

Abstract
In the live animal, tissue autofluorescence arises from a number of biologically important metabolites, such as the reduced form of nicotinamide adenine dinucleotide. Because autofluorescence changes with metabolic state, it can be harnessed as a label-free imaging tool with which to study metabolism in vivo. Here, we used the combination of intravital two-photon microscopy and frequency-domain fluorescence lifetime imaging microscopy (FLIM) to map cell-specific metabolic signatures in the kidneys of live animals. The FLIM images are analyzed using the phasor approach, which requires no prior knowledge of metabolite species and can provide unbiased metabolic fingerprints for each pixel of the lifetime image. Intravital FLIM revealed the metabolic signatures of S1 and S2 proximal tubules to be distinct and resolvable at the subcellular level. Notably, S1 and distal tubules exhibited similar metabolic profiles despite apparent differences in morphology and autofluorescence emission with traditional two-photon microscopy. Time-lapse imaging revealed dynamic changes in the metabolic profiles of the interstitium, urinary lumen, and glomerulus—areas that are not resolved by traditional intensity-based two-photon microscopy. Finally, using a model of endotoxemia, we present examples of the way in which intravital FLIM can be applied to study kidney diseases and metabolism. In conclusion, intravital FLIM of intrinsic metabolites is a bias-free approach with which to characterize and monitor metabolism in vivo, and offers the unique opportunity to uncover dynamic metabolic changes in living animals with subcellular resolution.
Keywords: metabolism, podocyte, tubules, endothelium
Metabolic networks are characterized by dynamic changes that occur in the highly compartmentalized intracellular space, and can be challenging to study in vivo.1–3 In the past two decades, a number of fluorescent probes and genetically encoded biosensors have been developed to monitor metabolism in living cells and organisms. However, these exogenous probes are limited by problems associated with delivery, perturbation of intrinsic physiology, and difficulty in multiplexing of fluorophores.4 Therefore, there is a critical need for a label-free, noninvasive imaging tool that permits monitoring of metabolites in vivo with subcellular resolution.
One attractive approach is to exploit the properties of naturally occurring intrinsic fluorophores (autofluorescence) and their fluorescence lifetimes (excited-state decay rates).5 Several endogenous metabolites, such as nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD), fluoresce at detectible levels and this fluorescence has been used as an index of the cellular redox status.6,7 However, the emission wavelengths of many endogenous fluorophores overlap, and this limits the ability to identify individual species with certainty.8,9 In contrast, the fluorescence lifetimes of these same endogenous fluorophores are significantly different and can be separated by phasor analysis.5,10 Accordingly, fluorescence lifetime imaging microscopy (FLIM) offers the ability to distinguish a number of intrinsic fluorophores despite their overlapping emission spectra.
The kidney is a highly metabolic organ and is second only to the heart in oxygen consumption and mitochondrial abundance.11 However, because of the complexity of the tissue and the enormous flux of fluid and solutes, the spatiotemporal metabolic profiles of various renal cell populations are poorly understood.12 In this study, we combined two-photon intravital microscopy with FLIM to generate a powerful approach for investigating cell-specific metabolic profiles in the kidneys of living animals. This method exploits the phasor transformation of FLIM images, developed by Colyer et al.13 The phasor transformation allows rapid determination of the lifetimes of autofluorescence without the requirement of a priori knowledge of the fluorescent species in the tissue.14,15 We provide, for the first time, intravital FLIM images of kidneys with subcellular resolution in health and disease. Intravital FLIM revealed previously unsuspected cell-type specific metabolic signatures that could be key to understanding renal pathophysiology and developing targeted therapies.
Results
Traditional fluorescence microscopy, including intravital two-photon microscopy, acquires images on the basis of fluorescence intensity. The autofluorescence signals from representative S1 and S2 mouse proximal tubules imaged by intravital two-photon microscopy are shown in Figure 1A. We have previously verified the identity of these tubules as upstream S1 and downstream S2 by following the sequential luminal appearance of fluorescent inulin.16 Using traditional red, green, and blue palettes to represent the three RGB channels, S1 tubules exhibit a dark orange color and S2 tubules appear bright green (Figure 1A). This likely reflects some red-emitting metabolites that are more abundant in S1 (Supplemental Figure 1, A and B). However, different metabolites can have similar emission spectra, making it difficult to deconvolve the sources of the signals in tissues where unknown numbers of metabolites coexist.
Figure 1.

Intravital fluorescence lifetime imaging (FLIM) distinguishes the metabolic signatures of S1 and S2 tubules. (A) Representative S1 and S2 proximal tubules identified by traditional intensity based intravital two-photon microscopy (red, green, and blue channels combined). (B) FLIM blue channel intensity shown in grayscale. (C) The phasor distribution of lifetimes collected from the area shown in (B). Note that lifetimes decrease moving from left to right in the phasor plot. (D) Four lifetime regions are selected in the phasor plot (yellow, beige, orange, and brown circles). (E) The gated lifetime regions are back plotted with corresponding colors into the blue channel (grayscale) image. These lifetime regions are characteristic of S1 tubules. (F and G) Three lifetime regions characteristic of S2 tubules are shown in blue, red, and pink.
To overcome the limitations of intensity measurements of emission signals from label-free metabolite imaging, we applied the frequency-domain FLIM method to intravital two-photon kidney imaging. Frequency-domain FLIM measurements in intravital two-photon microscopy take advantage of the pulsed excitation light source (80 MHz). Because of the duration of the excited-state, there is a phase shift (Φ) and a change in the modulation (M) of the emission signals from endogenous fluorophores. The difference between the phase and modulation of the excitation and emission waveform is represented by the Fourier transform components G and S on the phasor plot (Figure 1C). The phasor plot maps the frequency characteristics from each image pixel to the G and S coordinates using vectors with angles specified by the Φ and lengths determined by M. Most importantly, phasor plots allow the visualization of complex lifetime decays without the need for a priori knowledge of the system.14 The distribution of lifetimes within the phasor transformation for an unlabeled tissue (the phasor fingerprint) represents the contributions from the endogenous fluorophores. The phasor fingerprint approach enables the direct visualization of cellular metabolism and is ideal for intravital imaging for multiple reasons, including rapid data processing time and the ability to interpret a mixture of lifetime components without fitting presumed metabolites in the tissue.
Here, intravital FLIM imaging of mouse kidney was achieved with 800-nm two-photon excitation, and the endogenous fluorescence signals were acquired using a 480-nm emission filter (blue channel). The fluorescence intensity signal detected in the blue channel by FLIM setup (shown in the gray palette) is shown in Figure 1B. This is the same field depicted in Figure 1A (combined three channels) acquired by conventional two-photon microscopy. Next, the fluorescence lifetimes determined for each image pixel are mapped by the phasor plot to generate the phasor fingerprint (Figure 1C). The lifetime determined at each pixel is an average lifetime for all photons emitted by various species present in that pixel. The phasor fingerprint for this region of the kidney can be compared with phasor plots obtained from a mixture of known endogenous fluorophore standards to assess the lifetime contributions from individual species (Supplemental Figure 2). Additionally, regions of the phasor fingerprint can be selected (circular cursors in Figure 1D) to generate a color-coded phasor image that can be superimposed on the grayscale intensity plot (Figure 1E). For example, In Figure 1D, we selected four lifetime regions (four colored circles) that mapped to S1 tubules (Figure 1E, note the consistent colors of the gating circles and the image overlay). Similarly, we were able to identify three phasor regions that were more characteristic of S2 tubules (Figure 1, F and G). Therefore, these data show S1 and S2 proximal tubules to be clearly distinguishable on the basis of their endogenous fluorophore lifetime characteristics, indicating that their metabolic microenvironments differ significantly.
Surprisingly, intravital FLIM revealed that S1 proximal tubules and distal tubules have remarkably similar metabolic signatures despite the marked differences in their biologic function and morphologic appearance (Figure 2, A–C). Moreover, intravital FLIM delineated distinctive metabolic signatures for the urinary lumen (Figure 2B, green overlay), endothelium (Figure 2E, turquoise overlay), and interstitium (Figure 2E, pink and purple overlay); structures that cannot be well visualized or contrasted with intensity-based two-photon microscopy, partly because of overlapping emission spectra. In addition, we show the feasibility of time-lapse imaging of the kidney in live animals over an extended period with gating that highlights circulating immune cells (Figure 2, G and H, Supplemental Video 1).
Figure 2.

Intravital FLIM identifies renal structures that are not well resolved with conventional two-photon microscopy. (A) The urinary lumen, interstitium, and glomeruli exhibit weak autofluorescence and are not resolved with intravital two-photon microscopy. The distal tubules also have a very weak autofluorescence signal. (B and C) With FLIM, distal tubules and S1 (but not S2) possess similar metabolic signatures (yellow and orange). The urinary lumen, especially in the distal tubule, exhibits metabolites with long lifetimes (pseudocolored in dark green and light green). (D–F) The blood compartment shows species with short lifetimes (light green). Interstitial structures are highlighted in pink and violet and the endothelium in blue. (G and H) Dynamic changes in metabolic signatures of the urinary lumen and interstitium were captured with time-lapse intravital FLIM. See Supplemental Video 1. DT, distal tubule.
Because endothelial biology is of great importance in a variety of renal diseases, we sought to validate the endothelial FLIM mapping described above. To this end, we took advantage of the endothelial-specific CreERT2 transgenic mouse, which exhibits red fluorescence in the endothelium. This red fluorescence has no bleed-through into the blue channel where the FLIM data are collected. Thus, the red channel FLIM, which primarily maps tdTomato in the endothelium, can be directly compared with the blue channel FLIM originating from endogenous endothelial species. As shown in Figure 3A, conventional intravital microscopy reveals homogeneous distribution of tdTomato red fluorescence in peritubular capillary endothelial cells. The FLIM red channel intensity fluorescence mapped accurately with the red channel of conventional intravital microscopy (Figure 3B). Using the phasor approach, the tdTomato lifetime could be easily identified and back-mapped to endothelial cells (Figure 3, D–F). In contrast, endothelial cells could not be visualized in the blue channel. However, by gating the lifetime of endogenous endothelial fluorophores described in Figure 2F, the resulting back-mapping did indeed overlay the endothelium with accuracy (Figure 3, E–G).
Figure 3.

Endothelial specific tdTomato fluorescence confirms FLIM mapping of endothelial cells. (A) Endothelial specific CreERT2 transgenic mouse kidney imaged with conventional two-photon microscopy. The structure of peritubular capillary endothelium is delineated with red fluorescence (tdTomato). (B and C) Red and blue channel FLIM intensity images are shown. (D and E) The phasor distributions of red and blue channel lifetimes obtained from the area shown in (B) and (C). (F and G) Lifetime regions gated on the phasor plots are back-mapped into the intensity images. Note the overlap of the FLIM blue channel and conventional two-photon imaging of tdTomato signal. Arrow points to the endothelium that was not induced by tamoxifen.
The podocyte is a specialized epithelial cell that is essential for the maintenance of the glomerular filtration barrier. In the mouse, most glomeruli are not within the reach of conventional intravital two-photon microscopy. However, the Munich–Wistar–Frömter rat does have superficial glomeruli accessible to the two-photon laser. We show in Figure 4A that conventional two-photon microscopy is not suitable for characterizing podocytes because of their weak autofluorescence emission and spectral overlap with other glomerular structures. In contrast, intravital FLIM has the ability to resolve glomerular structures on the basis of the blue channel lifetime fingerprint. We found that the podocyte lifetime is similar to that of adjacent tubular epithelial cells (Supplemental Figure 3). To validate the podocyte FLIM mapping, we made use of the transgenic Munich–Wistar–Frömter rat, which exhibits DsRed fluorescence in the podocyte. Similar to what we demonstrated for the endothelium, this approach allowed us to confirm the lifetime fingerprint for the podocyte in the blue channel (Figure 4, B–H).
Figure 4.

Podocyte specific DsRed fluorescence confirms FLIM mapping of podocytes. (A) Conventional two-photon intravital imaging of wild-type Munich–Wistar–Frömter rat glomerulus. (B) Podocyte-DsRed Munich–Wistar–Frömter rat glomerulus imaged with conventional two-photon microscopy. (C–H) FLIM imaging of red and blue channels of the glomerulus shown in (B). Note that the FLIM mapping of the podocyte in the blue channel [red overlay; (H)] compares well with the DsRed lifetime mapping in the red channel (G) and the podocyte fluorescence by conventional two-photon microscopy (B). In (H), we also show the glomerular capillary endothelium (turquoise) and blood compartment (light green). DT, distal tubule; Glom, glomerulus; PT, proximal tubule; WT, wild-type.
In summary, many renal structures exhibit unique autofluorescence lifetime signatures originating from endogenous species. Intravital FLIM allows for the visualization of these structures that are otherwise indistinguishable by conventional intensity-based microscopy (Supplemental Figure 4). Therefore, label-free intravital FLIM may be a useful approach to investigate the kidney in the living animal.
To explore further the utility of the intravital FLIM approach, we applied it to a model of endotoxemia. Systemically administered endotoxin causes oxidative damage to S2 proximal tubules even though endotoxin uptake occurs in S1 (Figure 5, A and B). We have previously made this observation using fluorescently-labeled endotoxin and the oxidative stress probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA).16,17 However, the possibility of uneven H2DCFDA delivery to S1/S2, or other factors such as quenching of probe fluorescence in S1 could not be completely excluded. The confounding effects of these factors can be avoided using label-free autofluorescence FLIM. Indeed, intravital FLIM of the endotoxin-treated mice revealed significant changes in the S2 signature. We observed a right-ward shift toward shorter lifetimes from the control S2 tubules (Figure 5, C and D) to the LPS-treated S2 tubules (Figure 5, E and F). This was consistent with a shift in FAD–NADH phasor axis (Figure 6A) toward bound and free NADH. This could be indicative of an increase in the NADH-to-FAD ratio, as well as NADH-to-NAD+ ratio, a pathologic state that alters metabolic fluxes and accelerates the generation of reactive oxygen species.18 Indeed, the levels of NAD+ and FAD (and its precursor riboflavin) directly measured with mass spectrometry were significantly reduced in the endotoxin-treated kidney (Figure 6, B–D). Surprisingly, S1 tubules also exhibited changes in their FLIM signature that were apparent in the physical distribution of metabolites (but not lifetime changes) in the cytoplasm. Such changes in S1 had no counterpart by regular intensity microscopy and their significance remains unknown (Supplemental Figure 5). In addition, we noted that the lifetime of luminal species in the endotoxin-treated distal tubules was longer than that in the lumen of control animals, and its biologic implication remains to be determined (Supplemental Figure 6).
Figure 5.

Endotoxin alters the S2 metabolic signature. (A and B) Regular two-photon intravital imaging reveals that systemically-administered endotoxin (Alexa-LPS; red) is filtered and internalized primarily by S1 proximal tubules. Oxidative stress (reflected by strong H2DCFDA green fluorescence) occurs in S2 proximal tubules. (C and D) FLIM image of control mouse kidney before endotoxin exposure. S1 FLIM signature is highlighted in orange and yellow, S2 in blue and red. (E and F) FLIM revealed the loss of S2 signature in the kidney 24 hours after endotoxin administration. DT, distal tubule.
Figure 6.

Endotoxin treatment increases the NADH/NAD+ ratio in S2 tubules. The phasor lifetime distributions of control (Figure 5D) and endotoxin-treated mice (Figure 5F) were superimposed onto phasor plots of standard metabolites as indicated in (A). Additional metabolites mapped are shown in Supplemental Figure 2. When a pixel has two lifetime components, the phasor location of the mixture falls onto a straight line between the two individual lifetime points (see Concise Methods). (A) The alteration of S2 signature after endotoxin treatment can be explained by an increase in the NADH/NAD+ ratio as well as decreases in FAD and its precursor riboflavin. (B–D) Metabolite levels of kidney tissues measured with liquid chromatography/mass spectrometry are shown. Endotoxin treatment reduced the levels of NAD+, FAD, and riboflavin, consistent with the S2 signature changes observed with FLIM. Horizontal bars represent mean values.
Finally, we investigated whether a single dose of furosemide could alter the lifetime fingerprint in the renal cortex. We found no notable acute changes (Supplemental Figure 7).
Discussion
In recent years, fluorescence microscopy has become an indispensable tool for kidney research. The primary goal of this study was to extend the power of intensity-based fluorescence microscopy by exploring the “dark side” of fluorophores. Other groups have successfully used FLIM to image ex vivo kidney tissues.15,19 However, to our knowledge, this is the first demonstration of label-free two-photon FLIM imaging of the kidney in the live animal.
The fluorescence lifetime, the average time a fluorophore spends in the excited state, is an intrinsic property of every fluorophore. The time-resolved measurements of lifetimes by FLIM are unaffected by extrinsic factors, such as excitation wavelength and laser power. Conversely, fluorescence lifetime is sensitive to local microenvironmental parameters, such as pH and ionic changes, as well as the protein-bound or -unbound state of the fluorophore. Collectively, these unique properties render fluorescence lifetime imaging a complementary approach to traditional fluorescence intensity measurements, and provide otherwise unattainable biologic information.5,20–25
Intravital imaging of autofluorescence lifetimes is a practical approach that enables measurements of detailed in vivo tissue structure and metabolic state in a label-free manner. Indeed, most tissue autofluorescence arises from unique metabolites, such as NADPH and flavins. However, except for readily accessible organs such as the skin or the eye, autofluorescence imaging has not been widely applied to inner organs.26,27 In this study, we describe autofluorescence lifetime imaging of the kidney using two-photon intravital microscopy. Our study provides insights into previously unappreciated cell-type specific kidney metabolic signatures in vivo. This is a significant advantage of FLIM because many kidney structures, including the glomerulus, interstitium, and urinary lumen, cannot be well visualized with traditional intensity-based autofluorescence measurements.
We acknowledge that there are two major limitations to studying metabolism with autofluorescence lifetime measurements: (1) nonfluorescent intrinsic metabolites cannot be accounted for, and (2) identification of specific metabolic species in each pixel is challenging. Despite these limitations, given the number of biologically important fluorescent metabolites, collective in vivo metabolic signatures obtained in a bias-free fashion can be more revealing and meaningful than targeted metabolite analysis ex vivo. For example, we demonstrated that FLIM may enable us to assess the cellular redox state via lifetimes of FAD and NADH, two key determinants of bioenergetics. NADH and NADPH, which have identical emission spectra, can be also distinguished using FLIM.28 Other intrinsic renal fluorophores that can be measured with FLIM include cholecalciferol, retinoids, folic acid, serotonin, tryptophan, and porphyrin.5,8 Conceivably, once a comprehensive mapping of fluorescent metabolites in the kidney is complete, one could improve specificity by using spectral unmixing or other deconvolution techniques.
With FLIM, we detected significant differences in the metabolic signature of S1 and S2 tubules. In addition to metabolite composition, these differences in lifetimes could also be influenced by factors such as pH, calcium, oxygen, and fluorescence quenching. Conversely, the identical metabolic signatures between S1 and distal tubules strongly suggest that these two subsegments share similar metabolites and microenvironments.
Similarly, label-free FLIM confirmed the dramatic changes in S2 segments after endotoxin exposure. These changes are possibly related to the severe oxidative stress we previously documented with traditional intravital microscopy using exogenous fluorophores such as H2DCFDA and dihydroethidium.29 As discussed above, these changes likely represent pathologic alterations in the NADH, NAD+, and FAD ratios. Importantly, FLIM also detected changes in S1 tubules that were not observed by conventional two-photon microscopy. The significance of these S1 changes remains unknown at present. In addition, the identification of the prolonged lifetime species observed in the endotoxin-treated distal lumen requires further investigation. Interestingly, Datta et al. reported that lipid oxidation by reactive oxygen species gives rise to long lifetime signature.23 Therefore, studies such as targeted lipidomics and FLIM of the urine could be potentially fruitful to identify the exact species involved.
Peritubular capillary endothelia and podocytes are structures that are extremely difficult to visualize by conventional two-photon microscopy. We showed here that FLIM can be a useful approach to visualize both cell types. We validated the FLIM mapping of these cell types using two newly developed transgenic animals. The combined use of conventional two-photon imaging and FLIM in these animals will allow better exploration of the structural and metabolic changes of these elusive renal cells.
In conclusion, we have shown for the first time the feasibility of two-photon FLIM of the kidney in live animals using the phasor approach. This latter allows for high spatial and temporal resolution within an operator-friendly platform. Therefore, FLIM can greatly complement traditional intravital microscopy. We believe that the FLIM technique will play a significant role in elucidating metabolic and environmental changes in the kidney in both health and disease.
Concise Methods
All animal protocols were approved by Indiana University Institutional Animal Care Committee and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male C57BL/6J mice (8–10 weeks of age) were obtained from The Jackson Laboratory and wild-type Munich–Wistar–Frömter rats were from our established colony. All animals were given water and food ad libitum throughout the study.
The endothelial-specific CreERT2 transgenic mouse model is described elsewhere (Y. Zheng, unpublished observations). Briefly, a Tie2-CreERT2 transgene comprising a version of Tie2 promoter,30 β-globulin intron sequence, CreERT2 fusion gene, 2 polyadenylation signal (pA) fragments, and Tie2 enhancer sequence was used for C57BL/6 pronuclear microinjection to generate transgenic mouse lines. C57BL/6 mice and Rosa26-TD-Tomato mice [B6; 129S6-Gt (ROSA) 26Sortm9(CAG-tdTomato)Hze/J stock number: 007905] were from The Jackson Laboratory. Tie2-CreER;TD-Tomato mice were generated by crossbreeding Tie2-CreER mice with Rosa26-TD-Tomato transgenic mice. Tamoxifen was injected intraperitoneally into Tie2CreER/TD-Tomato mice once per day for 3 days with 50 µg/g body wt (T5648; Sigma) dissolved in 250 μl sunflower oil. Imaging was performed 4 weeks after tamoxifen induction.
Rats expressing the DSRed transgene restricted to podocytes were produced in the Wiggins Laboratory at the University of Michigan, as follows. First, Munich–Wistar–Frömter rats containing superficial glomeruli were crossed with a transgenic Fischer344 rat strain carrying the human diphtheria toxin receptor (hDTR) driven by the human podocin promoter, as previously described.31,32 hDTR transgene expression by podocytes allows podocytes to be specifically deleted by a predetermined amount and at a predetermined time, using diphtheria toxin injection.32 Rats were back-crossed against the Munich–Wistar–Frömter strain for five generations, selecting for superficial glomeruli and the hDTR transgene at each back-cross. This rat strain is designated as Super(ficial glomeruli)-hDTR. Second, a transgene containing DSRed driven by the human podocin promoter was then introduced into the Super-hDTR strain using standard transgenic methodology. Rats were selected for expression of superficial glomeruli, the hDTR transgene, and the DSRed transgene, and designated Super-Red-hDTR rats. This rat strain has normal kidney structure and function unless podocytes are injured by injection of diphtheria toxin (not used in this study).
C57BL/6J mice were subjected to a single intraperitoneal dose of 5 mg/kg unlabeled Escherichia coli LPS (serotype 0111:B4; Sigma-Aldrich) and imaged 4 and 24 hours later. Untreated mice received an equivalent dose of sterile normal saline vehicle. In some experiments, Alexa Fluor 568 hydrazide (Life Technologies) was used to label LPS from Salmonella minnesota (Re 595; Sigma-Aldrich). Oxidative stress was measured in the live mouse with carboxy-H2DCFDA (Thermo Fisher Scientific) as previously described.16 Furosemide (Cayman Chemical) was dissolved in DMSO (100 µg/µl) and diluted with PBS. C57BL/6J mice were subjected to a single intravenous dose of 15 mg/kg furosemide and imaged immediately and 4 hours later.
Intravital Fluorescence Lifetime Imaging of the Kidney
Live animal imaging was performed using an Olympus FV1000-MPE confocal/multiphoton microscope equipped with a Spectra Physics Mai Tai DeepSee laser and gallium arsenide detectors, with signal digitized to 12-bit resolution. The system was mounted on an Olympus Ix81 inverted microscope stand with a Nikon 20× and 60× NA 1.2 water-immersion objective. The objective lens was heated using a wrap-around objective heater (Warner Instruments). Animals were placed on the stage with the exposed intact kidney placed in a coverslip-bottomed cell culture dish (HBSt-5040; Warner Instruments) bathed in isotonic saline, as previously described.17 The rectal temperature was monitored using a thermometer. Two ReptiTherm pads (Zoo Med) and a heated water jacket blanket were used to maintain the temperature at around 36°C.
Digital frequency-domain lifetime imaging was acquired using MiniTDU (ISS) with scan speed 12.5 µs/pixel and image size 256×256 pixels. Blue (480/100) and red (630/90) channels were used. The laser was tuned to 800-nm excitation with laser power of 12%. Calibration of FLIM was performed before each experiment using fluorescein and HPTS. Each intravital FLIM image consists of approximately 15–20 1-second scans, so that a typical pixel has at least 100 photons. Note that this is different from our traditional two-photon microscopy, where we use 15% laser power to detect three channels and each image consists of a single scan with scan speed of 2.0 µs/pixel and image size 512×512 pixels. Therefore, the photon count per pixel is lower than that acquired by FLIM. Note also that the degree of laser attenuation in the optical train of this microscope system is approximately 90% at the surface of the kidney. No morphologic photodamage was observed with the aforementioned FLIM set up. Compare Supplemental Video 1 (12% laser power) with Supplemental Video 2 (18% laser power; see also Supplemental Figure 8). Photodamage is apparent with 18% and absent with 12% laser power.
The mathematical theory of frequency-domain lifetime imaging is described elsewhere.13,33,34 Note that when a pixel has multiple lifetime components, the phasor position of the mixture is given by the following:
 |
where  and
and  are the real (x-axis) and imaginary (y-axis) components of the Fourier transformation,
are the real (x-axis) and imaginary (y-axis) components of the Fourier transformation,  and
and  are the fluorescence decay time and angular modulation frequency respectively.
are the fluorescence decay time and angular modulation frequency respectively. 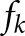 is the intensity-weighted fractional contribution of the component with lifetime
is the intensity-weighted fractional contribution of the component with lifetime  . Because the equations are linear, the phasor location of the mixture of lifetimes lies within a polygon, with vertices corresponding to each lifetime position in the phasor space. When a pixel has only two lifetime components, the mixture falls on the line between the two lifetime points.35
. Because the equations are linear, the phasor location of the mixture of lifetimes lies within a polygon, with vertices corresponding to each lifetime position in the phasor space. When a pixel has only two lifetime components, the mixture falls on the line between the two lifetime points.35
Lifetime Standards
Lifetime standard solutions were prepared as follows: flavin mononucleotide in MOPS buffer (pH 7) at a concentration of 1.5 mg/ml; riboflavin in MOPS at a concentration of 0.5 mg/ml; retinol in DMSO (pH 8) at concentration of 1 mg/ml; retinoic acid in DMSO (pH 8) at 3 mg/ml; NADH in MOPS at a concentration of 17.7 mg/dl; and protoporphyrin IX in DMSO/methanol (1:1, pH 7) at a concentration of 1.5 mg/ml. To prepare protein-bound NADH,14 LDH (1000 U/ml) and the NADH solution were mixed at a volume of 1:1. FAD was diluted in water (pH 7) at a concentration of 2 mg/ml. All reagents were purchased from Sigma-Aldrich.
Metabolite Analysis
Kidneys from vehicle-treated and endotoxin-treated mice were snap frozen in liquid nitrogen. Metabolomic analysis was performed at Metabolon on their ultra-HPLC tandem mass spectroscopy metabolomics platform.
Disclosures
None.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01-DK107623 and R01-DK099345 and Veterans Affairs Review award 1I01BX002901 (to P.C.D), NIH O’Brien Center grant P30-DK079312 (to B.A.M.), Paul Teschan Research grant (Dialysis Clinics Inc.), and Indiana Clinical and Translational Sciences Institute grant KL2TR001106 (to T.H.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016101153/-/DCSupplemental.
References
- 1.Shamir M, Bar-On Y, Phillips R, Milo R: Snapshot: Timescales in cell biology. Cell 164: 1302, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE, DeBerardinis RJ: Metabolic heterogeneity in human lung tumors. Cell 164: 681–694, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM: Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159: 1603–1614, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen EC: Use of fluorescent probes: Their effect on cell biology and limitations. Anat Rec (Hoboken) 295: 2031–2036, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Berezin MY, Achilefu S: Fluorescence lifetime measurements and biological imaging. Chem Rev 110: 2641–2684, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostrander JH, McMahon CM, Lem S, Millon SR, Brown JQ, Seewaldt VL, Ramanujam N: Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res 70: 4759–4766, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I: Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep 3: 3432, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW: Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA 100: 7075–7080, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Small DM, Sanchez WY, Roy S, Hickey MJ, Gobe GC: Multiphoton fluorescence microscopy of the live kidney in health and disease. J Biomed Opt 19: 020901, 2014 [DOI] [PubMed] [Google Scholar]
- 10.Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N: In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci USA 104: 19494–19499, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z: Mitochondrial function and disturbances in the septic kidney. Semin Nephrol 35: 108–119, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hato T, Friedman AN, Mang HE, Plotkin Z, Dube S, Hutchins GD, Territo PR, McCarthy BP, Riley AA, Pichumani K, Malloy CR, Harris RA, Dagher PC, Sutton TA: Novel application of complimentary imaging techniques to examine in vivo glucose metabolism in the kidney [published online ahead of print January 13, 2016]. Am J Physiol Renal Physiol doi: 10.1152/ajprenal.00535.2015 [DOI] [PMC free article] [PubMed]
- 13.Colyer RA, Lee C, Gratton E: A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc Res Tech 71: 201–213, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Stringari C, Cinquin A, Cinquin O, Digman MA, Donovan PJ, Gratton E: Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci USA 108: 13582–13587, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranjit S, Dobrinskikh E, Montford J, Dvornikov A, Lehman A, Orlicky DJ, Nemenoff R, Gratton E, Levi M, Furgeson S: Label-free fluorescence lifetime and second harmonic generation imaging microscopy improves quantification of experimental renal fibrosis. Kidney Int 90: 1123–1128, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC: Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol 22: 1505–1516, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hato T, Winfree S, Kalakeche R, Dube S, Kumar R, Yoshimoto M, Plotkin Z, Dagher PC: The macrophage mediates the renoprotective effects of endotoxin preconditioning. J Am Soc Nephrol 26: 1347–1362, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Georgakoudi I, Quinn KP: Optical imaging using endogenous contrast to assess metabolic state. Annu Rev Biomed Eng 14: 351–367, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Becker W: Fluorescence lifetime imaging--Techniques and applications. J Microsc 247: 119–136, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Becker W, Bergmann A, Biskup C: Multispectral fluorescence lifetime imaging by TCSPC. Microsc Res Tech 70: 403–409, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Goiffon RJ, Akers WJ, Berezin MY, Lee H, Achilefu S: Dynamic noninvasive monitoring of renal function in vivo by fluorescence lifetime imaging. J Biomed Opt 14: 020501, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abulrob A, Brunette E, Slinn J, Baumann E, Stanimirovic D: In vivo time domain optical imaging of renal ischemia-reperfusion injury: Discrimination based on fluorescence lifetime. Mol Imaging 6: 304–314, 2007 [PubMed] [Google Scholar]
- 23.Datta R, Alfonso-García A, Cinco R, Gratton E: Fluorescence lifetime imaging of endogenous biomarker of oxidative stress. Sci Rep 5: 9848, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Almeida RF, Loura LM, Prieto M: Membrane lipid domains and rafts: Current applications of fluorescence lifetime spectroscopy and imaging. Chem Phys Lipids 157: 61–77, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Stöckl MT, Herrmann A: Detection of lipid domains in model and cell membranes by fluorescence lifetime imaging microscopy. Biochim Biophys Acta 1798: 1444–1456, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Schweitzer D, Deutsch L, Klemm M, Jentsch S, Hammer M, Peters S, Haueisen J, Müller UA, Dawczynski J: Fluorescence lifetime imaging ophthalmoscopy in type 2 diabetic patients who have no signs of diabetic retinopathy. J Biomed Opt 20: 61106, 2015 [DOI] [PubMed] [Google Scholar]
- 27.Patalay R, Talbot C, Alexandrov Y, Lenz MO, Kumar S, Warren S, Munro I, Neil MA, König K, French PM, Chu A, Stamp GW, Dunsby C: Multiphoton multispectral fluorescence lifetime tomography for the evaluation of basal cell carcinomas. PLoS One 7: e43460, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, Duchen MR: Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun 5: 3936, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hato T, El-Achkar TM, Dagher PC: Sisters in arms: Myeloid and tubular epithelial cells shape renal innate immunity. Am J Physiol Renal Physiol 304: F1243–F1251, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY: Suppression of notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435: 98–104, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC: Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Wagner MC, Campos-Bilderback SB, Chowdhury M, Flores B, Lai X, Myslinski J, Pandit S, Sandoval RM, Wean SE, Wei Y, Satlin LM, Wiggins RC, Witzmann FA, Molitoris BA: Proximal tubules have the capacity to regulate uptake of albumin. J Am Soc Nephrol 27: 482–494, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clayton AH, Hanley QS, Verveer PJ: Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J Microsc 213: 1–5, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Redford GI, Clegg RM: Polar plot representation for frequency-domain analysis of fluorescence lifetimes. J Fluoresc 15: 805–815, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Digman MA, Caiolfa VR, Zamai M, Gratton E: The phasor approach to fluorescence lifetime imaging analysis. Biophys J 94: L14–L16, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.