells.
(B) Median (error bars denote IQR) frequencies of tetramer+ CD4 T cells in QFT− or QFT+ adolescents who received two vaccinations of H1:IC31. The arrows indicate vaccine administration time-points.
(C) Supervised heat map of 11 mRNA transcripts differentially expressed (Mann-Whitney U test p < 0.05 and False discovery rate (FDR) < 10%) between tetramer-sorted Ag85B-specific and ESAT-6-specific CD4 T cells from QFT+ adolescents, 14 days after H1:IC31 vaccination. Undetected mRNAs are depicted in grey.
(D) CCR7 and CD45RA expression by total CD4 T cells (grey background), Ag85B-specific (orange), or ESAT-6-specific (red) tetramer+ CD4 T cells. FDRs were calculated using the Benjamini-Hochberg method and p-values were calculated using the Mann-Whitney U test. See also Figure S5.
Frequencies of Ag85B and ESAT-6-specific cells were boosted 14 days after H1:IC31 vaccination in QFT+ adolescents, allowing us to sort ESAT-6 and Ag85B-specific T cells with MHCII tetramers (Figure 4A and B) and profile mRNA expression of 91 genes by microfluidic qRT-PCR. Importantly, we utilized tetramers containing epitopes common to Ag85A and Ag85B, to assess the role of Mtb-infection in driving the differentiation of T cells recognizing both antigens. Principal component analysis showed differences between Ag85B and ESAT-6-specific cells (Figure S5A), and 11 genes exhibited differential expression between Ag85B and ESAT-6-specific CD4 T cells (p <0.05; FDR <10%). Amongst these, IL12RB1, STAT4 and GZMA were upregulated in ESAT-6-specific CD4 T cells relative to Ag85B-specific cells (Figure 4C). IL12RB1 and STAT4 encode molecules in the IL-12 signaling pathway (Bacon et al., 1995) that are central to the generation of effector responses (Stamm et al., 1999). In contrast, GATA3, CCR4, ICOS, LEF-1, TCF7L2 and CAMK4 were lower in ESAT-6-specific than Ag85B-specific CD4 T cells (Figure 4C). Lower expression of the Th2 associated genes GATA3 and CCR4 is also consistent with IL-12 signaling in ESAT-6-specific CD4 T cells (Zielinski et al., 2011). In fact, STAT4 and GATA3 expression were inversely correlated (Figure S5B). ICOS is typically associated with long-lived memory CD4 T cells (Moguche et al., 2015), while LEF-1, CAMK4, and TCF7L2 expression are upregulated by Wnt signaling (Arrazola et al., 2009; Zhu et al., 2015) which is involved in T cell memory generation (Zhao et al., 2010). Thus, ESAT-6-specific cells appeared more differentiated and resembled effector cells, while Ag85B cells appeared less differentiated. We therefore measured co-expression of CD45RA and CCR7 expression to analyze the memory phenotype of tetramer+ CD4 T cells in QFT+ adolescents. Human memory T cells lose CD45RA expression after priming, while re-expression of CD45RA on memory cells (CD45RA+CCR7−) indicates terminally differentiated T cells (Sallusto et al., 1999). Significantly higher proportions of ESAT-6-specific CD4 T cells displayed a terminally differentiated phenotype than Ag85B-specific cells (Figure 4D). Ag85B CD4 T cells in QFT− individuals were phenotypically similar to those in QFT+ individuals (data not shown), but we were unable to recover sufficient numbers of ESAT-6-specific T cells by tetramer from QFT− controls to complete this assessment.
Next, we compared Th1 cytokine-expressing CD4 T cells from QFT+ and QFT− adolescents boosted by H1:IC31 vaccination in the THYB-04 vaccine trial (Figure S5C). Virtually all ESAT-6 specific CD4 T cells from QFT+ adolescents receiving the vaccine expressed IFN-γ (Figure 5A), consistent with highly differentiated T cells (Seder et al., 2008). In contrast, Ag85B-specific cells from QFT+ adolescents (Figure 5A), and both ESAT-6 and Ag85B-specific cells in uninfected controls (Figure S5D), frequently expressed TNF or IL-2 in the absence of IFN-γ, consistent with a less differentiated phenotype. IFN-γ+ cells displayed a more differentiated CD45RA−CCR7− effector/effector memory phenotype, compared to IFN-γ− cells (Figure 5B), and the comparison of ESAT-6 and Ag85B-specific T cells again supported greater differentiation of ESAT-6-specific T cells (Figure 5B and S5E). By contrast, proportions of Ag85B and ESAT-6-specific IFN-γ+ CD4 T cells and their memory phenotype were not different in QFT− adolescents (Figure S5D and S5F).
Figure 5. Human ESAT-6-specific CD4 T cells have a cytokine expression profile that reflects greater differentiation than Ag85B-specific cells.

(A) Intracellular IFN-γ, TNF, and IL-2 staining following 12hr whole blood stimulation with media alone, Ag85B, or ESAT-6 peptide pools. Summary graph below depicts the proportions of ESAT-6 or Ag85B-specific CD4 T cells co-expressing IFN-γ, TNF and/or IL-2 in latently infected adolescents 14 days after primary H1:IC31 vaccination. Horizontal lines indicate medians, boxes the IQR and whiskers the range. The blue box and pie arcs denote functional T cell subsets that express IL-2 and/or TNF, but not IFN-γ. The purple box and arcs denote functional T cell subsets that express IFN-γ alone or in combination with IL-2 and TNF.
(B) Correlation between the proportions of CCR7−CD45RA− effector/effector memory (EM) Ag85B and ESAT-6-specific CD4 T cells and proportions of IFN-γ+ CD4 T cells 14 days after H1:IC31 vaccination in QFT+ adolescents.
(C) The FDS of ESAT-6 or Ag85B-specific CD4 T cells in each adolescent 14 days after primary H1:IC31 vaccination. P-values were calculated using the Wilcoxon signed-rank test (a and c) and Spearman’s rank correlation (b). See also Figure S5.
As the intracellular expression of IFN-γ correlated with the level of differentiation, we devised a “functional differentiation score” (FDS), calculated as the ratio of the proportion of IFN-γ-expressing antigen-specific Th1 cytokine+ CD4 T cells over the proportion of cells not expressing IFN-γ (i.e. expressing TNF and/or IL-2). As expected, ESAT-6-specific CD4 T cells had a significantly higher FDS than Ag85B-specific CD4 T cells on day 14 (Figure 5C). ESAT-6 and Ag85B-specific CD4 T cells from QFT− adolescents displayed comparatively low FDS (Figure S4G), confirming that the high level of differentiation of ESAT-6-specific cells is dependent on Mtb infection.
We then compared the differentiation of Ag85B and ESAT-6-specific CD4 T cells during longitudinal follow-up in the clinical trial to day 224. At all H1:IC31 post-vaccination time points in QFT+ adolescents, ESAT-6-specific CD4 T cells displayed higher proportions of IFN-γ+TNF+IL-2+ and lower proportions of TNF+IL-2+ and single IL-2+ subsets than Ag85B-specific cells (Figure 6A–C). By contrast, in QFT− adolescents proportions of IFN-γ+TNF+IL-2+ and TNF+IL-2+ ESAT-6 and Ag85B-specific CD4 T cells were not different (Figure 6D–E), while proportions of single IL-2+ ESAT-6-specific cells were also lower than Ag85B-specific cells in QFT− adolescents (Figure 6F). However, ESAT-6-specific CD4 T cells displayed consistently higher FDS compared to Ag85B-specific cells throughout study follow-up (Figure 6G). This was also true in QFT+ adolescents from a different group of the THYB-04 trial, who received two 50 μg doses of H1:IC31 (Figure 6H). Our data suggest that during Mtb infection, ESAT-6-specific CD4 T cells exist in a more differentiated state than Ag85B-specific cells. These results suggest that ESAT-6 is more highly expressed than Ag85B, and likely also Ag85A, during human Mtb infection, consistent with findings in mice (Figure 1A and B).
Figure 6. Vaccine-expanded human ESAT-6-specific CD4 T cells exhibit a more differentiated phenotype than Ag85B-specific cells.

(A–C) Median (error bars denote IQR) proportions of ESAT-6 and Ag85B-specific CD4 T cells expressing (A) IFN-γ+TNF+IL-2+, (B) TNF+IL-2+ or (C) only IL-2, in QFT+ adolescents who received two 15μg doses of H1:IC31.
(D–F) Median proportions of ESAT-6 and Ag85B-specific CD4 T cells expressing (D) IFN-γ+TNF+IL-2+, (E) TNF+IL-2+ or (F) only IL-2, in QFT− adolescents who received two 15μg doses of H1:IC31.
(G–I) Median FDS’s of ESAT-6 and Ag85B-specific CD4 T cells in (G) QFT+ and QFT−+ adolescents who received two 15μg doses of H1:IC31 or (H) QFT+ adolescents who received two 50μg doses of H1:IC31. The Wilcoxon signed-rank test was used to compare ESAT-6 and Ag85B-specific CD4 T cells. After Bonferroni correction p-values below 0.01 were considered significant. The arrows indicate vaccine administration time-points.
Functional exhaustion restricts the protective capacity of CD4 T cells recognizing ESAT-6
Next, we returned to the mouse model to determine whether functional exhaustion restricts the protective capacity of ESAT-6-specific CD4 T cells. Serial infusions of Ag85B peptide into Mtb-infected mice has previously been shown to reduce the lung bacterial burden (Bold et al., 2011), but we hypothesized that infusions of ESAT-6 would not have this effect. Mtb-infected mice received weekly infusions of Ag85B, ESAT-6, or OVA peptides beginning on day 25 post-infection. Two weeks after the third treatment (day 55 post-infection), we found that peptide infusion resulted in increased PD-1 expression by both Ag85B and ESAT-6-specific CD4 T cells, and surprisingly, a loss of the KLRG1 subset for both specificities (Figure S6A). Furthermore, Ag85B-specific T cells exhibited diminished memory markers after in vivo peptide stimulation. These results show that Ag85B-specific T cells recognize their cognate antigen in a limited manner during chronic Mtb infection, but peptide infusion triggers their activation and differentiation into an effector phenotype.
Mice receiving Ag85B peptide infusion had increased numbers of Ag85B-specific T cells in their lungs, as measured by tetramer-binding (Figure 7A) and IFN-γ production (Figure S6B). In contrast, mice receiving ESAT-6 infusions showed no increase of ESAT-6-specific T cells. In addition, serial Ag85B peptide infusions induced a ~five-fold reduction in lung bacterial burdens compared to controls receiving OVA peptide (Figure 7B). In contrast, lung bacterial burdens in mice receiving ESAT-6 peptide were not significantly reduced. Importantly, Ag85B peptide infusions did not induce Ag85B CD4 T cell expansion or reduce the lung bacterial burdens in mice infected with recombinant Mtb constitutively expressing Ag85B (Figure S7). Taken together, Ag85B-specific T cells possessed the capacity to mediate protection above that provided by ESAT-6-specific T cells despite ongoing antigenic stimulation of the latter, but only if Ag85B peptide was provided exogenously. Mtb-specific CD4 T cells can thus fail to provide optimal protection for two distinct reasons, either because their cognate antigen is not expressed during chronic infection or because their cognate antigen is continuously expressed, resulting in T cell exhaustion.
Figure 7. Ag85B-specific CD4 T cells exhibit greater protective capacity than ESAT-6 specific CD4 T cells following peptide infusion.

(A) Percentage of tetramer+ Ag85B or ESAT-6-specific CD4 T cells isolated from the lungs of mice injected with control (OVA, left) or the respective Ag85B or ESAT-6 peptides (right). The graph depicts the absolute numbers of Ag85B or ESAT-6-specific CD4 T cells.
(B) Lung bacterial burdens for individual mice after three weekly peptide injections analyzed at 42 days post-infection. Data are represented as mean ± SD, and statistical significance was first determined by ANOVA (p=0.0001), then between individual groups with a t-test with n=5 for A and n=10 for B. Data are representative of two independent experiments with 5–10 mice per group. See also Figures S6 and S7.
DISCUSSION
Our data have important implications for the rational design of TB vaccines tailored to optimize protection conferred by specific CD4 T cells recognizing antigens expressed at distinct stages of Mtb infection. We report definitive evidence that some antigens, like Ag85A and Ag85B, are expressed almost exclusively during early infection, whereas other antigens, like ESAT-6, are expressed throughout infection. Although prior Mtb transcript profiles from lungs of Mtb-infected mice have suggested this (Rogerson et al., 2006; Shi et al., 2004) it has not gained widespread acceptance, perhaps in part because the relevance to human Mtb infection has not been clear. Our studies in mice showed that CD4 T cells recognizing persistently expressed antigens accumulate in high numbers in the lung parenchyma and are driven towards terminal differentiation/functional exhaustion that compromises their protective capacity. In contrast, T cells recognizing antigens expressed primarily during early infection contract in numbers, assume a less differentiated phenotype akin to memory cells, and retain the capacity to produce multiple cytokines. The finding that human CD4 T cells recognizing Ag85A and Ag85B are less differentiated than those recognizing ESAT-6 strongly suggests that Ag85A and B exhibit low levels of expression during chronic stages of human Mtb infection, as in mice, whereas ESAT-6 is expressed persistently in both species.
How do these findings shape the protective capacities of the T cells that recognize them? The murine data suggest that Ag85B and ESAT-6-specific CD4 T cells may contribute to Mtb control. However, the protection provided by each appears sub-optimal, but for different reasons. As shown previously (Bold et al., 2011), serial infusions of Ag85B peptide can reduce the lung bacterial burden. There are two possible explanations for this: 1) T cells recognize the infused peptide directly presented by MHCII molecules on the surface of Mtb-infected cells in the lung, facilitating a cognate interaction that does not otherwise occur without peptide infusion, and/or 2) T cells recognize the infused peptide on uninfected cells and the bolus of cytokine produced activates infected cells and promotes some Mtb-killing in a bystander manner. We now show that while this is true for the Ag85B peptide, infusions of ESAT-6 peptide do not reduce bacterial numbers. Thus, ongoing recognition of infection-derived ESAT-6 antigen is probably stimulating ESAT-6-specific T cells to their full capacity to eradicate Mtb. The observation that Ag85B peptide infusion, which promotes the activation of T cells with robust capacity to produce cytokines, can improve clearance despite ongoing recognition of ESAT-6 suggests that this “full capacity” of ESAT-6-specific T cells is still sub-optimal. In other words, protective immunity conferred by Ag85B-specific T cells is limited by antigen availability during chronic infection, whereas immunity by ESAT-6-specific T cells is limited by functional exhaustion. The role of antigen availability in shaping the differentiation, function, and protective capacity of Mtb-specific CD8 T cells needs to be addressed in future studies.
Understanding the distinct limitations of specific T cell subsets suggests unique vaccination strategies to enhance the protection that each confers. It is likely that vaccines based solely on antigens like Ag85B, which are primarily expressed in early infection, would confer only transient protection and may possess a limited capacity to promote Mtb containment after infection has been established. These problems may be exacerbated with systemically administered vaccines because peripheral T cells are delayed in their recruitment to the lungs after aerosol Mtb infection (Urdahl, 2014). Could reduced expression of Ag85A during chronic stages of Mtb infection underlie the lack of protection afforded by the MVA85A vaccine in two recent Phase IIb trials (Ndiaye et al., 2015; Tameris et al., 2013)? Antigens like Ag85B may yet be useful in vaccines that induce lung-resident T cells that confer early Mtb control after aerosol infection, especially if used in conjunction with late-stage antigens, like ESAT-6, that can act in concert to promote containment once infection is established.
Our findings help explain why vaccines containing ESAT-6, but not several other antigens, including Ag85B, were capable of conferring protection when administered to mice after Mtb infection had been established (Hoang et al., 2013). Yet our studies in mice also suggest that functional exhaustion can restrict the protective capacity of CD4 T cells recognizing persistently expressed antigens. While recent murine studies suggest that lung resident T cells can become functionally exhausted (Behar et al., 2014; Jayaraman et al., 2016), our study shows that T cells recognizing distinct Mtb antigens are differentially prone to this. Although functional exhaustion was not observed in ESAT-6-specific T cells isolated from the blood of Mtb-infected humans, this could reflect differences between the blood and lung, or between latent and active TB, rather than differences between species. Two recent murine studies have shown that Mtb-specific T cells in the blood have a higher cytokine-producing capacity than those in the lung parenchyma (Moguche et al., 2015; Sakai et al., 2014a). Likewise, in cynomologous macaques, ESAT-6-specific T cells in the blood were capable of producing multiple cytokines, but T cells isolated from lung granulomas produced multiple cytokines only after polyclonal stimulation, but not after stimulation with ESAT-6 peptides (Gideon et al., 2015). Furthermore, in rhesus macaques, LAG3, a cell surface inhibitory receptor associated with CD4 T cell exhaustion in this species (Bosinger et al., 2009; Chew et al., 2016; Fromentin et al., 2016; Rotger et al., 2011), was expressed during TB by lung T cells, but not the blood (Phillips et al., 2015). Thus, antigen-specific T cells may have greater functional capacity in the blood than at the site of high antigenic exposure in the lung. This is even more likely to be true during active TB disease when bacterial burdens are highest.
The challenge of devising a T cell-targeted vaccine against Mtb is heightened by the recent understanding that antigen-specific memory CD4 T cells that are less differentiated exert greater control of murine Mtb infection than highly differentiated effector cells (Lindenstrom et al., 2013; Moguche et al., 2015; Sakai et al., 2014a; Torrado et al., 2015; Vogelzang et al., 2014). The protective capacity of these “less differentiated” CD4 T cells reflects their superior ability to proliferate, maintain their cytokine producing capacity, and home to infected tissues (Andersen and Urdahl, 2015; Sakai et al., 2014b; Woodworth et al., 2016). Thus, while choosing persistently expressed vaccine antigens is likely to be important, these same antigens may drive T cells towards terminal differentiation and dysfunction. These considerations must be heeded in choosing the adjuvants and the antigen doses utilized in the next generation of TB vaccines. In some regions endemic for TB, where an effective TB vaccine is most needed, most individuals have already acquired Mtb infection by late adolescence (Andrews et al., 2014; Mahomed et al., 2011). The properties of a post-exposure vaccine that primes T cells to the optimal state of differentiation in these endemic regions are probably very different than those for vaccines that achieve this in naive individuals. These considerations are usually overlooked; most TB vaccines are tested pre-clinically in animals that are naïve to mycobacteria. Vaccines eliciting T cells that recognize subdominant Mtb antigens and possess TCRs that interact with low avidity to Mtb-infected cells may be less prone to terminal differentiation and promote more durable immunity. Such an approach has shown promise in inbred mice, but may be difficult to achieve in human populations because subdominant antigens will vary between individuals due to HLA diversity.
Overall, our studies demonstrate the value of crosstalk between mouse and human TB research. Currently human Mtb infection is categorized in a binary fashion into either latent infection or active disease based on immune reactivity (e.g., QFT status) and the presence or absence of clinical symptoms and/or detection of bacilli in sputum. We propose that using FDS of CD4 T cells to measure the relative availability of distinct subsets of Mtb antigens in different strata along the spectrum of Mtb infection may facilitate more precise classifications. For example, FDS determinations may help identify individuals classified by a memory T cell response as having latent Mtb infection, who have actually cleared infection; conversely, individuals with subclinical disease, or active TB disease with a high degree of ongoing Mtb replication may also be identified. Future studies are needed to determine if this approach could have prognostic value and shape clinical decision-making.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kevin Urdahl (kevin.urdahl@cidresearch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 mice were purchased from Jackson Laboratories. ESAT-6 TCRtg (C7) mice were provided by Eric Pamer and have been described previously (Gallegos et al., 2008). Ag85B TCRtg (P25) mice have been previously described (Tamura et al., 2004). All animals were housed and maintained in specific-pathogen-free conditions at the Center for Infectious Disease Research (CIDR). All animal studies were performed in compliance with the CIDR Animal Care and Use Committee. Both male and female mice between the ages of 8–12 weeks were used.
Human participants
We utilized samples collected from adolescents enrolled into the THYB-04 phase II clinical trial (Mearns et al., 2016), performed at the Field Site of the South African TB Vaccine Initiative (South African National Clinical Trials Register NHREC number: DOH-27-0612-3947 and Pan African Clinical Trial Registry number: PACTR201403000464306). Healthy adolescents who received routine BCG vaccination at birth were recruited from the Worcester district of the Western Cape, South Africa. Latent Mtb infection was diagnosed by Quantiferon Gold In-tube assay (Qiagen). The Human Research Ethics Committee of the University of Cape Town and the Medicines Control Council of South Africa reviewed the study protocols and approved the study. All participants provided signed informed assent. Signed informed consent was also obtained from their parents/legal representatives. We focused our analyses on adolescents in Group 1 (Table S1). To be eligible, participants aged 12 to 18 years had to be healthy based on medical examination. Evidence of current or prior active tuberculosis disease, any vaccination 3 months before screening, participation in other experimental tuberculosis vaccine trials, administration of immune modulating drugs or HIV seropositivity, or positive pregnancy test constituted exclusion from the study.
METHOD DETAILS
Aerosol infections and CFU determination
Most infections were done with a stock of Mtb H37Rv, as previously described (Urdahl et al., 2003). Some experiments utilized a virulent clinical isolate of Mtb (Beijing SA161) (Ordway et al., 2011) or a strain of Mtb H37Rv that constitutively expresses Ag85B. The constitutive Ag85B-expressing Mtb strain was generated using the pMSP12::GFP plasmid obtained from Lalita Ramakrishnan (Cosma et al., 2004). Using Mtb genomic DNA as the template, the Ag85B gene was amplified by PCR and inserted into the plasmid to replace GFP. The plasmid was then used to transform Mtb H37Rv after the Ag85B sequence was confirmed by sequencing, and kanamycin-resistant colonies were selected.
In brief, mice were infected in an aerosol infection chamber (Glas-Col) with ~100 CFU deposited into the lungs of each mouse. Two mice were sacrificed immediately after infection to determine the infectious dose for each experiment. To determine viable numbers of CFUs at time-points post-infection, the left lung of each mouse was homogenized in PBS with 0.05% Tween 80. Ten-fold serial dilutions were made in 0.05% Tween 80 and plated onto 7H10 plates. Colonies were counted after 21 d of incubation at 37°C to determine CFUs.
In vivo peptide injections
For in vivo peptide injections, mice were infused intravenously (I.V.) with 100 μg of Ag85B240-254 or ESAT-64–17 peptides and sacrificed 2 h later. Control mice were injected with OVA323–339 peptide. For protection experiments, mice were injected with 100 μg of Ag85B, ESAT-6 or OVA peptides I.V. once a week for a total of three injections and sacrificed three days after the last injection.
Preparation of single-cell suspensions
Murine lungs were harvested into HEPES buffer containing Liberase Blendzyme 3 (70 μg/ml; Roche) and DNase I (30 μg/ml; Sigma-Aldrich). The lungs were then minced into small pieces using the gentleMacs dissociator (Miltenyi Biotec), incubated at 37°C for 30 min, and further homogenized with the gentleMacs dissociator. The cells were filtered using cell strainers and suspended in FACS buffer (PBS containing 2.5% fetal bovine serum and 0.1% NaN3). In experiments involving intracellular cytokine detection, incubation steps were performed in media/buffers containing Brefeldin A (10 μg/ml; Sigma-Aldrich).
In vitro peptide stimulation
After single-cell preparations were made, murine lung cells were stimulated with ESAT-64–17, ESAT-62–17, or Ag85B240–254 (5 μg/ml final concentration) for 4 h in complete growth media (RPMI 1640 supplemented with 10% FCS, 2 mM L-glutamine, 10 mM Hepes, 0.5 μM 2-ME, 100 U/ml penicillin, and 100 μg/ml streptomycin) in the presence of monensin at 37°C with 5% CO2. Cells were then washed and stained with antibodies as described below.
CFSE labeling and transgenic T cell transfer
Spleens from C7 or P25 mice were enriched for CD4 T cells using the EasySep Mouse CD4 T cell Isolation Kit (Stemcell Technologies). Transgenic CD4 T cells were then labeled with CFSE using the CellTrace CFSE cell proliferation kit following the manufacturer’s protocol (Life Technologies). In brief, single-cell suspensions were incubated with CFSE at a final concentration of 10 μM for 10 min before quenching with complete growth medium supplemented with 10% fetal bovine serum. CFSE-labeled transgenic cells were then transferred I.V. into infected mice 5 days before sacrificing.
MHCII tetramer and antibody staining
MHCII tetramers containing amino acids 4–17 of Mtb ESAT-6 (made in house using a construct generated by Dr. Marc Jenkins) or 240–254 of Mtb Ag85B (NIH Tetramer Core Facility) were used to detect Mtb-specific murine CD4 T cells. Single-cell suspensions were stained at saturating concentrations with the tetramers and incubated at room temperature for 1 h. For direct ex vivo intracellular cytokine staining (ICS), tissues were processed in the presence of Brefeldin A (10 μg/ml; Sigma-Aldrich).
All surface staining was done at 4°C for 30 min. After tetramer and/or surface staining, the cells were fixed, permeabilized, and stained with antibodies against intracellular markers using eBioscience fixation/permeabilization and permeabilization buffers. Antigen-specific CD4 T cells were identified by gating on CD3+, CD4+, dump- (CD11b, CD11c, B220, F4/80, CD8) and tetramer+ cells.
For experiments with I.V. antibody labeling for identification of intravascular T cells, mice were injected with 1 μg of CD4-PE (RM4-4, Biolegend) antibody 5–10 min prior to sacrificing. This method has been previously described (Sakai et al., 2014).
Human H1:IC31 vaccination
H1:IC31 was administered intramuscularly into the deltoid area of alternate arms on days 0 and 56. H1:IC31 is a subunit vaccine comprised of a fusion protein of Ag85B and ESAT-6 (H1) formulated in the adjuvant, IC31. The adjuvant, IC31, consists of ODN1a (20 nmol) and KLK (500 nmol), a TLR9-stimulatory oligodeoxynucleotide and an antibacterial cationic peptide, respectively. H1:IC31 was developed and manufactured by the Statens Serum Institut (SSI) in Copenhagen, Denmark.
Whole blood intracellular cytokine staining
Blood was collected from human adolescents and isolated by density gradient centrifugation using BD Vacutainer CPT tubes. PBMC were cryopreserved in 10% DMSO and 40% fetal calf serum (FCS) in RPMI enriched with L-Glutamine.
Whole blood intracellular cytokine staining was performed as previously described (Kagina et al., 2015). Briefly, one mL of whole blood was stimulated with 15mer peptides spanning Ag85B (2 μg/mL Staten Serum Institute), ESAT-6 (2 μg/mL Staten Serum Institute), PHA (5 μg/mL Bioweb), or media alone for 12 h in the presence of the co-stimulatory antibodies anti-CD28 and CD49d (0.25μg/mL BD). After 7 h, Brefeldin A (10 μg/mL, Sigma Aldrich) was added, and blood was incubated for a further 5 h. After stimulation, red blood cells were lysed, and white cells were fixed with FACS Lysing solution (BD). Cells were frozen to allow for batch intracellular cytokine staining. Cells were thawed and permeabilized with Perm/Wash buffer (BD) and stained with CD3-BV421 (BD Clone: UCHT1), CD4-QDot605 (Life Technologies Clone: S3.5), CD8-PerCP-Cy5.5 (BD Clone: SK1), IFN-γ-Alexa700 (BD Clone: B27), TNF-α-PE-Cy7 (eBioscience Clone: MAb11), IL-2-FITC (BD Clone: 5344.111), IL-17-Alexa647 (BD Clone: SCPL1362), CCR7-PE (BD Clone: 150503), and CD45RA-BV570 (BioLegend Clone: HI100). Anti-Mouse Ig compensation beads (BD) were acquired prior to acquiring stained fixed white blood cells on a BD LSR II.
HLA typing of adolescents
High resolution (4-digit) HLA typing was performed at the La Jolla Institute for Allergy and Immunology using allele-specific primer amplification and/or DNA sequencing.
Ag85B, ESAT-6, and antibody staining
DPB1*04:01 and DQB1*06:02 HLA class II tetramers were kindly supplied by the NIH Tetramer Core Facility at Emory University in Atlanta, GA. For each Mtb-specific tetramer, irrelevant antigen control HLA-class II tetramers, loaded with an epitope from the HIV envelope protein, or the self-antigen, CHIP, were also supplied by the NIH Tetramer Core Facility (Table S2). DRB1*03:01 HLA tetramers loaded with an Ag85 or the human apolipoprotein B-100 peptide (irrelevant antigen control) were from Beckman Coulter. Cryopreserved PBMC were thawed and incubated in 1 mL of the viability dye LIVE/DEAD (Invitrogen) for 30 min at room temperature. PBMC were washed with 2% FCS and 2 mM ethylenediaminetetraacetic acid (EDTA) in PBS and then stained with the fluorescently labeled CCR7-PerCP-Cy5.5 antibody (BD Clone: 150503) for 30 min at 37°C. PBMC were washed again and incubated with HLA class II tetramers (1–2 μg/mL) for 60 min at room temperature. Following another wash, cells were incubated for 30 min at room temperature with the following fluorescently labeled antibodies: CD3-FITC (BD Clone: UCHT1), CD4-BV421 (Biolegend Clone: RPA-T4), CD45RA-PE-Cy7 (BD Clone: UCHL1), CD8-BV510 (Biolegend Clone: RPA-T8), CD19-BV510 (Biolegend Clone: HIB19) and CD14-BV510 (Biolegend Clone: M5E2). CD8, CD19, and CD14 staining was performed to exclude cell populations that expressed these surface markers (dump gate). Inclusion of the dump gate improves specificity when detecting rare tetramer+ T cells by eliminating aberrant tetramer binding events (Moon et al., 2009). Anti-Mouse Ig compensation beads (BD) and stained cells were acquired on a BD FACS Aria II.
Selection of the TaqMan GE assays
We initially selected 101 TaqMan GE assays specific for mRNA transcripts involved in T cell function (Table S3). The amplification efficiencies of all TaqMan GE assays should be close to 100% (±10%) to allow use of the comparative Ct method to quantify relative gene expression (Schmittgen and Livak, 2008). To determine the amplification efficiencies of each TaqMan GE assay, we modified a previously described method (Dominguez et al., 2013). A total of twelve 2-fold serial dilutions of PBMC were made, and the slope and R2 value for each TaqMan GE assay were determined. Assays had to achieve a slope between 3.1 to 3.6 and a very strong correlation (r2 > 0.97) across at least 5 consecutive dilution points. A slope between 3.1 and 3.6 corresponds to efficiencies ranging from 90% to 110%. Using these criteria, we determined that 95 of 101 TaqMan GE assays had efficiencies ranging from 90% to 110%. However, four TaqMan GE assays (CXCL10, GAPDH, IL-5, and PTPRC) in our panel could potentially detect genomic DNA (gDNA) according to Life Technologies. While we included 96 TaqMan GE assays on each 96.96 Dynamic Array chip, we did not analyze data obtained from TaqMan GE assays that failed qualification or detected gDNA. Therefore, analysis was restricted to 91 TaqMan GE assays.
High throughput microfluidic RT-qPCR
Specific transcript amplification (STA) cDNA was synthesized from RNA isolated from HLA class II tetramer sorted ESAT-6 and Ag85B-specific CD4 T cells from PBMC using the Invitrogen CellsDirect One-Step qRT-PCR kit. Briefly, HLA tetramer+ cells were sorted directly into a 200 μL PCR tube containing 5 μL CellsDirect 2x reaction mix, 0.5 μL SuperScript™ III RT/Platinum® Taq mix, 2.5 μL of pooled TaqMan GE assays at a concentration of 0.2x per assay, and 1 μL TE buffer (10 mM Tris, pH 8.0, 0.1 mM EDTA). PCR tubes were placed on a thermal cycler for 20 min at 50°C to lyse cells and synthesize specific transcript cDNA. Tubes were then heated to 95°C for 2 min to denature the reverse transcriptase, and specific transcripts were amplified following 18 cycles of cDNA denaturing at 95°C for 15 seconds and annealing and amplification at 60°C for 4 min. Five -fold diluted STA-cDNA and TaqMan GE assays were pipetted on a 96.96 Dynamic Array (Fluidigm) and placed in a BioMark HD System (Fluidigm) according to the manufacturer’s protocol. The BioMark Real-time PCR Analysis software determined Ct values using linear derivative background correction and an amplification curve quality threshold of 0.65. Samples with undetectable levels (i.e. Ct=40) of CD4, B2M, G6PD, and HPRT mRNA transcripts were removed from the analysis. Relative mRNA transcript levels (delta Et) in ESAT-6 and Ag85B CD4 T cells were determined by subtracting B2M Et (40-Ct) values from the gene of interest Et value. Delta Et values were calculated for each triplicate cDNA sample, and the average delta Et value was calculated. Our experimental approach was first optimized and could readily identify different transcriptomic signatures in FACS-sorted naïve (CCR7+CD45RA+), central memory (CCR7+CD45RA−) and effector/effector memory (CCR7−CD45RA−) CD4 T cells (data not shown).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data from stained murine cells were acquired using the BD LSRII flow cytometer. Flow data were analyzed using FlowJo V9 software (Tree Star). Statistical analysis and graphical representation of data were done using GraphPad Prism v6.0 software. Statistical significance was determined using a t-test or ANOVA. Statistical details, including the significance and the n value, can be found in the figure legends.
Human flow cytometry data were analyzed using FlowJo V9.2 or V10. Frequencies of IL-17 expressing cells were low or undetectable (Mearns et al., 2016) and therefore IL-17 expression was not analyzed here. To derive a frequency cut-off value for HLA class II tetramer experiments, we calculated the median frequency of control tetramer+ CD4 T cells plus 4 median absolute deviations (MAD) from the median frequencies of control tetramer+ CD4 T cells. The median frequency observed for control tetramer+ CD4 T cells was 0.001% and the MAD was 0.001%. Therefore, the minimum frequency (median + 4 MADs) required for transcriptomic analysis of Ag85B or ESAT-6-specific tetramer+ CD4 T cells was 0.005%. To ensure reliable measurement of FDS and memory phenotypes of antigen-specific T cells measured by ICS assay, we applied a positivity cut-off value of 0.023%, derived from the median frequencies of unstimulated controls plus 3 median absolute deviations. Thus, only antigen-specific T cell frequencies above 0.023% were included in these analyses. In addition, a minimum of 3 donors per group had to meet these positivity cut-off criteria for FDS and/or memory phenotype data to be included in group comparisons.
Statistical analyses were performed using R and GraphPad Prism v6.0. The Mann Whitney U test was used to compare QFT− and QFT+ adolescents, while the Wilcoxon signed-rank test was used to compare results within each group of adolescents. P values describing differences in the frequency, proportion of cytokine-producing subsets, and proportion of memory phenotypes were corrected using the Bonferroni correction, while p values describing differences in mRNA expression levels were corrected using the Benjamini Hochberg method (FDR). To visualize the multidimensional transcriptomic data from ESAT-6 and Ag85B-specific CD4 T cells, the heatmap.2 and prcomp functions in R were used to generate heatmap and PCA plots, respectively.
Supplementary Material
Highlights.
Mtb antigen Ag85B-specific CD4 T cells maintain memory cell features during infection.
Antigen availability limits immunity conferred by Ag85B-specific CD4 T cells.
Mtb antigen ESAT-6-specific CD4 T cells are driven towards terminal differentiation.
Functional exhaustion restricts ESAT-6-specific CD4 T cell-mediated immunity against Mtb.
Acknowledgments
We thank M. Bevan, S. Cohen, and L. Ramakrishnan for comments on the manuscript. The murine work was funded by grants to K.B.U. from the National Institutes of Health (U19 AI106761 and R01 AI076327) and from the Paul G. Allen Family Foundation. We also thank the adolescents who participated in the THYB-04 trial and acknowledge the contributions of the SATVI clinicians, nurses, and technicians. Also, thanks to Valneva GMBH, Austria for generously supplying ODN1a and KLK for the H1:IC31 vaccine. We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of MHC class II tetramers. Human work was supported by the European Developing Countries Clinical Trials Partnership (EDCTP) [IP.2007.32080.001] (http://www.edctp.org/), the Statens Serum Institut, and by grants to T.J.S. from the European Commission funded TBVAC2020 consortium (H2020-PHC-643381), and the Bill and Melinda Gates Foundation (BMGF) Global Health grant OPP1066265. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. A.O.M was supported by an award from the National Institutes of Health (T21-CA009537) and by a Bank of America Dissertation Fellowship. M.M. was supported by The Carnegie Corporation of New York.
Footnotes
Competing Financial Interests
Peter Andersen is co-inventor on a patent application covering the use of H1 in vaccines. All rights have been assigned to Statens Serum Institut, a Danish not for profit governmental institute. None of the other authors have a financial interest to declare.
Author Contributions
Conceptualization, A.O.M., C.R.P., R.P.L, P.S., P.A., W.A.H., T.J.S., and K.B.U.; Methodology, H.G., S.T.H., I.K., M.R., P.B., S.S., W.A.H., M.H., P.A.; Investigation, A.O.M., M.M., C.R.P., H.M., E.S., D. A., V.R., O. D., R.P.L., S.S., S.M., and D.R.S.; Resources, A.S., C.S.L.A., D.M.K., H.M.; Writing – Original Draft, A.O.M., M.M., C.R.P., P.A., T.J.S., and K.B.U.; Writing – Review & Editing, A.O.M., M.M., A.P.N., C.R.P., W.A.H., P.A., T.J.S., and K.B.U.; Supervision, A.P.N., H.G., P.A., T.J.S., and K.B.U.; Funding Acquisition, W.A.H., M.H., P.A., T.J.S., and K.B.U.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen P, Urdahl KB. TB vaccines; promoting rapid and durable protection in the lung. Curr Opin Immunol. 2015;35:55–62. doi: 10.1016/j.coi.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JR, Morrow C, Walensky RP, Wood R. Integrating social contact and environmental data in evaluating tuberculosis transmission in a South African township. J Infect Dis. 2014;210:597–603. doi: 10.1093/infdis/jiu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrazola MS, Varela-Nallar L, Colombres M, Toledo EM, Cruzat F, Pavez L, Assar R, Aravena A, Gonzalez M, Montecino M, et al. Calcium/calmodulin-dependent protein kinase type IV is a target gene of the Wnt/beta-catenin signaling pathway. J Cell Physiol. 2009;221:658–667. doi: 10.1002/jcp.21902. [DOI] [PubMed] [Google Scholar]
- Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar SM, Carpenter SM, Booty MG, Barber DL, Jayaraman P. Orchestration of pulmonary T cell immunity during Mycobacterium tuberculosis infection: immunity interruptus. Semin Immunol. 2014;26:559–577. doi: 10.1016/j.smim.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS pathogens. 2011;7:e1002063. doi: 10.1371/journal.ppat.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest. 2009;119:3556–3572. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014;159:1497–1509. doi: 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J Immunol. 1999;162:5407–5416. [PubMed] [Google Scholar]
- Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, Hammond KB, Clayton KL, Ishii N, Abdel-Mohsen M, et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS pathogens. 2016;12:e1005349. doi: 10.1371/journal.ppat.1005349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Humbert O, Ramakrishnan L. Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol. 2004;5:828–835. doi: 10.1038/ni1091. [DOI] [PubMed] [Google Scholar]
- Dominguez MH, Chattopadhyay PK, Ma S, Lamoreaux L, McDavid A, Finak G, Gottardo R, Koup RA, Roederer M. Highly multiplexed quantitation of gene expression on single cells. J Immunol Methods. 2013;391:133–145. doi: 10.1016/j.jim.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity. 2011;34:807–819. doi: 10.1016/j.immuni.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, Killian M, Epling L, Hoh R, Sinclair E, et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS pathogens. 2016;12:e1005761. doi: 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med. 2008;205:2359–2368. doi: 10.1084/jem.20080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos AM, van Heijst JW, Samstein M, Su X, Pamer EG, Glickman MS. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS pathogens. 2011;7:e1002052. doi: 10.1371/journal.ppat.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, Maiello P, Rutledge T, Marino S, Fortune SM, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS pathogens. 2015;11:e1004603. doi: 10.1371/journal.ppat.1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill WP, Harik NS, Whiddon MR, Liao RP, Mittler JE, Sherman DR. A replication clock for Mycobacterium tuberculosis. Nat Med. 2009;15:211–214. doi: 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AM, Difazio R, Flynn JL. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. 2013;190:270–277. doi: 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T, Aagaard C, Dietrich J, Cassidy JP, Dolganov G, Schoolnik GK, Lundberg CV, Agger EM, Andersen P. ESAT-6 (EsxA) and TB10.4 (EsxH) based vaccines for pre- and post- exposure tuberculosis vaccination. PLoS One. 2013;8:e80579. doi: 10.1371/journal.pone.0080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev. 2015;264:74–87. doi: 10.1111/imr.12274. [DOI] [PubMed] [Google Scholar]
- Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, Anderson AC, Kuchroo VK, Behar SM. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PLoS pathogens. 2016;12:e1005490. doi: 10.1371/journal.ppat.1005490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagina BM, Mansoor N, Kpamegan EP, Penn-Nicholson A, Nemes E, Smit E, Gelderbloem S, Soares AP, Abel B, Keyser A, et al. Qualification of a whole blood intracellular cytokine staining assay to measure mycobacteria-specific CD4 and CD8 T cell immunity by flow cytometry. J Immunol Methods. 2015;417:22–33. doi: 10.1016/j.jim.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer L, Maughan WN, Wilson RA, Dover LG, Besra GS. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett Appl Microbiol. 2002;34:233–237. doi: 10.1046/j.1472-765x.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Lawn SD, Myer L, Edwards D, Bekker LG, Wood R. Short-term and long-term risk of tuberculosis associated with CD4 cell recovery during antiretroviral therapy in South Africa. AIDS. 2009;23:1717–1725. doi: 10.1097/QAD.0b013e32832d3b6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenstrom T, Knudsen NP, Agger EM, Andersen P. Control of chronic mycobacterium tuberculosis infection by CD4 KLRG1- IL-2-secreting central memory cells. J Immunol. 2013;190:6311–6319. doi: 10.4049/jimmunol.1300248. [DOI] [PubMed] [Google Scholar]
- Lindestam Arlehamn CS, McKinney DM, Carpenter C, Paul S, Rozot V, Makgotlho E, Gregg Y, van Rooyen M, Ernst JD, Hatherill M, et al. A Quantitative Analysis of Complexity of Human Pathogen-Specific CD4 T Cell Responses in Healthy M. tuberculosis Infected South Africans. PLoS pathogens. 2016;12:e1005760. doi: 10.1371/journal.ppat.1005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahomed H, Hawkridge T, Verver S, Abrahams D, Geiter L, Hatherill M, Ehrlich R, Hanekom WA, Hussey GD. The tuberculin skin test versus QuantiFERON TB Gold(R) in predicting tuberculosis disease in an adolescent cohort study in South Africa. PLoS One. 2011;6:e17984. doi: 10.1371/journal.pone.0017984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearns H, Geldenhuys HD, Kagina BM, Musvosvi M, Little F, Ratangee F, Mahomed H, Hanekom WA, Hoff S, Ruhwald M, et al. H1:IC31 vaccination is safe and induces long-lived TNF-α+IL-2+ CD4 T cell responses in M. tuberculosis infected and uninfected adolescents: a randomized trial. Vaccine. 2016 doi: 10.1016/j.vaccine.2016.11.023. In press. [DOI] [PubMed] [Google Scholar]
- Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, Cambier CJ, Sissons JR, Gallegos AM, Fink PJ, Urdahl KB. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med. 2015;212:715–728. doi: 10.1084/jem.20141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2001;193:271–280. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, Zell T, Jenkins MK. Tracking epitope-specific T cells. Nat Protoc. 2009;4:565–581. doi: 10.1038/nprot.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dieye S, Dieye TN, Esmail H, Goliath R, Huygen K, et al. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85A in healthy adults infected with HIV-1: a randomised, placebo- controlled, phase 2 trial. Lancet Respir Med. 2015;3:190–200. doi: 10.1016/S2213-2600(15)00037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway DJ, Shang S, Henao-Tamayo M, Obregon-Henao A, Nold L, Caraway M, Shanley CA, Basaraba RJ, Duncan CG, Orme IM. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin Vaccine Immunol. 2011;18:1527–1535. doi: 10.1128/CVI.05127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan AJ, Ramakrishnan L. Immunity and Immunopathology in the Tuberculous Granuloma. Cold Spring Harb Perspect Med. 2015:5. doi: 10.1101/cshperspect.a018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat Immunol. 2011;12:467–471. doi: 10.1038/ni.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EJ, Reiss DJ, Turkarslan S, Minch KJ, Rustad T, Plaisier CL, Longabaugh WJ, Sherman DR, Baliga NS. A high-resolution network model for global gene regulation in Mycobacterium tuberculosis. Nucleic Acids Res. 2014;42:11291–11303. doi: 10.1093/nar/gku777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BL, Mehra S, Ahsan MH, Selman M, Khader SA, Kaushal D. LAG3 expression in active Mycobacterium tuberculosis infections. Am J Pathol. 2015;185:820–833. doi: 10.1016/j.ajpath.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, Urdahl KB, Winslow GM, Woodland DL. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19408–19413. doi: 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson BJ, Jung YJ, LaCourse R, Ryan L, Enright N, North RJ. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen-specific T cells during stationary level lung infection in mice. Immunology. 2006;118:195–201. doi: 10.1111/j.1365-2567.2006.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, Sandler NG, Roque A, Liebner J, Battegay M, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest. 2011;121:2391–2400. doi: 10.1172/JCI45235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, Barber DL. CD4 T Cell-Derived IFN-gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS pathogens. 2016;12:e1005667. doi: 10.1371/journal.ppat.1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, Barber DL. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. 2014a;192:2965–2969. doi: 10.4049/jimmunol.1400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Mayer-Barber KD, Barber DL. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol. 2014b;29:137–142. doi: 10.1016/j.coi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- Shi L, North R, Gennaro ML. Effect of growth state on transcription levels of genes encoding major secreted antigens of Mycobacterium tuberculosis in the mouse lung. Infect Immun. 2004;72:2420–2424. doi: 10.1128/IAI.72.4.2420-2424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ernst JD. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J Immunol. 2013;191:1016–1020. doi: 10.4049/jimmunol.1301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm LM, Satoskar AA, Ghosh SK, David JR, Satoskar AR. STAT-4 mediated IL- 12 signaling pathway is critical for the development of protective immunity in cutaneous leishmaniasis. Eur J Immunol. 1999;29:2524–2529. doi: 10.1002/(SICI)1521-4141(199908)29:08<2524::AID-IMMU2524>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, Shea JE, McClain JB, Hussey GD, Hanekom WA, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381:1021–1028. doi: 10.1016/S0140-6736(13)60177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Ariga H, Kinashi T, Uehara S, Kikuchi T, Nakada M, Tokunaga T, Xu W, Kariyone A, Saito T, et al. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol. 2004;16:1691–1699. doi: 10.1093/intimm/dxh170. [DOI] [PubMed] [Google Scholar]
- Torrado E, Fountain JJ, Liao M, Tighe M, Reiley WW, Lai RP, Meintjes G, Pearl JE, Chen X, Zak DE, et al. Interleukin 27R regulates CD4+ T cell phenotype and impacts protective immunity during Mycobacterium tuberculosis infection. J Exp Med. 2015;212:1449–1463. doi: 10.1084/jem.20141520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdahl KB. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin Immunol. 2014;26:578–587. doi: 10.1016/j.smim.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdahl KB, Liggitt D, Bevan MJ. CD8+ T cells accumulate in the lungs of Mycobacterium tuberculosis-infected Kb−/−Db−/− mice, but provide minimal protection. J Immunol. 2003;170:1987–1994. doi: 10.4049/jimmunol.170.4.1987. [DOI] [PubMed] [Google Scholar]
- Vogelzang A, Perdomo C, Zedler U, Kuhlmann S, Hurwitz R, Gengenbacher M, Kaufmann SH. Central memory CD4+ T cells are responsible for the recombinant Bacillus Calmette- Guerin DeltaureC::hly vaccine’s superior protection against tuberculosis. J Infect Dis. 2014;210:1928–1937. doi: 10.1093/infdis/jiu347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzl G, Ronacher K, Hanekom W, Scriba TJ, Zumla A. Immunological biomarkers of tuberculosis. Nat Rev Immunol. 2011;11:343–354. doi: 10.1038/nri2960. [DOI] [PubMed] [Google Scholar]
- Woodworth JS, Cohen SB, Moguche AO, Plumlee CR, Agger EM, Urdahl KB, Andersen P. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 2016 doi: 10.1038/mi.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, Xue HH. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol. 2010;184:1191–1199. doi: 10.4049/jimmunol.0901199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang W, Wang X. Roles of transcriptional factor 7 in production of inflammatory factors for lung diseases. J Transl Med. 2015;13:273. doi: 10.1186/s12967-015-0617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski CE, Corti D, Mele F, Pinto D, Lanzavecchia A, Sallusto F. Dissecting the human immunologic memory for pathogens. Immunol Rev. 2011;240:40–51. doi: 10.1111/j.1600-065X.2010.01000.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.