

Abstract
Mesenchymal stem/stromal cells (MSCs) are the predominant source of bone and adipose tissue in adult bone marrow and play a critical role in skeletal homeostasis. Age‐induced changes in bone marrow favor adipogenesis over osteogenesis leading to skeletal involution and increased marrow adiposity so pathways that prevent MSC aging are potential therapeutic targets for treating age‐related bone diseases. Here, we show that inositol hexakisphosphate kinase 1 (Ip6k1) deletion in mice increases MSC yields from marrow and enhances cell growth and survival ex vivo. In response to the appropriate stimuli, Ip6k1 −/− versus Ip6k1+/+ MSCs also exhibit enhanced osteogenesis and hematopoiesis‐supporting activity and reduced adipogenic differentiation. Mechanistic‐based studies revealed that Ip6k1 −/− MSCs express higher MDM2 and lower p53 protein levels resulting in lower intrinsic mitochondrial reactive oxygen species (ROS) levels as compared to Ip6k1+/+ MSCs, but both populations upregulate mitochondrial ROS to similar extents in response to oxygen‐induced stress. Finally, we show that mice fed a high fat diet exhibit reduced trabecular bone volume, and that pharmacological inhibition of IP6K1 using a pan‐IP6K inhibitor largely reversed this phenotype while increasing MSC yields from bone marrow. Together, these findings reveal an important role for IP6K1 in regulating MSC fitness and differentiation fate. Unlike therapeutic interventions that target peroxisome proliferator‐activated receptor gamma and leptin receptor activity, which yield detrimental side effects including increased fracture risk and altered feeding behavior, respectively, inhibition of IP6K1 maintains insulin sensitivity and prevents obesity while preserving bone integrity. Therefore, IP6K1 inhibitors may represent more effective insulin sensitizers due to their bone sparing properties. Stem Cells 2017;35:1973–1983
Keywords: Mesenchymal stem cells, Mesenchymal stromal cells, Inositol hexakisphosphate kinase 1, High fat diet, Adipogenesis, Osteogenesis, Bone
Significance Statement.
This study is the first to identify a role for inositol hexakisphosphate kinase 1 (IP6K1) in regulating primary mouse mesenchymal stem/stromal cell (MSC) fitness. Our data show that genetic deletion of Ip6k1 results in increased MSC yields from bone marrow, and enhances the growth, survival, hematopoiesis‐supporting activity, and osteogenic potential of culture‐expanded cells. Increased fitness correlated with lower intrinsic p53 protein expression and mitochondrial reactive oxygen species levels, consistent with the known sensitivity of MSCs to oxidative stress. Furthermore, pharmacological inhibition of IP6K1 in mice reversed high fat diet (HFD)‐induced decreases in bone volume. Together, these studies indicate that IP6K1 influences MSC frequency and function in bone marrow by modulating responsiveness to systemic changes induced by HFD feeding, and identify IP6K1 as a therapeutic target to prevent skeletal involution in response to these conditions.
Introduction
Mesenchymal stem cells (MSCs) are multipotent stem/progenitors first identified in bone marrow based on their ability to generate heterotopic osseous tissue with an architecture capable of supporting hematopoiesis when serially transplanted into mice (reviewed in Ref. 1). Subsequently, MSCs have been shown to localize adjacent to blood vessels in vivo, which has led to their characterization as vascular pericytes or perivascular stromal cells 2. Most recently, several groups have identified specific MSC subsets within bone marrow that comprise the hematopoietic stem cell niche and regulate hematopoietic stem cell self‐renewal and differentiation 3, 4, 5, 6. Included among the latter is a population of leptin receptor (LEPR) expressing perivascular cells, which are the principle source of chemokines that regulate hematopoietic stem cell self‐renewal and retention in bone marrow 7. Importantly, LEPR‐MSCs also represent the main source of osteoblasts and adipocytes that form bone and fat within adult bone marrow 8, and recent studies indicate that the LEPR acts as a systemic energy sensor in MSCs promoting adipogenesis and inhibiting osteogenesis in response to high fat diet (HFD) feeding 9. Therefore, MSC fate determination in marrow is modulated by nutritional status as a consequence of LEP/LEPR signaling. However, LEP/LEPR signaling is also involved in satiety. Therefore, targeting LER/LEPR signaling to reverse HFD‐induced trabecular bone loss and increased marrow adiposity is not feasible. Similarly, peroxisome proliferator‐activated receptor gamma (PPARG) agonists like rosiglitazone used to treat type II diabetes inhibit insulin resistance but induce osteoporosis 10. Therefore, a novel drug target that can reverse HFD‐induced weight gain and insulin resistance while preserving bone integrity remains to be identified.
Recent studies indicate that mice with a germline deletion of inositol hexakisphosphate kinase 1 (Ip6k1), the major isoform of three known inositol hexakisphosphate kinases 11, are protected against HFD‐induced weight gain and insulin resistance 12, 13, 14. A family of three mammalian IP6 kinases (IP6Ks) primarily generates the inositol pyrophosphate 5‐IP7 from IP6 15, 16, 17, 18, 19, 20. However, at a lower ATP/ADP ratio, IP6Ks dephosphorylate IP6 to a distinct form of IP5 21. IP6 and 5‐IP7 bind protein targets to modulate their functions 15, 16, 17. In addition, inositol pyrophosphate‐mediated pyrophosphorylation alters functions of target proteins 15, 16. HFD‐fed Ip6k1 −/− mice maintain insulin sensitivity by sustaining activity of the insulin sensitizing protein kinase AKT 12. Moreover, IP6K1 reduces adipose tissue browning mediated thermogenesis by inhibiting AMPK (AMP stimulated protein kinase) activity 13. Accordingly, the pan IP6K inhibitor TNP [N2‐(m‐Trifluorobenzyl), N6‐(p‐nitrobenzyl)purine] 22 reduces body weight and insulin resistance in diet induced obese mice via AMPK and AKT activation 13, 23. Although a previous study demonstrated that TNP‐mediated AKT activation protects aged MSCs from hypoxic injury 24 the precise function of IP6K1 in MSCs remains indeterminate. Since age‐dependent increases in cellular oxidative stress are strongly linked to skeletal aging and have been shown to negatively impact MSC proliferation, survival and differentiation by favoring adipogenesis at the expense of osteogenesis 25, 26, we questioned if IP6K1 may function in driving skeletal aging. Results from genetic and pharmacological studies demonstrate a role for IP6K1 in promoting an aging phenotype in MSCs, which is largely reversed by pharmacological inhibition of this kinase. Since the latter also preserves bone integrity in response to HFD feeding, these results identify IP6K1 as an attractive drug target for preventing obesity, insulin resistance, and osteoporosis.
Materials and Methods
MSC Isolation, Transfection, and Differentiation
MSCs were isolated from Ip6k1 −/− and congenic wild type mice via immunodepletion as described previously 27. Briefly, whole bone marrow obtained from the long bones of mice was suspended in complete culture media (CCM; α minimal essential media containing l‐glutamine and supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin), plated at a density of 1.4 × 106 cells per square centimeter, and cultured for 72 hours after which the non‐adherent cells were removed by aspiration. Adherent cells were cultured an additional 5–7 days with a single media change and then harvested by gentle scraping after a 5‐minutes incubation in 0.25% trypsin with 1 mM EDTA. The cell pellet was dispersed by gentle agitation, suspended in 20 ml of Hank's balanced salt solution (HBBS), and filtered through a 70‐μm filter. Cells were suspended in HBSS at a maximum density of 40 × 106 cells per milliliter and incubated on a rotator for 1 hour at 4°C, then successively for 50 minutes intervals with streptavidin‐conjugated Dynabeads (five beads per cell) (ThermoFisher Scientific, Waltham, MA, www.thermofisher.com) conjugated to anti‐CD11b, anti‐CD34, and anti‐CD45 antibodies (10 μg/mg beads) (BD Biosciences, San Jose, CA, www. Bdbiosciences.com). All cell isolation and culture manipulations were performed in a modular airtight chamber (BioSpherix Ltd., Lacona, NY, www.biospherix.com/) flushed with 5% O2 balanced with N2. MSCs were cultured at 37°C in a humidified chamber with 5% CO2 and 5% O2 unless stated otherwise. The total yield of MSCs per mouse was calculated based on the total number of MSCs obtained following immunodepletion. Population doubling (PD) times were calculated as PD = t log2/(log Nt – log No) where t is time period, Nt is the number of cells at time t, and No is the initial number of cells plated. Cumulative cell numbers and total PDs for each passage were determined from initial plating densities post‐immunodepletion. Delivery of siRNA into MSCs was accomplished using the Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific). Briefly, transfection reagent was prepared by mixing Lipofectamine RNAiMAX (1:2,000) and the appropriate RNAi duplex (10nM, Thermo Fisher Scientific) in Opti‐MEM® Reduced Serum Medium (1:5). MSCs (10,000 cells per square centimeter) were plated onto the transfection reagent, cultures were fed with fresh media 2 days later, and every 2–3 days thereafter for up to 7 days. Cultures were photographed using a Leica DMI3000 B upright fluorescent microscope attached to a DFC295 digital camera (Micro Optics of Florida, Inc., Davie, FL, www.microopticsfl.com).
Adipogenic and osteogenic potential of MSCs (1 × 104 cells per square centimeter) was quantified as described previously 28 with the following exceptions. Adipogenic induction media consisted of CCM supplemented with 0.5 μM dexamethasone, 0.5 mM isobutylxanthine, and 50 μM indomethacin, and lipid accumulation was quantified by staining cells with AdipoRed Assay Reagent (Lonza Rockland, Inc., Rockland, ME, www.lonza.com) for 10 minutes and quantifying fluorescence using a fluorescent plate reader (excitation 485 nm; emission 535). In all cases, AdipoRed and Alizarin Red S staining was normalized to cell number quantified by 4′,6‐diamidino‐2‐phenylindole staining.
Colony Assays
To quantify CFU‐Fs, MSCs (10,000 cells) were plated in 100 mm dishes and were cultured for 7–10 days with media changes every 2–3 days. On day 7, the monolayers were washed with phosphate‐buffered saline, stained with 0.5% crystal violet (Sigma‐Aldrich, St. Louis, MO, www.sigmaaldrich.com) in 25% methanol for 10 minutes at room temperature, and colonies ≥ 0.01 square centimeter in area were counted and their total area quantified using ImagJ software. To perform standard colonies assays for hematopoiesis, conditioned media (serum free) was collected over a 24‐hour period from confluent monolayers of wild type and Ip6k1 −/− MSCs, filtered, then used to suspend whole bone marrow cells isolated from wild type mice. The cells were then mixed with growth factor supplemented MethoCult media (StemCell Technologies, Vancouver, BC, www.stemcell.com) and plated in 60 mm dishes at 100,000 cells per dish. The plates were incubated 37°C in a humidified chamber in 5% CO2 and 5% O2 balanced with N2 for 8–10 days after which colonies were enumerated based on morphology.
Flow Cytometry
MSCs (2.5 × 105 cells per milliliter) were suspended in HBSS and incubated with antibodies against CD11b, CD19, CD29, CD31, CD34, CD44, CD45, CD73, CD105, CD106, CD146, CD166, SCA1, F4/80, SSEA‐1, and SSEA‐4 (R&D Systems Inc., Minneapolis, MN, www.rndsystems.com) for 30 minutes at 4°C. Cells were washed twice with HBSS, incubated with the appropriate fluorochrome‐conjugated secondary antibody for 30 minutes at 4°C, and analyzed using an Epics FC 500 FACS scanner equipped with CXP software (Beckman Coulter, Inc., Indianapolis, IN, www.beckmancoulter.com). Alternatively, MSCs were stained with MitoSOX Red (Thermo Fisher Scientific) to quantify mitochondrial superoxide production. For microscopic analysis, 10 µM MitoSOX Red was used while 100 nM was used for fluorescent quantification. To confirm mitochondrial localization of the dye, cells were counterstained with 5 µM Mito‐Tracker Green (Thermo Fisher Scientific) for 10 minutes. Normalization of fluorescent data was carried out using counterstaining with Hoechst 33342 (1 µM) for 5 minutes. Fluorescent wavelength pairs for the individual dyes were 510 nm/580 nm for MitoSOX Red, and 490 nm/516 nm for Mito‐Tracker Green.
RNA Sequencing
Total RNA was quantified on a Qubit 2.0 Fluorimeter (Thermo Fisher Scientific) and run on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, www.agilent.com) for quality assessment. Samples with an RNA integrity number > 8.0 were advanced for further analysis. Briefly, mRNA was isolated from total RNA (100–200 ng input) using poly‐dT oligos attached to magnetic beads, and the purified mRNA processed using the TruSeq Stranded mRNA sample prep kit (Illumina, San Diego, CA, www.illumina.com). RNA samples were chemically fragmented in buffer containing divalent cations at 94°C for 8 minutes, primed with random hexamers, and reverse transcribed to generate the first strand cDNA. The second strand was synthesized after removing the RNA template and incorporating dUTP in place of dTTP. The ds cDNA was then repaired and adenylated at its 3′‐ends. Adaptor‐ligated DNA was purified using magnetic Ampure XP beads and polymerase chain reaction (PCR) amplified using 13 cycles to generate the final libraries, which were size selected and purified using 1.0× Ampure XP beads to remove any primer dimers. The final library size was typically 200–600 bp with insert sizes ranging from 80 to 450 bp. Libraries were validated using bioanalyzer DNA chips and quantitative PCR (qPCR) using primers that recognize the adaptors, pooled at equimolar ratios, loaded onto a NextSeq 500 flow cell at 1.8 pM final concentration, and sequenced using 2 × 75 bp paired‐end chemistry. On average 18–20 million reads are generated (base quality score >Q30 suggesting less than 1 error in 1,000 bp). Data (fastq) generated from the NextSeq 500 were trimmed using Flexbar 2.4 and aligned to the Human reference database (Hg19) using TopHat version 2.0.9 29. HTseq‐count version 0.6.1 was used to generate gene counts and differential gene expression analysis was performed using Deseq2 30.
Quantitative Polymerase Chain Reaction
Total RNA was isolated using the RNeasy Kit (Qiagen, Valencia, CA, www.qiagen.com), converted to cDNA using the Transcriptor First Strand cDNA Synthesis Kit (F. Hoffmann‐La Roche, Basel, Switzerland, www.roche.com), and amplified by PCR using the TaqMan® EZ RT‐PCR kit (Applied Biosystems, Carlsbad, CA, www.appliedbiosystems.com) according to the manufacturer's instructions. Reactions were performed on a 7900 HT sequence detector (Applied Biosystems) using the FastStart Universal SYBR Green Master Mix (F. Hoffman‐La Roche) and transcript levels quantified using the relative Ct method with Gapdh as an internal control using the following primer pairs: Ip6k2, 5′‐ggaccatggaactgtgaatatag‐3′ and 5′‐cccagtccttgggattcttt‐3′; Ip6k3, 5′‐cgctggtggaaagtgaagat‐3′ and 5′‐tctcttgtcctctgggtactg‐3′; and Gapdh, 5′‐tcaacagcaactcccactcttcca‐3′ and 5′‐accctgttgctgtagccgtattca‐3′.
Western Blot
Protein lysates were prepared using the Qproteome Mammalian Protein Prep Kit (Qiagen) and protein concentrations were determined using the Bradford assay (Bio‐Rad, Hercules, CA, www.bio-rad.com). Protein samples (20 µg) were prepared in Laemmli sample buffer (Bio‐Rad) containing 5% β‐mercaptoethanol, denatured at 95°C for 5 minutes, electrophoresed on NuPAGE 10% Bis‐Tris gels using 1× NuPAGE MES SDS Running Buffer (Thermo Fisher Scientific), and then transferred to 0.45 μm nitrocellulose membranes in 1× NuPAGE transfer buffer containing 10% methanol. Membranes were washed with Tris‐buffered saline (TBS) for 5 minutes, incubated in TBS with 0.1% Tween‐20 (TBST) and Odyssey® blocking buffer (LI‐COR Biosciences, Lincoln, NE, www.licor.com) overnight at 4°C, washed an additional 3× in TBST, and then incubated with anti‐IP6K1(1:500, GeneTex, Irvine, CA, www.genetex.com), anti‐p53, anti‐MDM2, anti‐p16, and anti‐GAPDH (1:200, Santa Cruz Biotechnology Inc., Dallas TX, www.scbt.com) antibodies in Odyssey® blocking buffer for 2 hours at room temperature with gentle agitation. Membranes were washed 5× in TBST and probed with a Infrared (IR) dye‐labeled secondary antibody at a 1:10,000 dilution in Odyssey® blocking buffer for 1 hour. Blots were scanned using Odyssey® infrared image system (LI‐COR Biosciences).
Chemokine Quantification
Confluent monolayer cultures of Ip6k1 −/− and wild type congenic MSCs were maintained in serum‐free media for 24 hours, the conditioned media collected, and concentrated 50‐fold using Amicon Ultra‐15 centrifugal filters (Millipore, Sigma, Billerica, MA, www.emdmillipore.com). Media was then analyzed using the Mouse Pre‐mixed Multi‐Analyte Kit (R&D Systems) for simultaneous detection of the indicated analytes. Quantification of analyte abundance was performed using a Luminex 200 dual‐laser, flow‐based detection platform (Luminex Corp., Austin, TX, www.luminexcorp.com). Alternatively, media was analyzed using the mouse SCF ELISA Kit per the manufacturer's instructions (Thermo Fisher Scientific).
TNP Administration In Vivo
Diet induce obesity was generated in C57BL/6 mice by feeding a HFD for 8‐weeks as described previously 23. Subsequently, TNP was injected (10 mg/kg BW; daily) for 10 weeks during which time mice were maintained on the HFD 23. Glucose tolerance was assessed after 5‐weeks of TNP treatment and published elsewhere 23. After 10‐weeks, mice were killed and bones were harvested for MSC isolation and histology.
Histology
Long bones were fixed in 4% neutral buffered formalin overnight followed by decalcification in 10% EDTA for 7 days and dehydration in 30% sucrose for 2 days. Bones were sectioned using a CryoJane tape‐transfer system. Longitudinal sections of bones were stained with H&E/Safranin‐O for qualitative analysis of bone. Pictures were acquired using a Leica DMI3000 B inverted microscope attached with a Leica DFC295 camera and LAS V3.6 software (Micro Optics of Florida, Inc.). The area of bone and bone marrow was quantified in 11 histological sections from vehicle treated mice (n = 6) and 10 sections from TNP treated mice (n = 6) using Fiji software. Cartilage containing growth plates and joints visualized by Safranin‐O staining were excluded from the analysis.
Statistical Analysis
All data were expressed as mean ± SD. Data from experiments with two groups were compared using the Student's t test and data from three or more groups were compared using analysis of variance (ANOVA). Differences between treatment groups were considered significant if the p value was <.05.
Results
Ip6k1 −/− MSCs Exhibit Enhanced Growth and Survival Resulting in Increased Yields from Bone Marrow
To predict whether deletion of Ip6k1 impacts MSC function, we first demonstrated by RNA sequencing that Ip6k1 is the major isoform of three known family members (Ip6k1–3) detected in primary MSCs isolated from the bone marrow of wild type mice (Fig. 1A). Moreover, we confirmed by qPCR that genetic deletion of Ip6k1 did not significantly alter expressed levels of Ip6k2 and Ip6k3 in wild type MSCs (Fig. 1B). Subsequently, we found that marrow from the long bones of Ip6k1 −/− mice yielded significantly more MSCs on a per mouse basis following enrichment by immunodepletion as compared to age‐matched, wild type littermates (Fig. 1C), and that yields of Ip6k1 −/− versus Ip6k1+/+ MSCs were also significantly higher after short term (1 week) culture expansion based on cell counting (Fig. 1D). Consistent with these results, Ip6k1 −/− versus Ip6k1+/+ MSCs underwent significantly more PDs (Fig. 1E) following long‐term (1 month) culture resulting in a ∼40‐fold increase in total cell yield over three passages (Fig. 1F). Two‐way ANOVA revealed a significant effect of genotype [F(1,8)=160.63, p < .0001] and passage number [F(3,8)=42.89, p < .0001] on the number of PDs. Although MSC yields following immunodepletion were lower in aged (18 months old) versus young (2 months old) Ip6k1+/+ mice (Fig. 1C, 1G), overall MSC yields were significantly higher from aged Ip6k1 −/− mice as compared with aged‐matched Ip6k1+/+ littermates (Fig. 1G). MSCs from Ip6k1 −/− versus Ip6k1+/+ aged mice also exhibited enhanced growth in short‐term culture (Fig. 1H). Despite differences in growth and survival, staining with a panel of established cell surface markers failed to detect significant differences in the phenotype of Ip6k1+/+ versus Ip6k1 −/− MSCs (Supporting Information Fig. S1). Together these data suggest that Ip6k1 deletion enhances the overall fitness of primary MSCs from bone marrow.
Figure 1.
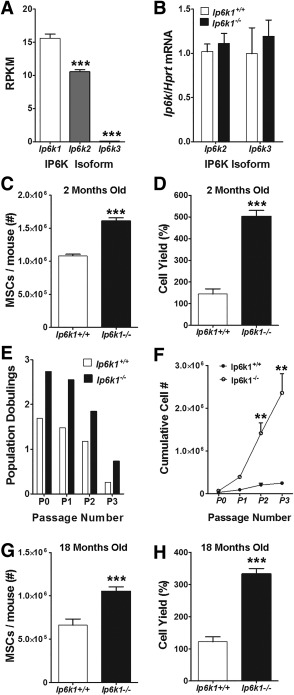
Deletion of inositol hexakisphosphate kinase 1 (Ip6k1) enhances yield and proliferative potential of primary, marrow‐derived mouse mesenchymal stem/stromal cells (MSCs). (A): RNA‐Seq analysis of Ip6k isoforms in mouse MSCs. Note that Ip6k1 is the predominate isoform. (B): Quantitative polymerase chain reaction analysis of Ip6k2 and Ip6k3 in Ip6k1 −/− MSCs. (C, G): Total yield of marrow‐derived MSCs following immunodepletion from young (2 months, C) versus old mice (18 months, G). (D, H): Yield of MSCs (percentage of initially plated cells) from young (D) and old (H) mice after 7 days of culture expansion. (E, F): Total population doublings (E) and cumulative cell yield (F) of immunodepleted MSCs from 2 months old wild type and Ip6k1 −/− mice over four cell passages. All data represent mean ± SD (n = 3). (A, C, D, G), and (H) are Student's t test (***, p < .001), and (F) is two‐way analysis of variance with Tukey post hoc test (**, p < .01). Abbreviations: Ip6k1, inositol hexakisphosphate kinase 1; MSC, mesenchymal stem/stromal cell; RPKM, Reads per Kilobase of transcripts per Million mapped reads.
Ip6k1 −/− MSCs Exhibit Enhanced Osteogenic Differentiation at the Expense of Adipogenesis, and Enhanced Hematopoiesis‐Supporting Activity
MSCs are best characterized by their differentiation potential and capacity to support hematopoiesis. Therefore, we evaluated how Ip6k1 deletion impacted these functions, which serve as metrics to assess overall MSC fitness. As shown in Figure 2, Ip6k1 −/− MSCs isolated from both young (Fig. 2A) and old (Fig. 2C) mice exhibited a significantly reduced capacity for stimulus driven adipogenic differentiation as compared with MSCs obtained from wild type, age‐matched littermates. This decline in adipogenic potential was offset by a modest but significant increase in stimulus‐driven osteogenic differentiation of Ip6k1 −/− MSCs (Fig. 2B, 2D). Next, we assessed the hematopoiesis‐supporting activity of Ip6k1+/+ versus Ip6k1 −/− MSCs using standard colony forming assays. This analysis revealed a significant difference [F(12,35) = 148.9, p = 2.6 × 10−19] in the ability of conditioned media from Ip6k1 −/− versus Ip6k1+/+ MSCs to support growth of hematopoietic progenitors from the bone marrow of wild type mice in vitro (Fig. 3A). Post hoc analyses of these data confirmed that conditioned media from Ip6k1 −/− versus Ip6k1+/+ MSCs yielded significantly more CFU‐GM, CFU‐E, CFU‐G, and CFU‐M colonies. Consistent with these findings, Luminex‐based multiplex arrays further revealed that conditioned media from Ip6k1 −/− versus Ip6k1+/+ MSCs contained significantly higher levels of SDF1 (Fig. 3B) and much lower levels of interleukin 6 (Fig. 3C) although the latter difference failed to reach statistical significance (Fig. 3C). Enzyme linked immunosorbent assay analysis further showed that Ip6k1 −/− versus Ip6k1+/+ MSC conditioned media contained significantly higher levels of SCF (Fig. 3D). As perivascular MSCs are the principle source in marrow of the HSC maintenance factors SDF1 and SCF 6, these findings further illustrate how IP6K1 inactivation enhances MSC fitness.
Figure 2.
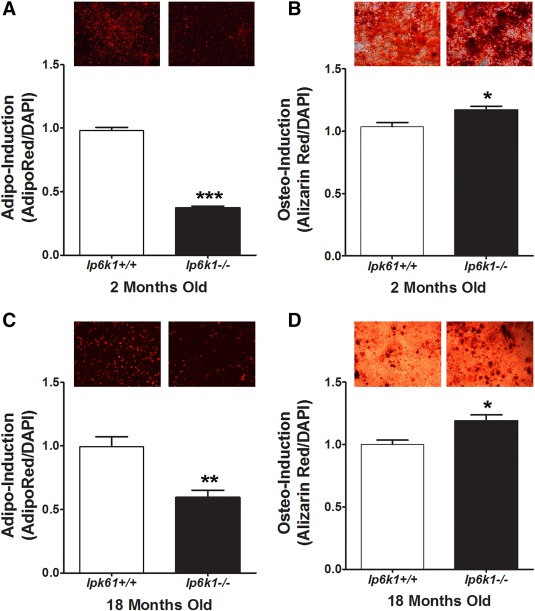
Enhanced osteogenic and reduced adipogenic differentiation of inositol hexakisphosphate kinase 1 (Ip6k1) null mesenchymal stem/stromal cells (MSCs). (A, C): Quantification of adipogenic differentiation by MSCs isolated from young (2 months, A) and old mice (18 months, C). Photomicrographs (top panels) are cell monolayers stained with AdipoRed at 3 days post‐induction. (B, D): Quantification of osteogenic differentiation by MSCs isolated from young (C) and old (D) mice. Photomicrographs (right panels) are cell monolayers stained with Alizarin Red at 10 days post‐induction. All data represent mean ± SD (n = 3) from experiments repeated three times. Student's t test (*, p < .05; **, p < .01; ***, p < .001). Magnification of photomicrographs is ×50. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; Ip6k1, inositol hexakisphosphate kinase 1.
Figure 3.
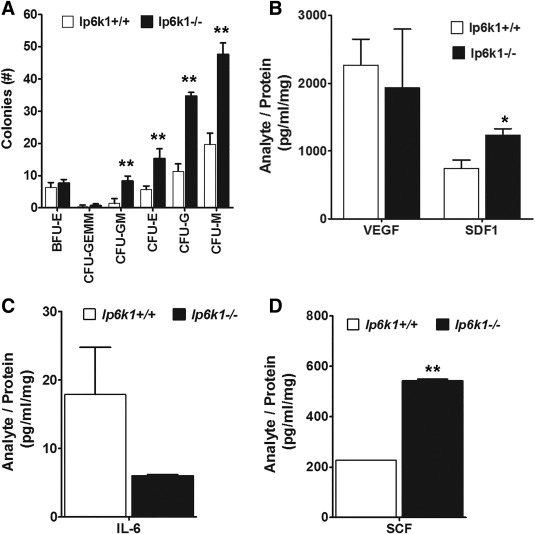
Inositol hexakisphosphate kinase 1 (Ip6k1) deletion enhances mesenchymal stem/stromal cell (MSC) hematopoiesis‐support activity. (A): Methylcellulose colony forming assays using conditioned media from Ip6K1+/+ versus Ip6k1 −/− MSCs. Data are mean ± SD (n = 3). **, p < .01 by analysis of variance (ANOVA) and Tukey post hoc test. (B): Conditioned media from Ip6K1+/+ versus Ip6k1 −/− MSCs was concentrated 50‐fold and analyzed using Luminex‐based multiplex bead arrays. Data represent mean ± SD (n = 2). (A) is two‐way ANOVA with Tukey post hoc test (**, p < .001) and (B–D) are Student's t test (*, p < .05; **, p < .01). Abbreviations: BFU, blast forming unit; CFU, colony forming unit; E, erythroid; G, granulocyte; GEMM, granulocyte, erythrocyte, monocyte, megakaryocyte; GM, granulcoyte, monocyte; M, monocyte; IL‐6, interleukin‐6; IP6K1, inositol hexakisphosphate kinase 1; SCF, stem cell factor; SDF, stromal derived factor; VEGF, vascular endothelial growth factor.
Pharmacological Inhibition of IP6K1 in Mice Reversed Adipogenesis in Bone Marrow
To explore whether IP6K1 inactivation protects bone and marrow from the effects of HFD feeding, we placed wild type mice on a HFD for 8 weeks and treated them with or without daily injections of TNP (10 mg/kg, IP), a pan‐inhibitor of IP6K activity. As shown in Figure 4A, the yield of MSCs from the bone marrow of HFD‐fed mice treated with TNP was significantly higher than compared with vehicle treated mice at the end of the 8 week feeding period. Additionally, bone marrow cells from TNP treated mice exhibited an enhanced capacity to generate CFU‐Fs (Fig. 4B), a measure of progenitor frequency within populations, as compared to cells from vehicle treated mice. CFU‐F's generated from the marrow of TNP treated mice were also significantly larger in surface area as compared to those obtained from the marrow of vehicle treated mice (Fig. 4C). Histological analysis of bone sections further revealed that TNP treatment resulted in conservation of trabecular bone (Fig. 4D). Morphometric analysis confirmed that the overall area of bone tissue was increased in TNP versus vehicle treated tissue sections (57.94 ± 9.8 vs. 48.89 ± 13.0) although differences between groups failed to reach statistical significance owing to wide variations in the two‐dimensional cross sections of the three‐dimensional trabecula (Fig. 4E). Furthermore, while tissue processing precluded a quantitative analysis of marrow adiposity, bone marrow parenchyma of TNP‐treated mice also contained fewer vacuoles as compared to vehicle treated mice based on visual inspection (Fig. 4D; Supporting Information Fig. S2), which was indicative of decreased marrow adiposity due to IP6K1 inhibition. Together, these data demonstrate that HFD feeding inhibits osteogenesis and promotes adipogenesis in adult bone marrow and that TNP treatment protects mice from these detrimental effects by inhibiting IP6K1 activity.
Figure 4.
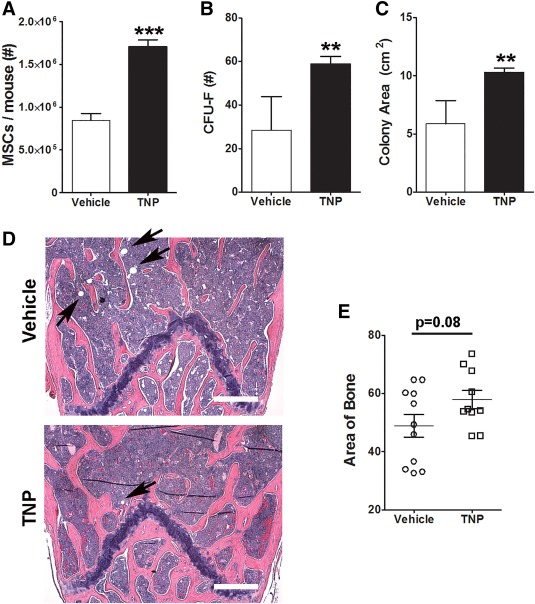
Pharmacological inhibition of inositol hexakisphosphate kinase 1 (IP6K1) preserves mesenchymal stem/stromal cell (MSC) integrity and inhibits high fat diet (HFD)‐induced adipogenesis and bone loss in adult bone marrow. (A): Total yield of immunodepleted MSCs from the bone marrow of mice fed a HFD for 2 months and treated with vehicle or N2‐(m‐trifluorobenzyl), N6‐(p‐nitrobenzyl)purine (10 mg/kg IP, n = 7 mice). Data represent mean ± SD (n = 3). (B): CFU‐F frequency measured in whole bone marrow from mice in (A). Data represent mean ± SD (n = 5). (C): Total area occupied by CFU‐Fs from (B). (D): Representative histological sections of long bones stained with H&E from mice in (A). Pink stained areas represent bone. Arrows demark lipid vacuoles. Magnification is ×50. Scale bar = 180 µm. (E): Total area of bone determined by morphometric analysis of tissue sections (two sections per mouse, n = 6 mice per group) from mice in (A). Data represent mean ± SD (n = 10–11). Student's t test (**, p < .01; ***, p < .001). Abbreviations: CFU, colony forming unit; F, fibroblast; MSC, mesenchymal stem/stromal cell; TNP, N2‐(m‐Trifluorobenzyl), N6‐(p‐nitrobenzyl)purine.
Ip6k1 −/− MSCs Are Resistant to Oxidative Stress
We previously reported that primary MSCs from mouse bone marrow are highly sensitive to oxidative stress, exposure to which induces growth arrest and cellular apoptosis via a p53 dependent mechanism 31. Therefore, we questioned if Ip6k1 deletion improved the fitness of MSCs by affecting the cellular redox balance. Flow cytometric analysis of unstained populations revealed that a higher proportion (58% vs. 46%) of Ip6k1 −/− versus Ip6k+/+ MSCs were characterized by low forward and side scatter (Fig. 5A; Supporting Information Fig. S3A), physical properties associated with low intrinsic mitochondrial reactive oxygen species (ROS) levels and resistance to oxygen‐induced stress 31. Consistent with this observation, flow cytometric analysis of MitoSOX Red stained cells revealed that Ip6k1 −/− MSCs exhibited lower intrinsic mitochondrial ROS levels when cultured in 5% oxygen saturation as compared to wild type MSCs (Fig. 5B; Supporting Information Fig. S3B). While we did not detect a significant difference in oxygen consumption rates between the two populations using Seahorse technology (data not shown) immunoblots of whole cell extracts revealed that p53 expression levels were markedly lower in Ip6k1 −/− versus Ip6k1+/+ MSCs cultured in 5% oxygen (Fig. 5C). Expression of p16INK4A was also downregulated whereas expression of the E3 ubiquitin ligase MDM2, which mediates p53 degradation, was upregulated. Owing to the fact that oxygen‐induced stress responses in primary MSCs are p53 dependent 31, lower intrinsic p53 levels likely account for lower overall mitochondrial ROS levels in Ip6k1 −/− MSCs. Flow cytometric analysis further revealed that Ip6k1 −/− versus Ip6k1+/+ MSC populations contain fewer pre‐apoptotic and apoptotic cells based on Annexin V and propidium iodide (PI) staining (Fig. 5D; Supporting Information Fig. 3C) and this difference in apoptosis persisted in populations through passage 4 (not shown). To evaluate whether Ip6k1 deletion conferred onto cells resistance to oxygen‐induced stress, Ip6k1+/+ and Ip6k1 −/− MSCs cultured in 5% oxygen were switched to atmospheric (∼21%) oxygen for 1 week then stained with MitoSOX Red. Flow cytometric analysis demonstrated that both populations exhibited a marked increase in mitochondrial ROS levels when exposed to 21% oxygen such that there was no significant difference in the overall abundance of MitoSOX Red high cells under these conditions (Fig. 5E). Together, these findings demonstrate that Ip6k1 −/− MSCs express intrinsically lower levels of mitochondrial ROS and p53 protein, which likely accounts for their enhanced growth and survival under physiological (∼5%) oxygen levels but still retain sensitivity to oxygen‐induced stress.
Figure 5.
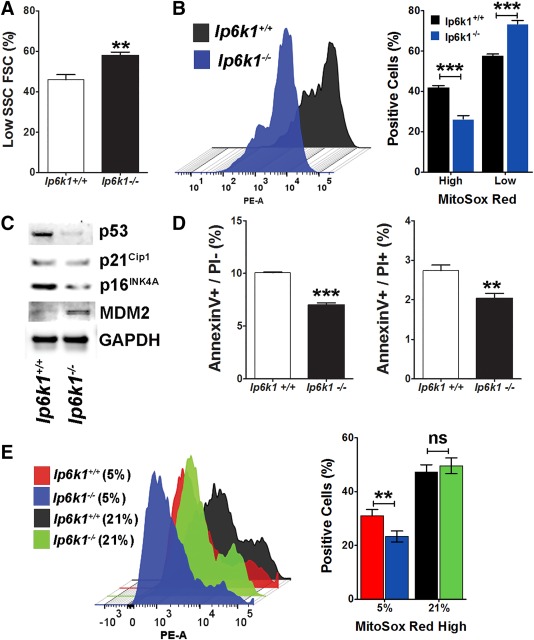
Inositol hexakisphosphate kinase 1 (Ip6k1) deletion enhances mesenchymal stem/stromal cell (MSC) survival and resistance to oxidative stress. (A): Flow cytometric analysis showing percentage of low forward scatter cells and side scatter cells in Ip6k1+/+ versus Ip6k1 −/− MSC populations isolated from 2 months old mice and cultured in 5% oxygen saturation. (B): Left panel; representative histogram from flow cytometric analysis of MSC populations from (A) after staining with MitoSOX Red. Right panel: Percentage of MitoSOX Red high and low expressing cells in each MSC population. Data represent mean ± SD (n = 2). (C): Western blot of MSC cell extracts isolated from femurs and tibias of 8‐week‐old Ip6k1 −/− and Ip6k1+/+mice. (D): Flow cytometric analysis of Annexin V and PI stained MSCs from (A) showing percentage of preapoptotic (AnnexinV+/PI‐) and apoptotic (AnnexinV+/PI+) cells. Data are mean ± SD (n = 2). (E): Left panel; representative histogram from flow cytometric analysis of MitoSOX Red stained MSCs cultured in 5% oxygen saturation or 21% oxygen saturation. Right panel: Percentage of MitoSOX Red high expressing cells in each MSC population. Data are mean ± SD (n = 2). Student's t test (**, p < .01; ***, p < .001). Abbreviations: FSC, forward scatter cell; Ip6k1, inositol hexakisphosphate kinase 1; PE‐A, phycoerythrin; SSC, side scatter cell.
Discussion
Skeletal homeostasis in adult organisms is controlled by a balance between bone formation and resorption, which is mediated by marrow resident osteoblasts and osteoclasts, respectively. During skeletal aging changes in the bone marrow microenvironment, such as increased oxidative stress and accumulation of advanced glycation end products, impairs osteogenesis resulting in reduced bone mineral density and increased marrow adiposity 32, 33, 34, 35. These age‐associated changes have been linked, in part, to impaired osteogenesis and enhanced adipogenic differentiation of marrow resident MSCs 26, 36. A number of disease states are also known to disrupt bone metabolism leading to defects in skeletal function. For example, patients with type 1 and type 2 diabetes have an increased risk of fracture due to diminished bone mineral density and bone fragility, and while the mechanisms responsible for these changes are varied 34, 37, these clinical observations demonstrate a strong link between nutritional status and skeletal homeostasis. Similarly, obesity induced by chronic steroid use is accompanied by rapid bone loss, indicating that obesity and bone metabolism are interrelated 38. Finally, genetic studies in rodents have shown that HFD feeding induces adipogenesis and reduces osteogenesis in bone marrow, and that HFD‐induced increases in leptin expression mediate these effects by acting on marrow‐resident, LEPR‐expressing MSCs 9. These findings are consistent with other studies showing that mouse models of type 2 diabetes mellitus exhibit delayed fracture healing 39. Nevertheless, as LEP/LEPR signaling exerts anorectic effects, its inhibition to prevent diabetes‐induced bone loss is contraindicated. Importantly, certain anti‐diabetic medications that have been developed that function as insulin sensitizers, such as the thiazolidinediones 40, have been shown to decrease bone mineral density by suppressing bone formation 41 and also increase marrow adiposity 42. Therefore, while new generation drugs that function as PPARG partial inverse agonists may reduce unwanted effects on bone metabolism 10, identification of proteins/pathways that restore insulin sensitivity while preserving bone integrity are needed.
Our findings reveal that IP6K1 inactivation increases overall MSC fitness as evidenced by increased cell yields from bone marrow and enhanced growth and survival ex vivo under physiological (5%) oxygen saturation levels. Moreover, MSCs from Ip6k1 null mice exhibit augmented osteogenic potential and reduced adipogenic potential as compared to those isolated from age‐matched congenic mice, consistent with an anti‐aging phenotype, and also secreted higher levels of hematopoiesis‐supporting cytokines. Furthermore, we found that Ip6k1 −/− MSCs express lower basal p53 and mitochondrial ROS levels as compared to wild type MSCs. We previously reported that primary mouse MSCs isolated from bone marrow via immunodepletion are highly sensitive to oxygen‐induced stress, that exposure to atmospheric oxygen rapidly induced mitochondrial ROS generation resulting in growth inhibition and increased cellular apoptosis, and that this stress response was p53‐dependent 31. Consistent with these results, we further demonstrated that exposure of p53 −/− MSCs to atmospheric oxygen failed to induce ROS generation, reduce cell viability or arrest growth. Therefore, considering the important role played by p53 in oxygen‐induced growth inhibition, these findings indicate that lower basal p53 levels in Ip6k1 −/− MSCs accounts in large part for their enhanced growth and survival at 5% oxygen saturation as compared to wild type cells. Rao et al. 43 showed that IP6K1 interacts with the Cullin‐RING ubiquitin ligase 4 (CLR4), which is activated in response to DNA damage and targets substrates involved in cell cycle regulation and cell death. In these studies, expressed levels of p21 and p53 were diminished in Ip6k1 knockout MEFs due to de‐repression of CLR4 activity. While we did not explore changes in this family of ubiquitin ligases in this study, IP6K1‐mediated repression of CLR4 could account for reduced basal p53 expression. Importantly, both Ip6k1 −/− and Ip6k1+/+ MSCs exhibited robust mitochondrial ROS generation in response to oxygen‐induced stress, indicating that Ip6k1 −/− MSCs retain oxygen sensitivity via p53 induction.
While we cannot confirm that IP6K1 inactivation results in expansion of the MSC progenitor pool in bone marrow, nor its effects specifically on LEPR+ MSCs, primary immunodepleted MSCs and LEPR+ MSCs both contain the majority of CFU‐F activity within bone marrow 9, 31. Therefore, while LEPR‐MSCs may represent a fraction of the total MSC pool obtained after isolation, culture expansion and immunodepletion, increased MSC yields suggest that IP6K1 inactivation may result in expansion of the MSC pool in marrow. Furthermore, our data suggest that TNP treatment protects bone marrow from the detrimental effects of HFD feeding by preserving trabecular bone and preventing fat deposition within marrow.
Previous studies from one of us showed that genetic deletion and pharmacological inhibition of IP6K1 protects mice from HFD‐induced weight gain and insulin resistance by enhancing insulin sensitivity via AKT activation, and decelerating weight gain by enhancing thermogenic energy expenditure in adipose tissue 12, 13, 14, 23. Our current findings together with previous studies demonstrate that IP6K1 inactivation/inhibition restores glucose homeostasis and insulin sensitivity in HFD fed mice 12 without adversely affecting bone metabolism. Indeed, IP6K1 inhibition appears to expand the MSC pool in bone marrow and alter the cellular redox balance in cells in a manner that increases overall fitness and osteogenic potential. For this reason, IP6K1 promises to be a better and consistent target in diseases of aging including insulin resistance, obesity, and osteoporosis as opposed to the widely studied PPARG or LEP/LEPR pathways.
Conclusion
MSCs play important roles in hematopoiesis and adult skeletal homeostasis, and both these processes are known to be impaired in response to systemic changes in energy metabolism, such as occurs in obesity and diabetes. The identification of LEPR expression by MSCs provided one mechanism by which systemic changes in energy metabolism may be sensed in bone marrow and impact hematopoiesis and skeletogenesis. Our findings that IP6K1 inactivation improves MSC fitness while promoting osteogenesis suggest a mechanism by which pharmacological inhibition of IP6K1 restores insulin sensitivity in HFD fed mice without promoting bone loss. Consequently, drugs that inhibit IP6K1 may be advantageous over other insulin sensitizers, such as the thiazolidinediones, which negatively impact bone metabolism resulting in increased risk of fracture in diabetic patients.
Author Contributions
S.V.B: performed research, collected and analyzed data, wrote the manuscript; S.G. performed research, collected and analyzed data, maintained mouse colony, generated young and aged IP6K1‐KO animals for experiments; C.N.B. performed research, collected and analyzed data; V.K.: performed research, collected and analyzed data; A.C.: conceived the study, designed research, collected and analyzed data, funded research, wrote the manuscript; D.G.P. conceived the study, designed research, analyzed data, funded research, wrote the manuscript, and final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Supporting information
Supporting Figure 1
Supporting Figure 2
Supporting Figure 3
Supporting Information
Acknowledgments
This work was supported by grants from the National Institutes of Health (R24‐OD018254) (to D.G.P.) and (R01‐DK103746) (to A.C.). The authors thank Prof. Solomon Snyder from Johns Hopkins University School of Medicine for sharing IP6K1‐KO mice. S.G. is currently affiliated with the Department of Pharmacology and Physiology, St. Louis University School of Medicine, St. Louis, MO.
References
- 1. Phinney DG. Building a consensus regarding the nature and origin of mesenchymal stem cells. J Cell Biochem 2002;38:7–12. [DOI] [PubMed] [Google Scholar]
- 2. Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008;3:301–313. [DOI] [PubMed] [Google Scholar]
- 3. Mendez‐Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010;466:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sugiyama T, Kohara H, Noda M et al. Maintenance of the hematopoietic stem cell pool by CXCL12‐CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006;25:977–988. [DOI] [PubMed] [Google Scholar]
- 5. Sacchetti B, Funari A, Michienzi S, et al. Self‐renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007;131:324–336. [DOI] [PubMed] [Google Scholar]
- 6. Ding L, Saunders TL, Enikolopov G et al. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012;481:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013;495:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou BO, Yue R, Murphy MM et al. Leptin‐receptor‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014;15:154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yue R, Zhou BO, Shimada IS et al. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 2016;18:782–796. [DOI] [PubMed] [Google Scholar]
- 10. Marciano DP, Kuruvilla DS, Boregowda SV, et al. Pharmacological repression of PPARgamma promotes osteogenesis. Nature Communications 2015;6:7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moritoh Y, Oka M, Yasuhara Y, et al. Inositol hexakisphosphate kinase 3 regulates metabolism and lifespan in mice. Sci Rep 2016;6:32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chakraborty A, Koldobskiy MA, Bello NT, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010;143:897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu Q, Ghoshal S, Rodrigues A, et al. Adipocyte‐specific deletion of Ip6k1 reduces diet‐induced obesity by enhancing AMPK‐mediated thermogenesis. J Clin Invest 2016;126:4273–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu Q, Ghoshal S, Tyagi R et al. Global IP6K1 deletion enhances temperature modulated energy expenditure which reduces carbohydrate and fat induced weight gain. Mol Metab 2017;6:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilson MS, Livermore TM, Saiardi A. Inositol pyrophosphates: Between signalling and metabolism. Biochem J 2013;452:369–379. [DOI] [PubMed] [Google Scholar]
- 16. Thota SG, Bhandari R. The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J Biosci 2015;40:593–605. [DOI] [PubMed] [Google Scholar]
- 17. Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signal 2011;4:re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barker CJ, Illies C, Gaboardi GC et al. Inositol pyrophosphates: Structure, enzymology and function. Cell Mol Life Sci 2009;66:3851–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas MP, Potter BV. The enzymes of human diphosphoinositol polyphosphate metabolism. FEBS J 2014;281:14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shears SB, Gokhale NA, Wang H et al. Diphosphoinositol polyphosphates: What are the mechanisms?. Advances Enzyme Regulation 2011;51:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wundenberg T, Grabinski N, Lin H et al. Discovery of InsP6‐kinases as InsP6‐dephosphorylating enzymes provides a new mechanism of cytosolic InsP6 degradation driven by the cellular ATP/ADP ratio. Biochem J 2014;462:173–184. [DOI] [PubMed] [Google Scholar]
- 22. Padmanabhan U, Dollins DE, Fridy PC et al. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: Use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem 2009;284:10571–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghoshal S, Zhu Q, Asteian A, et al. TNP [N2‐(m‐Trifluorobenzyl), N6‐(p‐nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol Metab 2016;5:903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z, Zhao C, Liu B, et al. Inositol pyrophosphates mediate the effects of aging on bone marrow mesenchymal stem cells by inhibiting Akt signaling. Stem Cell Res Ther 2014;5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atashi F, Modarressi A, Pepper MS. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim M, Kim C, Choi YS, et al. Age‐related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: Implication to age‐associated bone diseases and defects. Mech Aging Dev 2012;133:215–225. [DOI] [PubMed] [Google Scholar]
- 27. Phinney DG. Isolation of mesenchymal stem cells from murine bone marrow by immunodepletion. Methods Mol Biol 2008;449:171–186. [DOI] [PubMed] [Google Scholar]
- 28. Lai WT, Krishnappa V, Phinney DG. Fibroblast growth factor 2 (Fgf2) inhibits differentiation of mesenchymal stem cells by inducing Twist2 and Spry4, blocking extracellular regulated kinase activation, and altering Fgf receptor expression levels. Stem Cells 2011;29:1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA‐Seq. Bioinformatics 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boregowda SV, Krishnappa V, Chambers JW, et al. Atmospheric oxygen inhibits growth and differentiation of marrow‐derived mouse mesenchymal stem cells via a p53‐dependent mechanism: Implications for long‐term culture expansion. Stem cells 2012;30:975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Franke S, Ruster C, Pester J, et al. Advanced glycation end products affect growth and function of osteoblasts. Clin Exp Rheumatol 2011;29:650–660. [PubMed] [Google Scholar]
- 33. Alikhani M, Alikhani Z, Boyd C, et al. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 2007;40:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chuguransky SR, Cortizo AM, McCarthy AD. Alendronate can improve bone alterations in experimental diabetes by preventing antiosteogenic, antichondrogenic, and proadipocytic effects of AGEs on bone marrow progenitor cells. BioMed Res Int 2016;2016:5891925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Almeida M, Han L, Martin‐Millan M, et al. Skeletal involution by age‐associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 2007;282:27285–27297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou S, Greenberger JS, Epperly MW, et al. Age‐related intrinsic changes in human bone‐marrow‐derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 2008;7:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Napoli N, Chandran M, Pierroz DD, et al. Mechanisms of diabetes mellitus‐induced bone fragility. Nat Rev Endocrinol 2016; [DOI] [PubMed] [Google Scholar]
- 38. Moghadam‐Kia S, Werth VP. Prevention and treatment of systemic glucocorticoid side effects. Int J Dermatol 2010;49:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown ML, Yukata K, Farnsworth CW, et al. Delayed fracture healing and increased callus adiposity in a C57BL/6J murine model of obesity‐associated type 2 diabetes mellitus. PloS One 2014;9:e99656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yki‐Jarvinen H. Thiazolidinediones. New Engl J Med 2004;351:1106–1118. [DOI] [PubMed] [Google Scholar]
- 41. Grey A, Bolland M, Gamble G, et al. The peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: A randomized, controlled trial. J Clin Endocrinol Metab 2007;92:1305–1310. [DOI] [PubMed] [Google Scholar]
- 42. Grey A, Beckley V, Doyle A, et al. Pioglitazone increases bone marrow fat in type 2 diabetes: Results from a randomized controlled trial. Eur J Endocrinol 2012;166:1087–1091. [DOI] [PubMed] [Google Scholar]
- 43. Rao F, Xu J, Khan AB, et al. Inositol hexakisphosphate kinase‐1 mediates assembly/disassembly of the CRL4‐signalosome complex to regulate DNA repair and cell death. Proc Natl Acad Sci USA 2014;111:16005–16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure 1
Supporting Figure 2
Supporting Figure 3
Supporting Information