

Summary
Apoptosis has been implicated in Compensatory Proliferation Signaling (CPS), whereby dying cells induce proliferation in neighboring cells as a means to restore homeostasis. The nature of signaling between apoptotic cells and their neighboring cells remains largely unknown. Here we show that a fraction of apoptotic cells produce and release CrkI-containing microvesicles (distinct from exosomes and apoptotic bodies), which induce proliferation in neighboring cells upon contact. We provide visual evidence of CPS by videomicroscopy. We show that purified vesicles in vitro and in vivo are sufficient to stimulate proliferation in other cells. Our data demonstrate that CrkI inactivation by ExoT bacterial toxin or by mutagenesis blocks vesicle formation in apoptotic cells and inhibits CPS, thus uncoupling apoptosis from CPS. We further show that c-Jun amino-terminal kinase (JNK) plays a pivotal role in mediating vesicle-induced CPS in recipient cells. CPS could have important ramifications in diseases that involve apoptotic cell death.
Keywords: Apoptosis, Apoptotic Compensatory Proliferation Signaling (ACPS), Apoptotic Compensatory Proliferation Signaling Vesicle (ACPSV), CT10 Regulator of Kinase I (CrkI), c-Jun amino-terminal kinase (JNK), Pseudomonas aeruginosa, Exotoxin T (ExoT), Glomerulonephritis, Nephritis, Nephrotoxic serum (NTS)
Graphical abstract

Introduction
In the course of normal tissue turnover in humans, approximately one million cells die every second through a highly regulated process of programmed cell death (PCD), known as apoptosis (Danial and Korsmeyer, 2004; Krysko et al., 2008; Reed, 2006). Cell death must be swiftly balanced with compensatory proliferation to maintain homeostasis and tissue integrity. Apoptosis, in addition to its role in PCD, has been implicated in triggering compensatory proliferation signaling (CPS), whereby dying cells induce compensatory proliferation in their neighboring cells (Fan and Bergmann, 2008a, b; Fuchs and Steller, 2015; Huh et al., 2004; Kondo et al., 2006; Li et al., 2010; Perez-Garijo et al., 2009; Ryoo et al., 2004; Valentin-Vega et al., 2008). To date however, the nature of CPS and its molecular components remain largely unknown.
Dissecting the mechanisms by which Pseudomonas aeruginosa Exotoxin T (ExoT) induces apoptosis in target epithelial cells is an area of investigation in our laboratory (Goldufsky et al., 2015; Shafikhani et al., 2008a; Wood et al., 2015a; Wood et al., 2015b). In a recent study (Wood et al., 2015a), we demonstrated that ExoT, by ADPribosylating CrkI adaptor protein, disrupts focal adhesion and interferes with integrin/FAK/p130Cas/β-catenin survival signaling, inducing anoikis apoptosis in epithelial cells. During these studies, we have discovered what we believe to be the mediator of apoptotic CPS. Our data demonstrate that a fraction of apoptotic cells produce and release CrkI-containing microvesicles, (distinct from exosomes and apoptotic bodies), that stimulate proliferation in neighboring cells upon contact. Vesicle formation in apoptotic cells requires CrkI while compensatory proliferation signaling, induced by CrkI-microvesicles, is dependent on JNK activity in recipient bystander cells.
Results
Observation of apoptotic CPS
Recently, we reported that the ADPribosyltransferase (ADPRT) domain of ExoT - by ADP-ribosylating CrkI adaptor protein -induces anoikis apoptosis in epithelial cells (Wood et al., 2015a). In one experiment which was designed to examine the role of CrkI in ExoT-induced apoptosis, we found that ∼38% of HeLa cells transfected with the pIRES2 mammalian expression vector harboring wildtype CrkI-GFP succumbed to apoptosis (see Fig. 4 in (Wood et al., 2015a)). During these studies, we made a surprising observation and noted that ∼5% of the CrkI-GFP transfected apoptotic cells produced and released 1 to 3 small microvesicles containing CrkI-GFP which induced proliferation in neighboring cells upon contact (Fig. 1A, Suppl. Fig. 1 & Suppl. Movies 1A-1B). After contacting these vesicles, nearly 100% of recipient cells initiated mitosis and proliferated within ∼6 h. For simplicity, we will refer to these vesicles as ACPSVs (Apoptotic Compensatory Proliferation Signaling Vesicles). ACPSVs were not formed or released from healthy CrkI-transfected cells (identified by their spread-out morphology) or when cells, prior to transfection, were pre-treated with Z-VAD, a pan-caspase inhibitor which blocks apoptosis (Fig. 1B, Suppl. Movie 1C), indicating that death signal may be required for vesicle production. Furthermore, these vesicles were primarily produced in cells which had initiated apoptosis (exhibiting cell shrinkage), but prior to their death, as indicated by their negative propidium iodide (PI) staining (Fig. 1C). (The Committee on Cell Death has defined cell shrinkage as an early and reversible step in apoptosis, whereas PI uptake is designated as a late and an irreversible step which indicates cell death in apoptosis (Kroemer et al., 2009)).
Fig. 4. Proteomic analyses of ACPSV by LC- MS/MS.
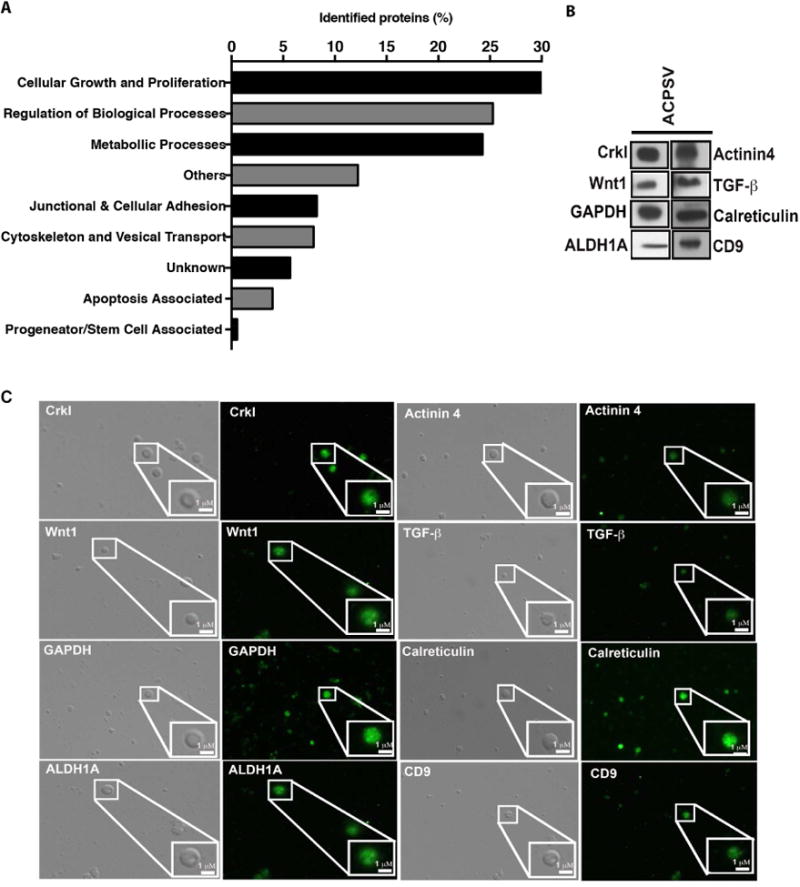
Peptides from the 16K fractions of serum-starved apoptotic HeLa cells were analyzed by LC-MS/MS. (A) Some of the notable protein groupings were plotted based on the number of proteins identified in each grouping. The results of LC-MS/MS analyses were further corroborated by evaluating the presence of representative proteins from the aforementioned categories within ACPSVs by Western blotting (B) and by IF microscopy (C).
Fig. 1. Apoptotic cells produce and release CrkI-containing vesicles, which appear to be capable of inducing proliferation in bystander cells.
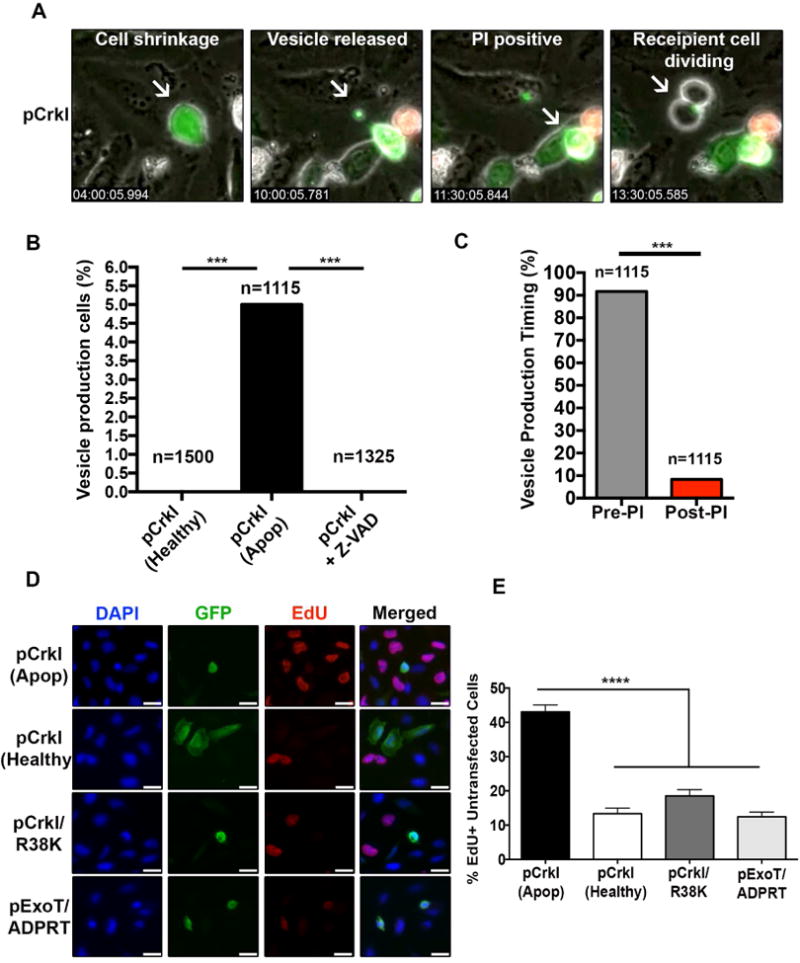
HeLa cells were transfected with CrkI-GFP in the presence or absence of Z-VAD and followed by IF time-lapse videomicroscopy. (A) Selected movie frames of a CrkI-transfected apoptotic cell are shown. This cell releases a CrkI-containing microvesicle, before it becomes PI positive, and the bystander cell initiates mitosis after contacting this vesicle. (B) The percentages of non-apoptotic, apoptotic, and Z-VAD-treated, CrkI-GFP transfected cells producing vesicle is shown. (C) The ability of CrkI-GFP transfected apoptotic cells to produce and release vesicles prior or post PI uptake was assessed by IF time-lapse microscopy and the tabulated results are shown. (For B & C, n≥1115, *** p<0.001, χ2 analysis). (D-E) Cells were transiently transfected with CrkI-GFP, CrkI/R38K-GFP, or ExoT/ADPRT-GFP expression vectors. 17h after transfection, cells were treated with EdU for 2h, fixed by 10% TCA, and analyzed for EdU incorporation in untransfected cells surrounding transfected apoptotic cells (identified by cell shrinkage/rounding) or healthy (identified by spread-out morphology). Representative images are shown in (D) and the tabulated data are shown in (E) (n≥3 independent experiments, 10 random fields/experiment, **** p<0.0001, One-way ANOVA).
To further corroborate these data, we transfected HeLa cells with CrkI-GFP expression vector and measured the proliferation rates in un-transfected cells (GFP-negative) surrounding healthy CrkI-GFP transfected cells (GFP-positive cells exhibiting spread-out morphology), or apoptotic CrkI-GFP transfected cells (GFP-positive cells exhibiting cell shrinkage/rounding morphology), by EdU incorporation which is a marker for cell proliferation (Salic and Mitchison, 2008). There was a significant increase in the number of EdU positive (proliferating) un-transfected cells surrounding apoptotic CrkIGFP transfected cells, as compared to healthy CrkI-GFP transfected cells (Fig. 1D-E), correlating apoptosis with increased proliferation in cells neighboring apoptotic cells.
Although, CrkI was contained in these vesicles (Fig. 1A), it remained unclear whether CrkI played any role in CPS or if it was a bystander protein that became trapped in these vesicles as they formed in apoptotic cells. CrkI is a splice variant of the Crk gene which is required for development and for cytokinesis (Guris et al., 2001; Shafikhani et al., 2008b). Since we have only been able to partially deplete CrkI protein in epithelial cells due to its required function in cell proliferation and cytokinesis (Shafikhani and Engel, 2006; Shafikhani et al., 2008b), we inactivated CrkI by a bacterial toxin and by mutagenesis to assess its possible role in CPS. The ADP-ribosyltransferase (ADPRT) domain of Pseudomonas aeruginosa ExoT inactivates CrkI and interferes with its known functions by ADP-ribosylating a conserved arginine residue in its SH2 domain (Deng et al., 2005; Shafikhani et al., 2008b; Sun and Barbieri, 2003; Sundin et al., 2004; Wood et al., 2015a). Similarly, the arginine (R) to lysine (K) substitution in the SH2 domain of CrkI at position 38 (CrkI/R38K) results in a dominant negative (DN) mutant which also inactivates CrkI and blocks its known activities in a manner that phenocopies ExoT/ADPRT activity (Lamorte et al., 2002; Shafikhani et al., 2008b; Wood et al., 2015a). Of note, transient transfection with ExoT/ADPRT or CrkI/R38K also results in apoptosis in epithelial cells (Wood et al., 2015a).
We transfected HeLa cells with CrkI/R38K-GFP, or ExoT/ADPRT-GFP, and measured the proliferation rates in un-transfected cells (GFP-negative) surrounding apoptotic transfected cells by EdU incorporation. All HeLa cells transfected with CrkI/R38K-GFP and ExoT/ADPRT-GFP were rounded, whereas only ∼35% HeLa cells transfected with CrkI-GFP were rounded. This is consistent with the degree of apoptosis induced by these constructs (Shafikhani et al., 2008a; Wood et al., 2015a). Importantly, there was a significant reduction in the number of EdU-positive un-transfected cells, surrounding ExoT/ADPRT-GFP and CrkI/R38K-GFP transfected apoptotic cells, as compared to CrkI-GFP transfected apoptotic cells (Fig. 1D-E). These data indicated that CrkI inactivation by ExoT or by CrkI/R38K DN interferes with apoptotic cells' ability to stimulate proliferation in surrounding cells. However, it remained unclear which step(s) of CPS (i.e., vesicle production, vesicle release, vesicle/recipient cell interaction, or vesicle-induced signaling in recipient cell) was blocked by ExoT/ADPRT and CrkI/R38K DN mutant.
To assess these possibilities, we repeated these transfection studies and evaluated vesicle formation and release, as well as vesicle interaction with recipient cells, by time-lapse IF videomicroscopy, as we described in Fig. 1 and Suppl. Movies 1A-B. Consistent with our previous report (Wood et al., 2015a), transfection with CrkI/R38K-GFP or ExoT/ADPRT-GFP resulted in apoptosis in HeLa cells (Suppl. Fig. 1A, Suppl. Movie 2A-2B). However, only 0.5% of CrkI/R38K-GFP transfected apoptotic cells and none of ExoT/ADPRT-GFP cells produced vesicles, as compared to 5.2% of CrkI-GFP transfected apoptotic cells which produced vesicles (Suppl. Fig. 1B, Suppl. Movie 2A-2B). Moreover, the few CrkI/R38K-GFP transfected cells which produced vesicles, never produced more than one vesicle, as opposed to CrkI-GFP transfected vesicle-producing cells, which on average produced 2.3 vesicles per vesicle-producing cell (Suppl. Fig. 1C, Suppl. Movies 1A-B). To further demonstrate that reduction in vesicle formation in CrkI/R38K or ExoT-transfected apoptotic cells interfered with their ability to induce proliferation in neighboring cells, we determined the proliferation-inducing capacity index (PCI) of the aforementioned transfected apoptotic cells, which we defined as the percentage of proliferating (mitotic) cells in the vicinity of CrkI-GFP, CrkI/R38K-GFP, or ExoT/ADPRT-GFP transfected apoptotic cells (Suppl. Fig. 1D). In these studies, we included all apoptotic transfected cells, as opposed to only the vesicle-producing apoptotic cells, to account for the reduced vesicle formation in the CrkI/R38K or ExoT-transfected apoptotic cells. Consistent with our hypothesis, the PCIs of CrkI/R38K-GFP and ExoT/ADPRT-transfected apoptotic cells were significantly lower than the PCI of CrkI-transfected apoptotic cells (Suppl. Fig. 1E). Collectively, these data highlight a role for CrkI in CPS and indicate that CrkI inactivation - by ExoT or by R38K DN mutation - blocks vesicle formation and inhibits CPS in apoptotic cells, thus uncoupling apoptosis from apoptotic CPS.
Purification and characterization of CrkI-containing microvesicles (ACPSVs)
To ensure that the observed CPS was not an artifact of CrkI overexpression in HeLa cells, we tested whether triggering apoptosis by another method would also result in production of CrkI-containing microvesicles capable of inducing compensatory proliferation in neigbouring cells. To this end, we induced apoptosis in HeLa cells by serum starvation (Chiarugi and Giannoni, 2008; Frisch and Screaton, 2001) which as expected, caused significant Z-VAD sensitive apoptosis within 24h (Fig. 2A-B). Supernatants from serum-starved apoptotic and serum-fed healthy (Mock) HeLa cells were then collected after 24h and subjected to differential centrifugation as described in the Experimental Proceedure; (Protocol: centrifugation at 1500× g (1.5K) for 5 min followed by passaging through 5-micron filter to remove cell debris and apoptotic bodies, followed by centrifugation at 16,000× g (16K fraction) for 30 min, followed by 100,000× g (100K fraction) for 30 min).
Fig. 2. Purification and characterization of CrkI-containing microvesicles (ACPSVs).
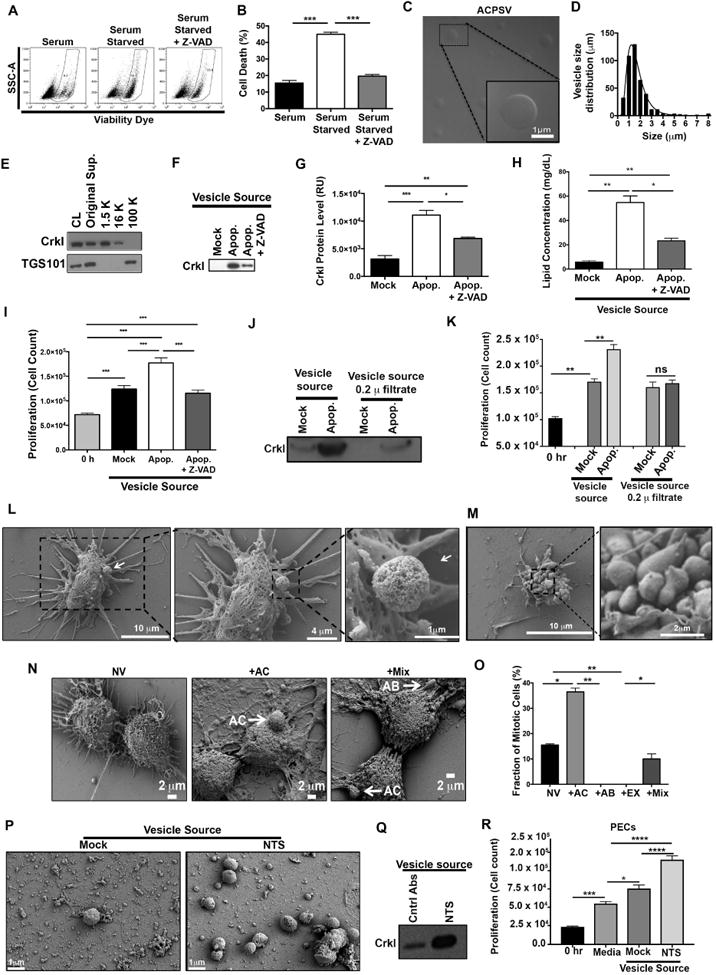
(A) HeLa cells either received 10% serum (Mock) or were induced to undergo apoptosis by serum-starvation in the presence or absence of Z-VAD. 24h after serum starvation, apoptotic cell death was assessed by flow cytometry and the tabulated results from 3 independent experiments are shown in (B). (*** p<0.001, One-way ANOVA). (C-I) Supernatants from the indicated cultures were fractionated by differential centrifugation and evaluated for their CrkI-containing microvesicles (ACPSVs) and CPS. (C) ACPSVs in the 16K fraction of apoptotic cells were visualized by DIC. (D) Vesicle size distribution was determined and plotted (Mean size ± S.D. = 1.76 ± 1.04 μm, n=822). (E) Peptides from the cell lysate (CL) and the indicated fractions of serum-starved apoptotic cells were probed for TGS101 (exosome marker) and CrkI (ACPSV marker) by Western blotting. (F) The 16K fractions of healthy (Mock), serum-starved (Apop.) and Z-VAD treated serum-starved (Apop. + Z-VAD) were probed for ACPSV content by Western blotting of CrkI and the results from 6 independent studies were plotted in (G). (* p<0.05, ** p<0.01, *** p<0.001, One-way ANOVA). (H) The lipid contents of the vesicles in the 16K fractions of the indicated cultures were measured and the tabulated data from 3 experiments are shown. (* p<0.01, ** p<0.005, One-way ANOVA). (I) HeLa cells were treated with 16K fractions of serum-fed healthy (Mock), serum-starved apoptotic (Apop.), or Z-VAD treated serum-starved (Apop. + Z-VAD) and the proliferation levels were determined by cell count 24h after treatment. The tabulated data from n≥6 independent experiments are shown as the Mean ± S.D. (*** p<0.001, One-way ANOVA). (J-K) The 16K fractions of mock and apoptotic (apop.) HeLa cells were passaged through 0.2 μ filter to remove vesicle. (L-M) HeLa cells were seeded on a coverslip and induced to undergo apoptosis by serum-starvation. 24h after serum starvation, cells were fixed and analyzed by SEM. (L) Representative images of a vesicle producing cell are shown. (M) Representative images of an apoptotic body-producing cell are shown. (N-O) Original supernatant from serum-starved apoptotic HeLa cell culture was spun down at 100,000× g (100K) to collect all vesicles, which were then added to adherent HeLa cells on a coverslip. Five hours after vesicle addition, the fractions of mitotic cells, with no vesicle (NV); with ACPSV only (+AC); with apoptotic body only (+AB); or with a combination of vesicles (+Mix) were assessed by SEM. Selected images representing each group is shown in (N) and the tabulated data from 3 experiments are shown in (O). (** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001; One-way ANOVA). (P-Q) C57BL/6 mice were injected with nephrotoxic serum (NTS) to induce nephritis or isotype antibody control (Mock). Vesicles were harvested from mock and NTS-injected mice glomeruli and imaged by SEM (P) and evaluated for their CrkI contents by Western blot (Q), and for their ability to induce proliferation in adherent kidney parietal epithelial cells (PECs) in (R). (3 independent experiment, 4 mice/group, each group was done in triplicates, * p<0.05, *** p<0.001, **** p<0.0001, One-way ANOVA).
Consistent with the time-lapse videomicroscopy data (Fig. 1 & Suppl. Movie 1A-B), microvesicles derived from serum-starved apoptotic cell cultures were globular, as visualized by Differential Interference Contrast (DIC) microscopy (Fig. 2C). These vesicles were distinct from exosomes in that they were primarily found in the 16K fraction as opposed to exosomes that are pelleted at speeds ≥ 50,000× g (Tauro et al., 2012), and they were also significantly larger in diameter (1.76 ± 1.04 μm, n=820), than exosomes which are reported to be 40-100 nm in diameter (Akers et al., 2013; Cocucci et al., 2009; Kastelowitz and Yin, 2014; Muralidharan-Chari et al., 2010; Raposo and Stoorvogel, 2013) (Fig. 2C-D). Although, the original supernatant (Original Sup.) from apoptotic cells and the 100K fraction contained exosomes - as they probed positive for the exosome marker TGS101 (Michael et al., 2010; Tauro et al., 2012) - the 16K fraction did not, indicating that 16K fraction was devoid of exosome contamination (Fig. 2E). As expected, CrkI was present in the original supernatant and in the 16K fraction, but not in the 100K exosome fraction (Fig. 2E). Furthermore, 16K fraction from apoptotic cell cultures contained significantly more CrkI-containing vesicles than 16K fractions from healthy (Mock) cell cultures, as assessed by Western blot analyses of their CrkI levels (Fig. 2F-G), and their lipid content measurements (Fig. 2H). Importantly, ACPSVs stimulated proliferation in adherent HeLa cells when added exogenously in the media (Fig. 2I). Removing ACPSV vesicles from the 16K fraction by passaging it through 0.2μ filter abrogated its ability to stimulate proliferation in adherent cells, indicating that the vesicles in the 16K fraction of apoptotic cells, but not a soluble factor contaminant in this fraction, are responsible for CPS (Fig. 2J-K). In line with videomicroscopy data, (Fig. 1B, Suppl. Movie 1C), pretreatment with Z-VAD pancaspase inhibitor significantly reduced apoptosis (Fig. 2A-B), and vesicle production in serum-starved cells (Fig. 2F-H), and abrogated CPS in recipient cells (Fig. 2I); thus, highlighting the need for cell death signal for vesicle production and CPS.
HeLa cells undergoing intrinsic, extrinsic, and anoikis apoptosis - (induced by UV radiation, by anti-Fas antibody, by serum-starvation, or by growth in suspension media (Almeida et al., 2000; Lee et al., 2013; Schulze-Osthoff et al., 1998; Waring and Mullbacher, 1999; Wood et al., 2011; Yonehara et al., 1989)) - also produced ACPSVs capable of stimulating proliferation in other cells (Ha and Snyder, 1999; Zong et al., 2004), indicating that other apoptotic programmed cell death can also lead to CPS (data not shown).
We next asked whether under apoptotic conditions, primary cells would also produce ACPSVs capable of stimulating proliferation in other cells. We induced apoptosis in human epithelial keratinocytes (HEK) by growing these cells in suspension media (Almeida et al., 2000; Wood et al., 2011). The 16K fraction from apoptotic HEK cells grown in suspension media also contained CrkI-containing vesicles, which were nearly identical in dimensions to vesicles produced by HeLa cells and significantly stimulated proliferation in other HEK adherent cells when added exogenously to the media (Suppl. Fig. 2A-D). As expected, Z-VAD significantly reduced apoptosis and prevented vesicle formation and CPS in HEK cells grown in suspension media (Suppl. Fig. 2A-D). Similarly, mouse epithelial keratinocyte (MEK) primary cells also produced CrkI-containing microvesicles under apoptotic conditions that stimulated proliferation in MEK adherent cells when added exogenously to the media (Suppl. Fig. 2E-G, Suppl. Movie 3). These data indicated that CPS is not restricted to transformed cells and it also occurs in primary cells in response to apoptosis. Of note, HeLa cells undergoing necrotic cell death, due to ATP depletion resulting from treatment with 0.5 mM MNNG (N-methylN-nitro-N-nitrosoguanidine) (Ha and Snyder, 1999; Zong et al., 2004) did not produce CrkI-containing ACPSVs and failed to induce compensatory proliferation in adherent HeLa cells (Suppl. Fig. 3), indicating that necrotic cell death does not lead to CPS.
Characterization of ACPSVs by Scanning Electron Microscopy (SEM)
To gain insights into the ultrastructure of ACPSVs, we seeded HeLa cells on coverslips, serum-starved them for 24h, and analyzed vesicle formation by SEM. We found a number of apoptotic cells (exhibiting cell shrinkage morphology) that appeared to be forming and releasing vesicles (Fig. 2L) that were very similar to the ACPS vesicles with respect to their size we observed in time-lapse videomicroscopy and by DIC imaging (Fig. 1A, Fig. 2C). We did not find vesicle-producing cells in serum-fed healthy HeLa cells (Mock), again highlighting the need for apoptosis signal for CPS. Of note, we also found many apoptotic cells exhibiting classical apoptotic bodies, but in contrast to ACPSVs which were globular in shape and had spongy surface textures, apoptotic bodies were irregularly shaped and had smooth surfaces (Fig. 2M). These morphological differences allowed us to unambiguously distinguish ACPSVs from apoptotic bodies.
There remained a possibility that contaminating apoptotic bodies or exosomes in the 16K fraction may be responsible for mediating CPS. To address this possibility, we spun down the original supernatant of serum-starved apoptotic HeLa cells, which contained ACPSVs, exosomes, and apoptotic bodies (Fig. 2C, 2E, & 2M), at 100× g to collect all vesicles. We then added these vesicles to HeLa cells seeded on coverslips. Five hours after vesicle addition, we fixed the cells, imaged them by SEM, and determined the fractions of proliferating (mitotic) cells with no vesicle (NV), those with ACPSV only (+AC), those with apoptotic bodies only (+AB), those with exosomes only (+EX), and those that had more than one type of vesicle (Mix), based on vesicle size and vesicle surface characteristics. Of note, 37.5% of mitotic cells were not included in these analyses because they could not be scored unambiguously due to image distortion resulting from sample preparation for SEM. Importantly, majority of proliferating (mitotic) cells had either ACPSV alone (36.5%) or both ACPSV and apoptotic body (10.5%) associated with their membranes (Fig. 2N-O). In contrast, there were no mitotic cells that had only apoptotic bodies alone (+AB) or exosomes alone (+EX) and ∼15.5% of mitotic cells had no vesicles (NV) associated with their membranes (Fig. 2N-O). These data correlated interaction with ACPSV, but not exosomes or apoptotic bodies, with increased proliferation.
Characterization of apoptotic CPS in vivo
To investigate apoptotic CPS in vivo, we used the nephrotoxic serum (NTS)–induced glomerulonephritis animal model (Dai et al., 2013; Ophascharoensuk et al., 1998). We chose this model because NTS-induced nephritis is characterized by increased apoptosis of glomerular cells, resulting in the formation of crescent glomeruli (Dai et al., 2013). Interestingly, NTS nephritis is also characterized with increased proliferation in kidney parietal epithelial cells (PECs) within the crescent glomeruli (Dai et al., 2013), suggesting that apoptotic CPS may also be responsible for the enhanced proliferation in these apoptotic crescent glomeruli.
To this end, we injected mice with NTS or an isotype antibody (Mock), collected glomeruli at the peak of apoptosis as determined by proteinuria, (∼6 days after injection), and evaluated them for crescent formation by hematoxylin and eosin (H&E), Periodic Acid-Schiff (PAS), and Masson's Trichrome histological analyses; for apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay; and for proliferation by Ki-67 staining (Dai et al., 2013; Ophascharoensuk et al., 1998). In line with the previous reports (Dai et al., 2013; Ophascharoensuk et al., 1998), NTS injection resulted in crescent formation in glomeruli, increased apoptosis, and increased proliferation in crescent glomeruli (Suppl. Fig. 4). We then assessed the mock and the crescent apoptotic glomeruli for their CrkI-containg ACPSV contents by differential centrifugation. Large microvesicles similar to ACPSV, (0.7-1.7 μm in diameter), were substantially enriched in the NTS-injected crescent glomeruli (Fig. 2P). Vesicles prepared from NTS-injected crescent apoptotic glomeruli contained substantially more CrkI than mock (Fig. 2Q) and were also significantly more effective in inducing proliferation in adherent PECs than vesicles from healthy glomeruli (Fig. 2R). Of note, there were also low-level ACPSV-like large microvesicles in mock-injected glomeruli, which could be due to low-level natural cellular turnover in healthy glomeruli (Suppl. Fig. 4B). Cell loss through apoptosis and through urination has been reported in kidney and kidney is known for its remarkable regenerative capacity to replenish and heal (Reviewed in (Dekel, 2016)). Collectively, these data indicated that vesicle-induced CPS also occurs in vivo in response to apoptosis and it may be an important and immediate response to injury.
Role of JNK in CPS
In Drosophila, c-Jun N-terminal kinase (JNK) becomes activated (phosphorylated) in healthy bystander cells neighboring apoptotic cells and JNK activity has been shown to be crucial in mediating CPS in bystander cells (Bergantinos et al., 2010; Fan et al., 2014; Ryoo et al., 2004). To determine whether JNK played a role in mediating ACPSV-induced proliferation in recipient cells, we first examined the impact of ACPSVs on JNK activation in recipient cells. ACPSVs treatment resulted in transient but significant increases in activated (phosphorylated) forms of JNK1/2 isoforms in recipient HeLa cells within 30 min of vesicle treatment but returned to normal levels by 1h post-treatment (Fig. 3A-B). Although, ACPSVs contained unphosphorylated JNK1/2, they did not contain phosphorylated JNK1/2 (p-JNK1/2), indicating that vesicles were not the source of increased p-JNK1/2 in the recipient cells (Fig. 3C). To assess the requirement for JNK activity in ACPSV-induced compensatory proliferation in recipient cells, we pre-treated HeLa cells with JNK-specific inhibitor SP600125 (SP) (Luo et al., 2014; Zhao et al., 2013), 2h prior to vesicle treatment. Pre-treatment with SP inhibited ACPSV-induced JNK activation (Fig. 3A-B) and significantly reduced ACPSV-induced compensatory proliferation in recipient HeLa cells (Fig. 3D). JNK protein depletion in recipient cells by siRNA also abrogated ACPSV-induced proliferation in recipient cells (Fig. 3E-F), ruling out the possibility of off-target effect by SP600125 and indicating that JNK activity in recipient cell is required to mediate proliferation induced by ACPSVs.
Fig. 3. JNK activity is required for ACPSV-induced compensatory proliferation in bystander cells.
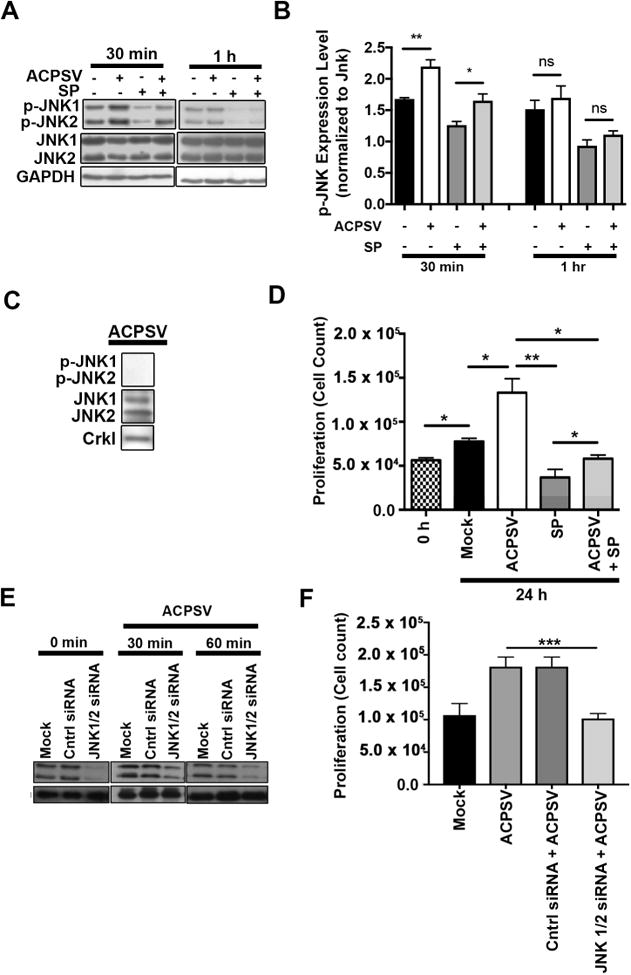
(A) The impact of ACPSVs on JNK1/2 activation (phosphorylation) in recipient HeLa cells in the presence and absence of 20μM JNK-specific inhibitor, SP600125 (SP), was assessed by Western blotting at 30 min and 1h after treatment with 16K fractions from serum-fed healthy (no ACPSV) or apoptotic (contains ACPSV) HeLa cells. Phospho JNK1/2 (p-JNK1/2) expression levels were normalized to total JNK1/2 protein levels and plotted in (B). Data are shown as Mean ± SEM (n=3, * p ≤ 0.05, ** p ≤ 0.01, One-way ANOVA). (C) ACPSVs were probed for phosphorylated and unphosphorylated JNK1/2. (D) HeLa cells were pre-treated with SP or DMSO (0.1%) for 2h prior to treatment with ACPSV or Mock. Proliferation was determined by measuring total cell counts at the time of treatment (0h) and 24h post treatment. Data are shown as Mean ± SEM (n=3, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, One-way ANOVA). (E-F) HeLa cells were treated with JNK1/2-specific siRNA or scrambled control (Mock), 24h prior to ACPS vesicle treatment. (E) JNK1/2 levels were evaluated by Western blotting in HeLa recipient cells prior to vesicle treatment (0 mins) or 30 or 60 mins after vesicle treatment. (F) The ability of ACPSVs to induce proliferation in control or JNK1/2-depleted HeLa cells was evaluated 24h after vesicle addition by cell counts. The data indicate that JNK function is required in recipient cells to mediate CPS induced by ACPSVs. (n=3, ** p ≤ 0.001).
Proteomic analyses of ACPSVs
To gain insight into the proteomic content of ACPSV, peptides (∼10 μg) extracted from the 16K fraction preparations from two independent experiments, were subjected to proteomics analyses, using the Filter-Aided Sample Preparation Protocol (FASP), as described previously (Wisniewski et al., 2009) and in the Experimental Procedure. Liquid chromatography tandem-mass spectrometry (LC-MS/MS) analyses of ACPSVs identified 2,167 proteins. Of notable proteins found in these vesicles (Fig. 4A), 30.8% have been annotated to function in cellular growth and proliferation processes (e.g., Wnt1, TGF-β1, JNK, Ras, Rac, CrkI); 25.3% in regulation of biological and housekeeping processes (e.g., Elongation factor 2, Protein disulfideisomerase A3, HSP90); 24.3% in metabolic processes (e.g., GAPDH, Phosphoglycerate kinase, ATP synthetase); 8.25% in junctional and adhesion structures (e.g., Integrin, Vinculin, Profilin-1, Ezrin, Integrin β4); 7.93% in cytoskeletal and transport processes (e.g., Cadherin-1, Actin, Microtubule, Myosin-9, Dynein); 3.96% in apoptosis-associated processes (e.g., Calreticulin, Apoptosis-inducing factor 1, Apoptosis-associated speck-like protein containing a CARD). We corroborated the LC-MS/MS data by evaluating the presence of representative proteins in each category within ACPSVs by Western blotting and by IF microscopy of ACPSVs (Fig. 4B-C).
Discussion
In this report, we provide compelling evidence to demonstrate that CPS is mediated by CrkI-containing microvesicles (ACPSVs). We provide visual evidence of CPS and show by live videomicroscopy, SEM imaging, and functional analyses how ACPSVs, (distinct from exosomes and apoptotic bodies), are generated and released from apoptotic cells and how they induce proliferation both in normal and transformed cells upon contact. It is not clear why only ∼5% of apoptotic cells have the capacity to produce ACPS vesicles under apoptotic conditions. Intriguingly, ACPSVs contain a number of proteins that have been associated with normal and malignant progenitor and stem cells, such as ALDH1, CD9, CD44, and CD166 (Chitteti et al., 2014; Ginestier et al., 2007; Karlsson et al., 2013), as revealed by proteomic analyses. Whether these cells are progenitor or stem-like cells or whether they are yet another unidentified specialized cell type, remains to be determined.
In Drosophila, Wingless (Wg; orthologue of mammalian Wnt1), and Decapentaplegic (Dpp; the ortholog of mammalian TGF-β and bone morphogenic proteins) mitogens, are secreted from apoptotic cells and have been postulated to initiate CPS in bystander cells in a manner that is dependent on bystander cell's JNK activity (Bergantinos et al., 2010; Fan et al., 2014; Fuchs and Steller, 2015; Ryoo et al., 2004). Our data reveal that ACPSVs contain Wnt1 and TGF-β mitogens and demonstrate the importance of JNK activity in mediating vesicle-induced CPS in recipient cells, suggesting that ACPS vesicles may be the means by which apoptotic cells deliver mitogens to recipient bystander cells during CPS in Drosophila. Future studies are needed to assess the role of ASPS vesicles in medaiating CPS in Drosophila. Why would vesicles be needed to deliver mitogen(s) to recipient cells? We posit that packaging mitogens in vesicles may increase their effective concentrations upon delivery to recipient cells, by possibly protecting them from proteolysis/degradation and/or diffusion. Vesicles could also enhance local responses by targeting a subset of cells as opposed to affecting all cells in the environment. These possibilities need to be further investigated.
ACPSV formation and release closely resembles microvesicle biogenesis in that vesicle formation initiates at the plasma membrane by membrane protrusion followed by vesicle dissociation in a manner that resembles cytokinesis (Akers et al., 2013; Lee et al., 2012; Muralidharan-Chari et al., 2009; Muralidharan-Chari et al., 2010). Although, CrkI has not been implicated in microvesicle biogenesis, our data indicate that it may play an important role in ACPSV biogenesis. Interestingly, CrkI is also required for mammalian cytokinesis and its inactivation by ExoT or mutagenesis interferes with the separation of daughter cells during the abscission step in cytokinesis (Shafikhani and Engel, 2006; Shafikhani et al., 2008b). Future studies are needed to determine whether CrkI functions in microvesicles biogenesis or whether alterations in CrkI (through ADPribosylation by ExoT or by mutagenesis) render CrkI disruptive to the process of microvesicles biogenesis.
Recently, we demonstrated that although CrkI is recruited to focal adhesion (FA) sites, it plays no role in FA assembly or FA-associated functions (Shafikhani et al., 2008b; Wood et al., 2015a). This begs the question as to why CrkI is recruited to FA sites in the first place and what possible physiological role it may play in this compartment? Since all apoptotic pathways eventually converge on disruption of FA sites, as manifested by cell shrinkage (Elmore, 2007), we propose that CrkI is perfectly positioned at FA sites to function as a sensor for apoptotic cell death and to initiate CPS. Our model (Graphical abstract) posits that in response to pro-apoptotic stimuli, executioner caspase-3 becomes activated (Porter and Jänicke, 1999). Activated caspase-3 then degrades paxillin and p103Cas (Chiarugi and Giannoni, 2008; Wei et al., 2004) (proteins that recruit and anchor CrkI at FA (Bouchard et al., 2008; Chiarugi and Giannoni, 2008; Vachon, 2011)), thus liberating CrkI from FA to initiate CPS. We also propose that ExoT, by ADP-ribosylating CrkI blocks CPS because ADP-ribosylated CrkI is no longer competent to initiate vesicle formation.
Apoptotic CPS may have important ramifications in health and disease. For example, over 95% of cancer drugs and radiation therapy eliminate tumor cells by apoptosis (Cummings et al., 2004; Pace et al., 2000; Rahmani et al., 2007; Reed, 2006; Spiro and Silvestri, 2005). By inducing proliferation in other cancer cells, CPS could potentially limit the effectiveness of apoptotic-inducing cancer drugs. The finding that ExoT and a mutation in CrkI block vesicle production and CPS in apoptotic cells suggests that by targeting the components of CPS, we may be able to enhance the effectiveness of current cancer therapeutics.
Unlike in cancer, where apoptotic CPS is undesirable, there may also be situations where CPS could have desirable therapeutic potential. For example, diabetic foot ulcers show decreased proliferative capacity and changes associated with cellular senescence (Darby et al., 1997; Hehenberger et al., 1997; Hehenberger et al., 1998; Mansbridge et al., 1999). In this context, CPS vesicles could potentially restore proliferative capacity to these cells and improve healing. Future studies are needed to address the positive and the negative impacts of CPS in health and disease.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Sasha Shafikhani (Sasha_Shafikhani@rush.edu).
Experimental Model and Subject Details
Animal Model of Experimental Glomerulonephritis (George et al., 2012; Verma et al., 2015) was used to assess the occurrence of CPS in vivo. All procedures were approved by Rush University Medical Center IACUC (No: 15-069) and followed the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. Nephritis was induced by nephrotoxic serum (NTS) injection as described previously (George et al., 2012; Verma et al., 2015). Briefly, 6 to 8 weeks old C57BL6 male mice (Jackson Laboratory) were injected retroorbitally with sheep anti-rat glomerular lysate antiserum (kindly provided by Dr. David Salant, Boston University) or control sheep IgG (Sigma) at a dose of 3 mg per mouse. Urine was collected at base line and on successive days. Urinary albumin and creatinine were measured by using mouse albumin ELISA (Bethyl Labs, E99-134), and creatinine assay (Cayman Chemical, 500701) kits. Albumin-to-Creatinine Ratio (ACR) was calculated and used as a parameter to determine proteinuria. Mice were sacrificed at 6 days after the NTS injection for histological and biochemical analyses.
Processing of glomeruli and isolation of vesicles
For processing of glomeruli and isolation of vesicles, kidneys of sheep IgG (mock)- and NTS-treated mice were minced into 1 to 3-mm tissue fragments with a scalpel blade while being maintained in ice-cold PBS. The tissue was gently pressed through a 100 μm-cell strainer (Falcon) using a flattened pestle, followed by washing with 5 ml of ice-cold HBSS (Life Technologies) to prevent the adverse effects of shear heating. Then, the filtered flow-through was passaged through 40-μm cell strainer without pressing to retain glomeruli and tubules. The flow-through was then subjected to differential centrifugation as described in Methods.
Renal parietal epithelial cells
Mouse renal parietal epithelial cells (PECs) were prepared as described (Ohse et al., 2008) and were cultured in RPMI 1640 (Life Technologies) containing 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies). The media was supplemented with 2% FBS and 0.1 mM sodium pyruvate (Sigma). PECs were cultured at 33°C and 5% CO2 in collagen I (BD Biosciences)-coated flasks in the presence of interferon-γ (Cell Sciences; 50 U/ml for both cells). For proliferation experiments, 25000 cells were seeded to each well of a 24-well plate and cultured at 33°C for 24h. Then, PECs were treated with microvesicles isolated from control and NTS-treated mice, respectively. After 24 h treatment, cells were washed with PBS (Life Technologies), detached by using trypsin-EDTA (Life Technologies) for 5 min at 37°C and harvested by ce ntrifugation at 1200 rpm for 5 min. The cell pellet was re-suspended in the complete medium for counting and analysis.
Histopathological Evaluations were performed as described previously (Dai et al., 2013; Ophascharoensuk et al., 1998). Briefly, freshly harvested mouse kidneys were fixed in 4% PFA (Electron Microscopy Sciences) and embedded in paraffin. The sections were cut at 4μm thickness and stained with hematoxylin and eosin (H&E), Periodic acid-Schiff (PAS), and Masson's Trichrome. Staining of sections with Ki-67 (Bethyl Laboratories, IHC-00375-T) was used as a marker of cell proliferation. Apoptotic cells were identified by in situ TdT-mediated dUTP nick end labeling (TUNEL) using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore, S7101).
Method Details
Cell Culture and Transient Transfection protocols have been described previously (Wood et al., 2015a; Wood et al., 2015b). Briefly, cells were cultured in complete DMEM (Life Technologies) media supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% L-glutamine at 37°C in the presence of 5% CO2. Human epithelial keratinocytes (HEK) and mouse epidermal keratinocytes (MEK) were cultured in Dermal cell growth media (ATCC PCS-200-030) supplemented with keratinocyte growth kit (ATCC PCS-200-040) at 37°C in the presence of 5% COM2. HEK cells were a generous gift from Dr. Mitchell Denning (Loyola Medicine, Chicago, IL.) MEK cells were harvested and cultured from BALB/c neonates, and cultured in Dermal cell growth media (ATCC PCS-200-030) supplemented with keratinocyte growth kit (ATCC PCS-200-040) at 37°C in the presence of 5% CO2 as described (Pirrone et al., 2004). The plasmids used in these studies, pIRES∷CrkI-GFP, pIRES∷CrkI/R38K-GFP, and pIRES∷ExoT/ADPRT-GFP, have been previously described (Shafikhani and Engel, 2006; Wood et al., 2015a).
Cytotoxicity Measurement by Time-lapse Videomicroscopy protocols were described previously (Wood et al., 2015a; Wood et al., 2013). Briefly, cells were seeded in 24-well plates (Falcon) at 6.0 × 104 cells per well and were grown overnight in DMEM without phenol red. These cells were then transfected with the indicated expression vectors as described (Wood et al., 2015a; Wood et al., 2013). One hour after transfection, 7 μg/ml propidium iodide (PI) (Sigma) was added to the cells which were then placed on an Axiovert Z1 microscope (Zeiss) fitted with a live-imaging culture box (Pecon) maintaining 37°C in the presence of 5% CO2. Time-lapse videos were taken using AxioVision 4.2.8 software. Cytotoxicity from each frame was determined at 15-min intervals, by analyzing the PI channel in ImageJ v1.46 (National Institutes of Health) software by setting an appropriate threshold to isolate PI-positive cells.
Chemical Inhibitor Treatments
SP600125 (JNK inhibitor) and Z-VAD (pan-caspase inhibitor) were obtained from R&D Systems. These inhibitors were reconstituted in DMSO per the manufacturer's instructions, and then added to cells at 20 μM (for SP600125) or 60 μM (for Z-VAD) final concentrations respectively, 2h prior to the indicated treatments. An equivalent amount of DMSO (vehicle) was added to control cells to account for the effect of the vehicle.
Generation and Purification of ACPSVs
7.5 × 106 cells were cultured in 150 mm2 flasks (TPP) overnight in 25 mL of indicated media. Next day, cells were induced to undergo intrinsic apoptosis by serum-starvation (Braun et al., 2011), extrinsic apoptosis apoptosis by treatment with anti-Fas mAb (abcam-ab11881) (Scholz and Cinatl, 2005), or anoikis apoptosis by growing the cells detached from the surface in suspension media in Thermo Scientific Nunclon Sphera plates (Wood et al., 2011), in the presence or absence of Z-VAD. 24 to 48h after induction of apoptosis, culture supernatants from serum-fed healthy (Mock) and apoptotic cells were collected and subjected to differential centrifugation. Protocol: Supernatants were centrifuged at 1,500× g (1.5K) for 5 min followed by passage through 5-micron filter (Sterlitech, PES502005) to remove cell debris and apoptotic bodies, followed by centrifugation at 16,000× g (16K) for 30 min to collect ACPSVs, followed by centrifugation at 100,000× g (100K) for 30 min for exosomes purification. Pellets at each centrifugation steps were re-suspended in 300 μL PBS. For functional analyses, 100 μL of PBS containing vesicles were mixed with 900 μL regular DMEM media containing 10% FBS and Penicillin-Streptomycin antibiotics and added to indicated adherent cells. The proliferation-inducing effect associated with each fraction was determined by cell counts after growing recipient cells for 24h at 37°C in the presence of 5% CO2, using a hemocytometer after trypsin digestion.
Anoikis Apoptosis
To assess whether anoikis apoptosis could lead to vesicle production and CPS, 7.5 × 106 of indicated cells were cultured for 24h at 37°C i n the presence of 5% CO2 in suspension media in 90 mm2 Nunclon Sphera surface plates (Thermo Scientific) in the presence or absence of 60 μm Z-VAD and 1% Pen-Strep antibiotics added to the appropriate media indicated for each cell line. After 24 h incubation under these conditions cells and ACPSVs were collected and processed as described.
Anti-Fas mediated extrinsic apoptosis
To induce extrinsic apoptosis, anti-Fas (anti-CD95) mAb (abcam-ab11881), was added at the final concentration of 10 mg/mL. Isoform antibody was used as control (abcam). 24h after treatment, cytotoxicity was assessed to ensure the effectiveness of anti-Fas induced apoptosis. The ACPSVs were then purified by differential centrifugation as described and visulised by DIC.
Necrotic Cytotoxicity was induced by treatment of HeLa cells with 1 μg/mL of N-methyl N′-nitro-N-nitrosoguanidine (MNNG) as described (Zong et al., 2004). Briefly, To assess the production of ACPSVs in necrotic cells, 7.5 × 106 indicated cells were grown overnight in 25 mL of indicated media in 150 mm2 flasks (TPP). Cells were washed four times with PBS. Cells were then treated with 1 μg/mL of N-methyl N′-nitro-Nnitrosoguanidine (MNNG) (resuspended in 25 mL of indicated media and antibiotics). Fifteen minutes after MNNG treatment, the cells were washed twice with PBS and grown in indicated media containing antibiotics at 37°C i n the presence of 5% CO2. After 24 h, culture supernatants were collected and subjected to differential centrifugation as discussed above.
Cytotoxicity Assessment by Flow Cytometry was previously described (Goldufsky et al., 2015). Briefly, cells were collected at 24 h following cytotoxic treatments used to yield ACPSVs. Cytotoxicity was assessed using Fixable Viability Dye (eBiosciences) stain to determine the percent cell death in treated and untreated cells. The data were analyzed by flow cytometry (FACSCanto II; BD Biosciences). FACS plots were generated using FlowJo version 8.8.7.
Western Immunoblotting was performed as we described previously (Goldufsky et al., 2015; Wood et al., 2013). Briefly, cells and vesicles were lysed with 1% Triton X-100 containing a protease inhibitor cocktail (Roche Diagnostics) and Halt protease and phosphatase inhibitor (Life Technologies). Lysates were mixed with 4× SDS loading buffer and loaded onto 10% sodium dodecyl sulfate (SDS) polyacrylamide gels. After resolving, gels were transferred to polyvinylidene fluoride (PVDF) membranes, blocked with 5% milk, and probed overnight with primary antibody at 4°C. After washing, blots were probed with HRP-conjugated secondary antibody (Cell Signaling Technologies). Blots were then developed with ECL or ECL+ reagent (GE Healthcare). Films were developed using a film processor (Alphatek). The sources of antibodies (either mouse (Ms), or rabbit (Rb)) used in these studies are as follows: Crk (BD Biosciences; 610036; Ms); GAPDH (Sigma Life Sciences; G9545; Rb). HRP-linked anti-rabbit (Cell Signaling Technology; 7074) or anti-mouse (Cell Signaling Technology; 7076) IgG secondary antibodies were used.
Lipid Measurements
Vesicles lipids were extracted using the lipid quantification kit from Cell Biolabs, Inc. (Cat no. STA-613) and quantified as described previously (Osteikoetxea et al., 2015), using Cytation3 Microplate Reader (BioTek). The lipid content of FBS-containing media was also measured and deducted from the total lipid measurements of 16K fractions to account for the media contribution.
Proliferation Assay
Cells were seeded at indicated densities (e.g., HeLa, HEK, and MEK cells at 6 × 104 cells, kidney podocytes at 1.5 × 104 cells per well) in a 24-well plate. The next day at the time of treatment with vesicles, the initial cell number per well (0 h) was determined by trypsin digestion and cell counts using a hemocytometer. Cells were then treated with ACPSVs and incubated for 24 h at 37°C (for HeLa, HEK, and MEK cells) and 33°C (for podocytes) in the presence of 5% CO2. Proliferation rates were determined blindly by cell counts determination using a hemocytometer.
Scanning Electron Microscopy (SEM) was previously described (Selvaraju et al., 2014) with some modifications described below. Cell and ACPSVs were grown or added on coverslips pretreated with poly-L-lysine and fixed on 4% PFA. The samples were then washed with 0.1 M sodium cacodylate buffer, post-fixed with 1% osmium tetroxide (Agar Scientific, UK), and placed in a modified histology cassette for automated gradient ethanol dehydration (25%-100% alcohol, Lynx II Automated tissue processor, Electron Microscopy Sciences) before they were critical point dryed (Bal-tec850 Critical Point Dryer, Electron Microscopy Sciences). After drying cells in a precise and controlled way, cells were mounted to aluminum stubs with carbon conductive tabs (Ted Pella). Finally, the specimens were gold-coated in a sputter coater (Cressington 108auto Sputter Coater, UK), prior to view under a SEM. Micrographs were collected with the Sigma HDVP electron microscope (Zeiss) via secondary electron detector.
5-ethynyl-2′-deoxyuridine (EdU) Labeling was performed as described previously (Salic and Mitchison, 2008). Briefly, 8 × 104 HeLa cells were seeded on coverslips, pre-treated with poly-L-lysine and human fibronectin (40 μg/mL). The following day cells were transfected as described for 24 h and then treated with 10 μM EdU for 2 h. Cells were fixed with 3.7% formaldehyde for 15 min and permeablized with 0.5% Triton X-100 for 20 min at room temperature. Next, cells were washed twice with 3% BSA in PBS, before treatment with Click-iT Plus reaction cocktail (Molecular Probes) for 30 min at room temperature protected from light. Following reaction, cells were washed once and blocked with 3% BSA for 1 h and then stained with anti-GFP antibody (Abcam; ab5450) overnight followed by anti-goat AF488 secondary antibody (Abcam; ab150129) for 1 h. Coverslips were washed three times with PBS and mounted on slides with ProLong Diamond Antifade Mountant with DAPI (Molecular Probes) and imaged using an AxioVert Z1 fluorescent microscope (Zeiss).
Proteomic Analysis
Vesicles were lysed in lysis buffer (25 mM Tris-HCl, 125 mM NaCl, 5 mM EDTA, 1% Triton X-100, pH 7.4) in the presence of a protease inhibitor mixture. The vesicle peptide content was determined by BCA assay. Filter-aided sample preparation (FASP) (Wisniewski et al., 2009) was applied to extract and digest proteins from vesicles. In brief, 10 μg of sample was diluted 50 times by 8 M urea in 0.1 M Tris/HCl pH 8.5 then filtered with a 0.22 μM membrane (Millipore, Billerica, MA). The flow-through was collected and transferred into a 1.5 mL MicroconYM-10 centrifugal unit (Millipore, Billerica, MA). Protein reduction, alkylation and tryptic digestion were performed step by step in the centrifugal unit, as described (Wisniewski et al., 2009). After overnight digestion at 37°C, the peptides were eluted twice with 150 μL 0.1% formic acid (FA). The concentration of proteins and peptides collected in each step was measured using a Nanodrop 1000 (Thermo Scientific, San Jose, CA). The digested peptides were then aliquoted, dried, and stored at −80°C until further use. Sample were further fractionated using offgel pH 3-10 immobiline dry strips (GE Healthcare, Pittsburgh, PA) into 24 fractions. Fractions were desalted using Nestgroup c18 tips (Southborough, MA). Fractionated peptides were dried and re-dissolved in 0.1% FA for LC–MS/MS analysis. Fractions were run on Thermo Fisher Orbitrap Velos Pro coupled with Agilent NanoLC system (Agilent, Santa Clara, CA) over a 60 min gradient. RAW files were converted into .mgf files using MSConvert (from ProteoWizard). Database search was carried out using Mascot server (from Matrix Science). Search results from 24 runs were imported into Scaffold (Proteome Software, Portland, OR) for quantitative analysis.
Immunofluorescence (IF) static microscopy was performed as described previously (Wood et al., 2011): Briefly, ACPSVs, (16K fraction from serum-starved HeLa cells), were seeded on poly-L-lysine and fibronectin coated coverslips, fixed with 4% PFA for 20 mins, blocked and permeabilized in permeabilization buffer (1× PBS + 5% FBS + 0.3% Triton™ X-100) for 60 mins at room temperature, and treated with primary antibodies for Crk (BD Transduction Laboratories-610035), Calreticulin (Abcamab2907), GAPDH (GenScript-A00191-40), Wnt1 (Abcam-ab85060), TGF-b (Cell signaling 3711), Actinin-4 (Abcam-ab108198), CD9 (Abcam-ab2215), or ALDH1 (Abcam-ab52492), at 1:100 or 1:200 dilutions as recommended by manufacturer in antibody dilution buffer (1× PBS/1% BSA/0.3% Triton™ X-100) overnight at 4°C. After three times wash in 1× PBS for 5 min each, coverslips were then incubated with fluorochrome-conjugated secondary antibody, Anti-mouse alexa 488 or Anit-rabbit alexa 488 procured from Jackson ImmunoResearch, for 2 h at room temperature in the dark. Coverslips were then rinsed with 1 × PBS three times for 5 min each and visualized under Immunofluorescence microscope.
siRNA transfection
For JNK1/2 siRNA tranfection, Hela cells were seeded at 5 × 105 in 24 well plate and transfected using Opti-MEM containing 5 μl/ml Lipofectamine 2000 and 50 nM JNK1/2 or scramble siRNA control (mock) for 24 h. siRNA sequences were decribed previously (Kitanaka et al., 2017). siRNA efficiency was determined by Western blotting using anti JNK antibody (Cell Signaling Technology; 9252).
Animal Model of Experimental Glomerulonephritis (George et al., 2012; Verma et al., 2015) was used to assess the occurrence of CPS in vivo. All procedures were approved by Rush University Medical Center IACUC (No: 15-069) and followed the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. Nephritis was induced by nephrotoxic serum (NTS) injection as described previously (George et al., 2012; Verma et al., 2015). Briefly, 6 to 8 weeks old C57BL6 male mice (Jackson Laboratory) were injected retroorbitally with sheep anti-rat glomerular lysate antiserum (kindly provided by Dr. David Salant, Boston University) or control sheep IgG (Sigma) at a dose of 3 mg per mouse. Urine was collected at base line and on successive days. Urinary albumin and creatinine were measured by using mouse albumin ELISA (Bethyl Labs, E99-134), and creatinine assay (Cayman Chemical, 500701) kits. Albumin-to-Creatinine Ratio (ACR) was calculated and used as a parameter to determine proteinuria. Mice were sacrificed at 6 days after the NTS injection for histological and biochemical analyses.
Processing of glomeruli and isolation of vesicles
For processing of glomeruli and isolation of vesicles, kidneys of sheep IgG (mock)- and NTS-treated mice were minced into 1 to 3-mm tissue fragments with a scalpel blade while being maintained in ice-cold PBS. The tissue was gently pressed through a 100 μm-cell strainer (Falcon) using a flattened pestle, followed by washing with 5 ml of ice-cold HBSS (Life Technologies) to prevent the adverse effects of shear heating. Then, the filtered flow-through was passaged through 40-μm cell strainer without pressing to retain glomeruli and tubules. The flow-through was then subjected to differential centrifugation as described in Methods.
Renal parietal epithelial cells
Mouse renal parietal epithelial cells (PECs) were prepared as described (Ohse et al., 2008) and were cultured in RPMI 1640 (Life Technologies) containing 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies). The media was supplemented with 2% FBS and 0.1 mM sodium pyruvate (Sigma). PECs were cultured at 33°C and 5% CO2 in collagen I (BD Biosciences)-coated flasks in the presence of interferon-γ (Cell Sciences; 50 U/ml for both cells). For proliferation experiments, 25000 cells were seeded to each well of a 24-well plate and cultured at 33°C for 24h. Then, PECs were treated with microvesicles isolated from control and NTS-treated mice, respectively. After 24 h treatment, cells were washed with PBS (Life Technologies), detached by using trypsin-EDTA (Life Technologies) for 5 min at 37°C and harvested by ce ntrifugation at 1200 rpm for 5 min. The cell pellet was re-suspended in the complete medium for counting and analysis.
Histopathological Evaluations were performed as described previously (Dai et al., 2013; Ophascharoensuk et al., 1998). Briefly, freshly harvested mouse kidneys were fixed in 4% PFA (Electron Microscopy Sciences) and embedded in paraffin. The sections were cut at 4 μm thickness and stained with hematoxylin and eosin (H&E), Periodic acid-Schiff (PAS), and Masson's Trichrome. Staining of sections with Ki-67 (Bethyl Laboratories, IHC-00375-T) was used as a marker of cell proliferation. Apoptotic cells were identified by in situ TdT-mediated dUTP nick end labeling (TUNEL) using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore, S7101).
Quantification and Statistical Analysis
Statistical Analyses were performed as described previously (Kroin et al., 2015; Kroin et al., 2016; Wood et al., 2014). For secondary outcomes, preplanned contrasts for between the groups at each timepoint were analyzed using a general linear model structured as an ANOVA with additional post-hoc testing using Tukey multiple comparison adjustment. Covariates were included in the model if necessary. To account for error inflation due to multiple testing, the Bonferroni stepdown adjustment was used. For experiments involving two categorical variables from a single population Chi Square (χ2) analysis was used. All analyses were performed using GraphPad Prism 6.0 software or SAS version 9.3 (SAS Inc. Cary, NC). Statistical significance threshold was set at 0.05.
Supplementary Material
Suppl. Movie 1. Apoptotic compensatory proliferation signaling appears to be mediated by CrkI-containing vesicles. Related to Figure 1. HeLa cells were transiently transfected with CrkI-EGFP expression vector. Video images were captured every 15 min. (A) Shown is a CrkI-GFP transfected apoptotic cell, which prior to becoming propidium iodide (PI) positive (red), forms and releases a CrkI-GFP containing vesicle that appears to induce proliferation in an untransfected neighboring cell upon contact. (B) Shown is a CrkI-GFP transfected apoptotic cell which forms and releases 3 vesicles that appear to induce proliferation in an untransfected neighboring cell upon contact. (C) HeLa cells were transiently transfected with CrkI-EGFP expression vector in the presence of Z-VAD pan-caspase inhibitor. This movie shows that Z-VAD inhibits apoptosis and vesicle formation in two representative CrkI-GFP transfected HeLa cells.
Suppl. Movie 2. Inactivation of CrkI by Pseudomonas aeruginosa ExoT/ADPRT or a null mutation in the SH2 domain of CrkI abrogates vesicle formation in apoptotic cells. Related to Figure 1. HeLa cells were transiently transfected with CrkI/R38K-GFP expression vector (A) or ExoT/ADPRT-GFP expression vector (B). Video images were captured ev. ery 15 min. As expected, transfection with CrkI/R38K-GFP or ExoT/ADPRT-GFP resulted in apoptosis, as indicated by cellular uptake of PI (red). Unlike CrkI-GFP (Movies 1A-B), CrkI/R38K-GFP or ExoT/ADPRT-GFP transfected apoptotic cells are impaired in vesicle production and in inducing CPS in surrounding bystander cells.
Suppl. Movie 3. Exogenous vesicles induce proliferation in other cells. Related to Figure 1. CrkI-containing microvesicles were purified from apoptotic MEK cells. These vesicles were then added to adherent MEK cells. This movie shows a bystander recipient cell that proliferates upon contacting one such vesicle.
Suppl. Table 1: ACPVs Mass spec data (Related to Figure 4).
Highlights.
Apoptotic cells release vesicles that stimulate proliferation in neighboring cells.
These vesicles are distinct from exosomes and apoptotic bodies.
CrkI inactivation inhibits vesicle formation in apoptotic cells and blocks CPS.
Vesicle-induced proliferation requires JNK activity in neighboring cells.
In Brief.
Apoptotic cells can induce proliferation in neighboring cells, a process known as compensatory proliferation. Gupta, Goldufsky, Wood et al. now show that a fraction of apoptotic cells produce and release CrkI-containing microvesicles, distinct from exosomes and apoptotic bodies, that stimulate proliferation in bystander cells upon contact.
Acknowledgments
We would like to thank Dr. Mitchell Denning (Loyola Medicine, Chicago, IL.) for providing us invaluable insights and his generous gift of HEK cells, used in these studies. We would also like to thank Dr. David Salant (Boston University) for providing us with anti-rabbit glomerular basement membrane (GBM) antibody used in animal studies. This work was supported by National Institutes of Health Grant R21 AI110685-01 (to S. H. S.); The Brian Piccolo Cancer Research Fund (to S. H. S.); and The Bears Care Grant 56613 (to S. H. S.).
Footnotes
Conflict of Interest: A US divisional patent has been filed by Rush University Medical Center, based on research data included in this manuscript.
Author Contributions: S. H. S. conceived and coordinated all aspects of the studies and wrote the paper. S. H. S. also contributed to Fig. 1. J. R. conceived and coordinated the in vivo studies. K. H. G., J. W. G., S. J. W., D. G., and J. P. Z. performed and/or contributed to in vitro and in vivo studies. N. J. T., S. J. W., and K. H. G. conducted and analyzed electron microscopy data. E. H., M. M. A, S. J. W., and K. H. G., performed the in vivo studies. All authors had the opportunity to review and edit the manuscript. All authors approved the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113:1–11. doi: 10.1007/s11060-013-1084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida EA, Ilic D, Han Q, Hauck CR, Jin F, Kawakatsu H, Schlaepfer DD, Damsky CH. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J Cell Biol. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137:1169–1179. doi: 10.1242/dev.045559. [DOI] [PubMed] [Google Scholar]
- Bouchard V, Harnois C, Demers MJ, Thibodeau S, Laquerre V, Gauthier R, Vezina A, Noel D, Fujita N, Tsuruo T, et al. B1 integrin/Fak/Src signaling in intestinal epithelial crypt cell survival: integration of complex regulatory mechanisms. Apoptosis: an international journal on programmed cell death. 2008;13:531–542. doi: 10.1007/s10495-008-0192-y. [DOI] [PubMed] [Google Scholar]
- Braun F, Bertin-Ciftci J, Gallouet AS, Millour J, Juin P. Serum-nutrient starvation induces cell death mediated by Bax and Puma that is counteracted by p21 and unmasked by Bcl-x(L) inhibition. PloS one. 2011;6:e23577. doi: 10.1371/journal.pone.0023577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochemical pharmacology. 2008;76:1352–1364. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Chitteti BR, Kobayashi M, Cheng Y, Zhang H, Poteat BA, Broxmeyer HE, Pelus LM, Hanenberg H, Zollman A, Kamocka MM, et al. CD166 regulates human and murine hematopoietic stem cells and the hematopoietic niche. Blood. 2014;124:519–529. doi: 10.1182/blood-2014-03-565721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Cummings J, Ward TH, Ranson M, Dive C. Apoptosis pathway-targeted drugs--from the bench to the clinic. Biochimica et biophysica acta. 2004;1705:53–66. doi: 10.1016/j.bbcan.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Dai Y, Gu L, Yuan W, Yu Q, Ni Z, Ross MJ, Kaufman L, Xiong H, Salant DJ, He JC. Podocyte-specific deletion of signal transducer and activator of transcription 3 attenuates nephrotoxic serum–induced glomerulonephritis. Kidney international. 2013;84:950–961. doi: 10.1038/ki.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Darby IA, Bisucci T, Hewitson TD, MacLellan DG. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. The international journal of biochemistry & cell biology. 1997;29:191–200. doi: 10.1016/s1357-2725(96)00131-8. [DOI] [PubMed] [Google Scholar]
- Dekel B. The Ever-Expanding Kidney Repair Shop. J Am Soc Nephrol. 2016;27:1579–1581. doi: 10.1681/ASN.2015111207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Sun J, Barbieri JT. Uncoupling Crk signal transduction by Pseudomonas exoenzyme T. The Journal of biological chemistry. 2005;280:35953–35960. doi: 10.1074/jbc.M504901200. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicologic pathology. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol. 2008a;18:467–473. doi: 10.1016/j.tcb.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008b;14:399–410. doi: 10.1016/j.devcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Wang S, Hernandez J, Yenigun VB, Hertlein G, Fogarty CE, Lindblad JL, Bergmann A. Genetic models of apoptosis-induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet. 2014;10:e1004131. doi: 10.1371/journal.pgen.1004131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Screaton RA. Anoikis mechanisms. Current opinion in cell biology. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–344. doi: 10.1038/nrm3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George B, Verma R, Soofi AA, Garg P, Zhang J, Park TJ, Giardino L, Ryzhova L, Johnstone DB, Wong H, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. The Journal of clinical investigation. 2012;122:674–692. doi: 10.1172/JCI60070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldufsky J, Wood S, Hajihossainlou B, Rehman T, Majdobeh O, Kaufman HL, Ruby CE, Shafikhani SH. Pseudomonas aeruginosa exotoxin T induces potent cytotoxicity against a variety of murine and human cancer cell lines. Journal of medical microbiology. 2015;64:164–173. doi: 10.1099/jmm.0.000003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–298. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehenberger K, Heilborn J, Brismar K, Hansson A. Increased lactate production in chronic diabetic wound fibroblasts showing decreased cellular proliferation. Diabetologia. 1997;40:A466. [Google Scholar]
- Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1998;6:135–141. doi: 10.1046/j.1524-475x.1998.60207.x. [DOI] [PubMed] [Google Scholar]
- Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Current biology: CB. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Karlsson G, Rorby E, Pina C, Soneji S, Reckzeh K, Miharada K, Karlsson C, Guo Y, Fugazza C, Gupta R, et al. The tetraspanin CD9 affords high-purity capture of all murine hematopoietic stem cells. Cell Rep. 2013;4:642–648. doi: 10.1016/j.celrep.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Kastelowitz N, Yin H. Exosomes and microvesicles: identification and targeting by particle size and lipid chemical probes. Chembiochem. 2014;15:923–928. doi: 10.1002/cbic.201400043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka T, Nakano R, Kitanaka N, Kimura T, Okabayashi K, Narita T, Sugiya H. JNK activation is essential for activation of MEK/ERK signaling in IL-1beta-induced COX-2 expression in synovial fibroblasts. Sci Rep. 2017;7:39914. doi: 10.1038/srep39914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26:7258–7268. doi: 10.1128/MCB.00183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell death and differentiation. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroin JS, Buvanendran A, Li J, Moric M, Im HJ, Tuman KJ, Shafikhani SH. Short-Term Glycemic Control Is Effective in Reducing Surgical Site Infection in Diabetic Rats. Anesthesia and analgesia. 2015 doi: 10.1213/ANE.0000000000000650. [DOI] [PubMed] [Google Scholar]
- Kroin JS, Li J, Goldufsky JW, Gupta K, Moghtaderi M, Buvanendran A, Shafikhani SH. Perioperative high inspired oxygen fraction therapy reduces surgical site infection with Pseudomonas aeruginosa in rats. Journal of medical microbiology. 2016 doi: 10.1099/jmm.0.000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44:205–221. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lamorte L, Royal I, Naujokas M, Park M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell. 2002;13:1449–1461. doi: 10.1091/mbc.01-10-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Wu SB, Hong CH, Yu HS, Wei YH. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int J Mol Sci. 2013;14:6414–6435. doi: 10.3390/ijms14036414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125–134. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. Apoptotic cells activate the “ phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Kohli MR, Yu Q, Kim S, Qu T, He WX. Biodentine Induces Human Dental Pulp Stem Cell Differentiation through Mitogen-activated Protein Kinase and Calcium-/Calmodulin-dependent Protein Kinase II Pathways. J Endod. 2014;40:937–942. doi: 10.1016/j.joen.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Mansbridge JN, Liu K, Pinney RE, Patch R, Ratcliffe A, Naughton GK. Growth factors secreted by fibroblasts: role in healing diabetic foot ulcers. Diabetes, obesity & metabolism. 1999;1:265–279. doi: 10.1046/j.1463-1326.1999.00032.x. [DOI] [PubMed] [Google Scholar]
- Michael A, Bajracharya SD, Yuen PS, Zhou H, Star RA, Illei GG, Alevizos I. Exosomes from human saliva as a source of microRNA biomarkers. Oral diseases. 2010;16:34–38. doi: 10.1111/j.1601-0825.2009.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D'Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Current biology: CB. 2009;19:1875–1885. doi: 10.1016/j.cub.2009.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. Journal of cell science. 2010;123:1603–1611. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohse T, Pippin JW, Vaughan MR, Brinkkoetter PT, Krofft RD, Shankland SJ. Establishment of conditionally immortalized mouse glomerular parietal epithelial cells in culture. J Am Soc Nephrol. 2008;19:1879–1890. doi: 10.1681/ASN.2007101087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophascharoensuk V, Pippin JW, Gordon KL, Shankland SJ, Couser WG, Johnson RJ. Role of intrinsic renal cells versus infiltrating cells in glomerular crescent formation. Kidney Int. 1998;54:416–425. doi: 10.1046/j.1523-1755.1998.00003.x. [DOI] [PubMed] [Google Scholar]
- Osteikoetxea X, Balogh A, Szabo-Taylor K, Nemeth A, Szabo TG, Paloczi K, Sodar B, Kittel A, Gyorgy B, Pallinger E, et al. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PloS one. 2015;10:e0121184. doi: 10.1371/journal.pone.0121184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace E, Melis M, Siena L, Bucchieri F, Vignola AM, Profita M, Gjomarkaj M, Bonsignore G. Effects of gemcitabine on cell proliferation and apoptosis in non-small-cell lung cancer (NSCLC) cell lines. Cancer chemotherapy and pharmacology. 2000;46:467–476. doi: 10.1007/s002800000183. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–1177. doi: 10.1242/dev.034017. [DOI] [PubMed] [Google Scholar]
- Pirrone A, Hager B, Fleckman P. Epidermal Cells : Methods and Protocols. 2004:3–14. [Google Scholar]
- Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell death and differentiation. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, Grant S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–5513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3:388–398. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M, Cinatl J. Fas/FasL interaction: a novel immune therapy approach with immobilized biologicals. Medicinal research reviews. 2005;25:331–342. doi: 10.1002/med.20025. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- Selvaraju TR, Khaza'ai H, Vidyadaran S, Abd Mutalib MS, Vasudevan R. The neuroprotective effects of tocotrienol rich fraction and alpha tocopherol against glutamate injury in astrocytes. Bosn J Basic Med Sci. 2014;14:195–204. doi: 10.17305/bjbms.2014.4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani SH, Engel J. Pseudomonas aeruginosa type III-secreted toxin ExoT inhibits host-cell division by targeting cytokinesis at multiple steps. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15605–15610. doi: 10.1073/pnas.0605949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani SH, Morales C, Engel J. The Pseudomonas aeruginosa type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cellular microbiology. 2008a;10:994–1007. doi: 10.1111/j.1462-5822.2007.01102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafikhani SH, Mostov K, Engel J. Focal adhesion components are essential for mammalian cell cytokinesis. Cell Cycle. 2008b;7:2868–2876. doi: 10.4161/cc.7.18.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro SG, Silvestri GA. One hundred years of lung cancer. Am J Respir Crit Care Med. 2005;172:523–529. doi: 10.1164/rccm.200504-531OE. [DOI] [PubMed] [Google Scholar]
- Sun J, Barbieri JT. Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. The Journal of biological chemistry. 2003;278:32794–32800. doi: 10.1074/jbc.M304290200. [DOI] [PubMed] [Google Scholar]
- Sundin C, Hallberg B, Forsberg A. ADP-ribosylation by exoenzyme T of Pseudomonas aeruginosa induces an irreversible effect on the host cell cytoskeleton in vivo. FEMS Microbiol Lett. 2004;234:87–91. doi: 10.1016/j.femsle.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012;56:293–304. doi: 10.1016/j.ymeth.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Vachon PH. Integrin signaling, cell survival, and anoikis: distinctions, differences, and differentiation. Journal of signal transduction. 2011;2011:738137. doi: 10.1155/2011/738137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega YA, Okano H, Lozano G. The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell death and differentiation. 2008;15:1772–1781. doi: 10.1038/cdd.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Venkatareddy M, Kalinowski A, Patel SR, Salant DJ, Garg P. Shp2 Associates with and Enhances Nephrin Tyrosine Phosphorylation and Is Necessary for Foot Process Spreading in Mouse Models of Podocyte Injury. Mol Cell Biol. 2015;36:596–614. doi: 10.1128/MCB.00956-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring P, Mullbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999;77:312–317. doi: 10.1046/j.1440-1711.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- Wei L, Yang Y, Zhang X, Yu Q. Cleavage of p130Cas in anoikis. J Cell Biochem. 2004;91:325–335. doi: 10.1002/jcb.10760. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009;6:359. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wood S, Goldufsky J, Shafikhani SH. Pseudomonas aeruginosa ExoT Induces Atypical Anoikis Apoptosis in Target Host Cells by Transforming Crk Adaptor Protein into a Cytotoxin. PLoS pathogens. 2015a;11:e1004934. doi: 10.1371/journal.ppat.1004934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S, Jayaraman V, Huelsmann EJ, Bonish B, Burgad D, Sivaramakrishnan G, Qin S, Dipietro LA, Zloza A, Zhang C, et al. Pro-inflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response. PloS one. 2014;9:e91574. doi: 10.1371/journal.pone.0091574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S, Pithadia R, Rehman T, Zhang L, Plichta J, Radek KA, Forsyth C, Keshavarzian A, Shafikhani SH. Chronic alcohol exposure renders epithelial cells vulnerable to bacterial infection. PloS one. 2013;8:e54646. doi: 10.1371/journal.pone.0054646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S, Sivaramakrishnan G, Engel J, Shafikhani SH. Cell migration regulates the kinetics of cytokinesis. Cell Cycle. 2011;10:648–654. doi: 10.4161/cc.10.4.14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Goldufsky JW, Bello D, Masood S, Shafikhani SH. Pseudomonas aeruginosa ExoT induces mitochondrial apoptosis in target host cells in a manner that depends on its GAP domain activity. The Journal of biological chemistry. 2015b doi: 10.1074/jbc.M115.689950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. The Journal of experimental medicine. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YF, Xu J, Wang WJ, Wang J, He JW, Li L, Dong Q, Xiao Y, Duan XL, Yang X, et al. Activation of JNKs is essential for BMP9-induced osteogenic differentiation of mesenchymal stem cells. BMB Rep. 2013;46:422–427. doi: 10.5483/BMBRep.2013.46.8.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Movie 1. Apoptotic compensatory proliferation signaling appears to be mediated by CrkI-containing vesicles. Related to Figure 1. HeLa cells were transiently transfected with CrkI-EGFP expression vector. Video images were captured every 15 min. (A) Shown is a CrkI-GFP transfected apoptotic cell, which prior to becoming propidium iodide (PI) positive (red), forms and releases a CrkI-GFP containing vesicle that appears to induce proliferation in an untransfected neighboring cell upon contact. (B) Shown is a CrkI-GFP transfected apoptotic cell which forms and releases 3 vesicles that appear to induce proliferation in an untransfected neighboring cell upon contact. (C) HeLa cells were transiently transfected with CrkI-EGFP expression vector in the presence of Z-VAD pan-caspase inhibitor. This movie shows that Z-VAD inhibits apoptosis and vesicle formation in two representative CrkI-GFP transfected HeLa cells.
Suppl. Movie 2. Inactivation of CrkI by Pseudomonas aeruginosa ExoT/ADPRT or a null mutation in the SH2 domain of CrkI abrogates vesicle formation in apoptotic cells. Related to Figure 1. HeLa cells were transiently transfected with CrkI/R38K-GFP expression vector (A) or ExoT/ADPRT-GFP expression vector (B). Video images were captured ev. ery 15 min. As expected, transfection with CrkI/R38K-GFP or ExoT/ADPRT-GFP resulted in apoptosis, as indicated by cellular uptake of PI (red). Unlike CrkI-GFP (Movies 1A-B), CrkI/R38K-GFP or ExoT/ADPRT-GFP transfected apoptotic cells are impaired in vesicle production and in inducing CPS in surrounding bystander cells.
Suppl. Movie 3. Exogenous vesicles induce proliferation in other cells. Related to Figure 1. CrkI-containing microvesicles were purified from apoptotic MEK cells. These vesicles were then added to adherent MEK cells. This movie shows a bystander recipient cell that proliferates upon contacting one such vesicle.
Suppl. Table 1: ACPVs Mass spec data (Related to Figure 4).