

Abstract
Multiplexing, labeling for multiple immunostains in the very same cell or tissue section in situ, has raised considerable interest. The methods proposed include the use of labeled primary antibodies, spectral separation of fluorochromes, bleaching of the fluorophores or chromogens, blocking of previous antibody layers, all in various combinations. The major obstacles to the diffusion of this technique are high costs in custom antibodies and instruments, low throughput, and scarcity of specialized skills or facilities. We have validated a method based on common primary and secondary antibodies and diffusely available fluorescent image scanners. It entails rounds of four-color indirect immunofluorescence, image acquisition, and removal (stripping) of the antibodies, before another stain is applied. The images are digitally registered and the autofluorescence is subtracted. Removal of antibodies is accomplished by disulfide cleavage and a detergent or by a chaotropic salt treatment, this latter followed by antigen refolding. More than 30 different antibody stains can be applied to one single section from routinely fixed and embedded tissue. This method requires a modest investment in hardware and materials and uses freeware image analysis software. Multiplexing on routine tissue sections is a high throughput tool for in situ characterization of neoplastic, reactive, inflammatory, and normal cells.
Keywords: antibody removal, epitope, immunofluorescence, multiplex
Introduction
An increasing percentage of diagnosis in pathology is finalized to the identification of specific proteins, relevant for the patient’s management, via antibodies and the deposition of a pigment or fluorochrome at the protein’s location in the tissue section. The variety of specific antibodies is of increasing numerosity and complexity, ranging in the hundreds for an average lab, targeting constitutive tissue elements, oncogenes, growth factor and hormone receptors, aberrant product of genomic aberrations, and so on. Some assays are aimed at identifying diseases or refine the pathological classification, others at guiding the therapy. The standard is to perform one stain at a time, on serial sections, until the amount of material allows it, and no competitive tests (e.g., genetic or extractive) are favored over the immunostain.1 In the life sciences realm, fluorescent reporters are used more often, not necessarily viewed under a microscope, but also with a variety of other quantitative methods (flow cytometry, image cytometry, confocal microscopy, etc.), which better suit the analysis of live or lightly fixed cells. The fundamental difference between immunohistochemistry (IHC) and immunofluorescence (IF) is the use of light: absorption for IHC, emission for IF. Connatural with IF are the diversification of fluorescent reporters, limited by the source of light, the overlap of fluorescence spectra, the available fluorochromes, and, ultimately, the cost of the reagents.
Surgical pathologists do not venture outside IHC and, if so, very rarely.2 Life science scientists follow the ground rules of IHC when dealing with fixed and embedded tissues.3
Recently, the interest in performing multiple assays on formalin fixed, paraffin embedded (FFPE) specimens has gained ground, both in pathology and in the life sciences fields, and is currently referred to as multiplexing. To set aside a plethora of multiple staining methods of this type of material already published,4–6 we consider only methods in which more than three different stains are performed on the very same slide.
A strategy to perform multiple stainings at the same time on the same section involves the use of animal-specific secondary antibodies linked with a reporter directed against antibodies raised in different animals (rabbit, goat/sheep, rat, and three to four mouse isotypes). Given the limited variety of antibody sources, it is inevitable to devise multiplexing strategies to stain antibodies raised in the same species with contrasting colors. This has been accomplished essentially in three ways: (1) bleaching directly conjugated primary antibodies before adding other layers, (2) blocking the access to a previously deposited antibody for a second staining round, or (3) removing antibodies from sections after staining and imaging.
The use of directly labeled primary antibodies has been pioneered by Schubert et al.,6 followed by others.7–9 After the tissue image has been acquired, the fluorophore is inactivated by ultraviolet (UV) light,6 alkaline solutions,9 or sodium borohydride (NaBH4).7 The major drawbacks of this technology are the cost of the directly labeled antibodies, the requirement for a customized conjugation, the loss of antigens,10 and the occasional lack of sensitivity for important makers.8,11 In addition, if a robotized solution is available for otherwise highly repetitive tasks, the number of staining stations is a limiting factor for throughput.6,8 Last, the steric hindrance of a previously deposited antibody against the subsequent deposition of the very same antibody has not been addressed at all.8,12
Blocking the access to the first antibody layer is an old technique,13 based on the blocking ability of an insoluble (and therefore hydrophobic) precipitate such as diaminobenzidine (DAB), which prevents another staining round to get access to the first, even if the antibodies in both rounds are raised in the same species. Because the amount of insoluble precipitate conditions the access of the second staining to the same subcellular or cellular structure, a variety of hybrid IHC−IF methods have been employed,14 using extensive antibody titrations and differential reporter sensitivity, for example, with tyramide (insoluble) precipitation.15
A purely immunological block by using Fab monomeric fragment dates back several years16,17 and has been used rarely for multiplexing in IF: Concentrations of the Fab fragments in excess of 500 µg/ml, required to achieve a noncomplete blocking, are costly and impractical.16,18
Removal of the previously deposited layer of antibodies is a strategy tested by multiple investigators9,14,19–23 and involves a broad range of solutions, chemical agents, and temperature. The protocols range from a very mild boiling in Antigen Retrieval solution15 (shown by us—Gendusa et al.19—and others—Tornehave et al.24—to be inefficient at removal of antibodies), to an acidic Glycine buffer,23 to strong chemicals,22 to proteases,9 and to a mixture of strong reducing agent and a detergent.19,21
The cyclic deposition of an immune layer, the capture of the image, the removal of the antibodies and reporters, and a new immunostaining is suitable both for IHC and IF methods.14,19,21
Alcohol-soluble precipitates and single color IHC are required for sequential IHC staining. In fluorescence, the limits are dictated by the fluorochromes and the filters. Additional colors may be added to the standard four (4′6-diamidino-2-phenylindole dihydrochloride [DAPI], fluorescein isothiocyanate [FITC], tetramethylrhodamine isothiocyanate [TRITC], Cy5) by using spectral deconvolution of the fluorescent signals.25,26
Most of these methods for multiplexing had a limited diffusion, and only a few could demonstrate more than a dozen staining on the same section; with the exception of two methods based on directly labeled antibodies,6,8 only one applied on routinely treated sections.
The landscape of multiplexing may change because of the introduction of the so-called “next generation IHC,” a multiplex technique based on isotope-tagged antibodies and in situ mass-spectrometry detection (reviewed in Rimm27). In addition, barcode labeled probes, including antibodies, may allow quite extensive multiplexing with the NanoString technology (http://NanoString.com). Although, the ability to visualize single cells in a whole slide image of both systems is unknown.
Having published a preliminary evidence for a potentially high-volume multiplexing by antibody removal19 and established the foundation for the reproducibility of the method,28 we embarked in a study to investigate antibody removal methods, one novel, and multiplexing for >30 antibodies on a single routinely processed section.
Materials and Methods
Tissues and Antigen Retrieval
FFPE fully anonymous human leftover material used was exempt from the San Gerardo Institutional Review Board (IRB) approval as per Hospital regulations (ASG-DA-050 Donazione di materiale biologico a scopo di ricerca e/o sperimentazione, May 2012).
Three micron sections were cut and placed on coated glass slides (SuperFrostRPlus; Bio-Optica, Milano, Italy), baked in a vertical position overnight at 40C or for 1 hr at 60C, dewaxed using xylene, rinsed in a graded alcohol series, rehydrated in distilled water29; antigen retrieval (AR) was performed as published30 with (ARx) or without (AR) as pretreatment. Sections in distilled water were inserted into radiotransparent slide holders (model #S2029; Dako, Glostrup, Denmark) and transferred to a glass container filled with 800 mL of the retrieval solution (10 mM EDTA in Tris-buffer pH 8; Sigma-Aldrich, Milan, Italy). The container was irradiated in a household microwave oven at full power for 8 min, followed by 20 min of intermittent electromagnetic radiation to maintain constant boiling. Sections were cooled to about 50C before transferring to buffer, being ~60C, a sort of landmark between two temperature ranges with quite different effects on antigens.30 The ARx procedure maximizes the antigen exposure in tissue, overcoming the requirement for a pH-dependent retrieval for individual antibodies.30
Sections not being stained for extended periods of time (typically >3 days) were stored at −20C in 50% glycerol (Sigma-Aldrich) in pH 7.5 Tris buffer and 300 mM sucrose (5% of a saturated solution).
Primary and Secondary Antibody Dilution and Incubation
Primary antibodies to be used for multiplexing (Supplemental Table 1) were screened for sensitivity and specificity31 on target positive tissue sections at 1 and 0.1 µg/ml dilution in TrisHCl buffered saline (TBS) to which 2% bovine serum albumin (BSA), 0.05% sodium azide, and 100 mM trehalose were added, counterstained with an alkaline-phosphatase conjugated secondary antibody and developed with nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyl phosphate (NBT-BCIP; Roche, Monza, Italy).32 If the antibody concentration was not known, two one-log dilutions of the recommended dilution were tested. One µg/ml was almost invariably the dilution of choice and a saturating concentration. To test for saturating concentration, a 12 ml, 1 µg/ml solution of CD79a in a vertical five-slide mailer (model 715409; Electron Microscopy Science, Hatfield, PA) stained 36 sequentially placed tissue sections with an average staining variation intensity of −4% ±6% from time 0.
The dilution and the diluent were found appropriate for both IF and IHC, for either 1 hr or overnight incubation. Compared with 1 hr, overnight incubation increases the staining strength of a factor between 30% and 200%, depending on the antibody (Supplemental Fig. 1A). Secondary antibodies (Supplemental Table 2) were diluted in the same diluent and used at the 1:300 dilution.
Incubation was at room temperature in a horizontal plastic slide box (Kartell, Milan, Italy) containing a moist paper towel. At least 100µl of antibody was applied for a section of 1 × 1 cm or less, and the volume multiplied accordingly for larger sections.
TBS buffer, to which 0.01% Tween-20 (Sigma-Aldrich) and 100 mM sucrose were added (TBS-Ts), was used throughout all the experiments for washing. Three 5 min changes of buffer were used throughout.
For IF, antibodies of different isotype and/or species were pooled, each at the final dilution and incubated for the designed amount of time.
After washing, pooled, non-cross-reactive conjugated secondary antibodies were applied for 30 min.
A shortened sequence of primary and secondary antibodies (30 min each, with three TBS-Ts washes in between), so-called double indirect IF staining, repeated once after the first IF staining cycle32 doubles the staining intensity (Supplemental Fig. 1B) and was used when appropriate.
The variability of the staining efficiency, measured on duplicate parallel staining in serial sections, averages 3.1% (minimum 0%, maximum 12.3%) of a given fluorescent channel and is shown in Supplemental Table 3.
Slides stained in IF were mounted with phosphate buffered (pH 7.5) 60% glycerol–40% distilled water mixture containing 0.2% N-propyl gallate and 584 mM sucrose (10% of a saturated solution), to which DAPI dilactate 5.45 µM (Sigma) was added, the latter from a 1.09 mM stock solution in phosphate-buffered saline (PBS). The concentration of DAPI was adjusted so that the DNA DAPI fluorescence did not bleed into the other channels.
Preliminary work showed that a Glycerol-based mounting medium containing sucrose maintains or increases the antigen availability, differently from a Polyvinyl alcohol mounting medium (Sigma) containing 10% sucrose or other mounting media (e.g., Gycerol-Gelatin, C0563, Dako; FluoroGel; Electron Microscopy Sciences, Hatfield, PA), which decreases antigenicity (Supplemental Fig. 1C). Hardening fluorescent mounting media and glycerol-gelatin mix were found to cause antigen remasking with repeated use (not shown).
Antibody Stripping
Coverslip was gently removed by soaking the slides in TBS or distilled water (no effect on subsequent stripping detected; not shown).
Beta-mercaptoethanol/sodium dodecyl sulfate (2ME/SDS) stripping was performed as published,19 modified by halving the concentration of the buffer with an equal volume of distilled water. After stripping, the slides were washed in TBS-Ts for at least 30 min with repeated buffer changes.
Chaotropic salt-dependent removal of antibodies and antigen renaturation was performed as published by Narhi et al.33,34 Sections were immersed for 10 min at 40C in 12-ml 6-M guanidinium hydrochloride (GnHCl, Sigma) solution in vertical slide mailers. The GnHCl solution was buffered with 0.05-M citric acid–sodium citrate buffer solution (0.12 g citric acid monohydrate, 0.17 g trisodium citrate dihydrate in 100 ml).
The 40C temperature was chosen over room temperature (RT) for consistency; no other temperatures were tested.
A second shorter passage in 6-M GnHCl to which 5% w/v sucrose (290 mM) was added, ensured the removal of eluted antibodies. Subsequently, the sections were transferred to 6-M urea solution containing 5% sucrose at 40C for 10 min, followed by another 10 min incubation in 3-M urea and 5% sucrose solution. Finally, the sections were left to re-equilibrate in TBS-Ts. In some experiments, sections were then transferred to 10-mM EDTA in Tris-buffer pH 8 and irradiated until reaching the boiling point for 1 min and left to cool below 50C.
A schematic depiction of the staining and stripping sequence is shown in Table 1.
Table 1.
Flowchart of cyclic antibody staining and removal with 2ME/SDS and GnHCl.
| 2ME/SDS staining and stripping method | |
|---|---|
| 1 | Perform ARx on dewaxed sections affixed to positively charged glass slides |
| 2 | Allow to cool to about 50C or lower |
| 3 | Acquire the AF image for all the channels deemed necessary (optional) |
| 4 | Perform the first IF stain in 100 mM trehalose-containing dilution buffer |
| 5 | Mount with 60% Glycerol in PBS, 0.2% N-propyl Gallate, and 584 mM sucrose mounting medium containing 5.45 µM DAPI |
| 6 | Label the slide, acquire the images for all channels, including DAPI and AF, if not acquired before |
| 7 | Unmount the slides in buffer/distilled water |
| 8 | Transfer to Tris buffer |
| 9 | Immerse for 30 min in preheated (56C) 2ME/SDS buffer with agitation |
| 10 | Transfer to Tris buffer and wash extensively with TBS-Ts buffer |
| 11 | Repeat from step 4 with additional positive and/or negative antibodies |
| 12 | Store in 50% glycerol at −20C/−80C for extended storage, before returning to step 4 or proceed as below |
| 13 | Perform H&E or insoluble stainings if final |
| GnHCl staining and stripping method | |
| 1 | Perform ARx on dewaxed sections affixed to positively charged glass slides |
| 2 | Allow to cool to about 50C or lower |
| 3 | Acquire the AF image for all the channels deemed necessary (optional) |
| 4 | Perform the first IF stain in 100 mM trehalose-containing dilution buffer |
| 5 | Mount with 60% Glycerol in PBS, 0.2% N-propyl Gallate, and 584 mM sucrose mounting medium containing 5.45 µM DAPI |
| 6 | Label the slide, acquire the images for all channels, including DAPI and AF, if not acquired before |
| 7 | Unmount the slides in buffer/distilled water |
| 8 | Transfer to Tris buffer |
| 9 | Immerse in GnHCl 6 M at 40C for 10 min |
| 10 | Transfer to GnHCl 6 M + 290 mM sucrose at 40C for 10 min |
| 11 | Transfer to 6 M urea + 290 mM sucrose at 40C for 10 min |
| 12 | Transfer to 3 M urea + 290 mM sucrose at 40C for 10 min |
| 13 | Transfer to Tris buffer and wash with TBS-Ts buffer |
| 14 | Transfer to 10 mM EDTA AR buffer pH 8, boil for 1 min, cool to 50C |
| 15 | Transfer to TBS-Ts buffer |
| 16 | Repeat from step 4 with additional positive and/or negative antibodies |
| 17 | Store in 50% glycerol at −20C/−80C for extended storage, before returning to step 4 or proceed as below |
| 18 | Perform H&E or insoluble stainings, if final |
The sequential steps for repeated antibody staining and removal with 2ME/SDS and GnHCl are listed. Abbreviations: 2ME/SDS, 2-mercaptoethanol/sodium dodecyl sulfate stripping buffer; GnHCl, guanidinium hydrochloride; ARx, two-step antigen retrieval method30; AF, autofluorescence; IF, immunofluorescence; PBS, phosphate-buffered saline; TBS-Ts, Tris-buffered saline containing Tween-20 and sucrose; DAPI, 4′6-diamidino-2-phenylindole, dihydrochloride; H&E, hematoxylin and eosin. For detailed composition of buffers and solutions, see text.
Removal of the bound immunoglobulins with an NaBH4 treatment35 was performed as follows: A stable 4.4-M NaBH4 (Sigma) stock solution was obtained by dissolving 1.67 g in 10 ml of 14-M NaOH (Rohm & Haas, https://www.scribd.com/document/326437122/Sodium-Borohydride-Digest).
For antibody removal, sections were equilibrated in 0.05-M Tris-buffer pH 9, exposed to various freshly made concentrations of NaBH4 in the same buffer for 10 min, with or without 0.5% SDS, followed by immersion in 0.1-M citrate buffer pH 4 to inactivate NaBH4.
Immunofluorescence Scanner and Virtual Whole Slide Acquisition
The Hamamatsu Nanozoomer S60 scanner (Nikon, Campi Bisenzio, Italia) is equipped with an Olympus 20×/0.75 PlanSApo objective, a Fluorescence Imaging Module equipped with a L11600 mercury lamp (Hamamatsu, Roma, Italy), a linear ORCA-Flash 4.0 digital CMOS camera (Hamamatsu) and two six-position filter wheels, one for excitation, the other for emission filters, and a three-cube turret. The excitation filters (all from Semrock, Inc.; Rochester, NY) are 387/11 (DAPI), 420/10 (AF) 480/17 (FITC), 556/20 (TRITC), and 650/13 (Cy5). The emission filters are, in the same order, 435/40, 530/55, 520/28, 617/73, and 694/44. Three dichroic mirrors are as follows: a triband FF403/497/574-Di01 and two single pass Di03-R488 and FF655-Di01. The AF and Cy5 dichroic and emission filters are housed in a cube each. The filter combinations and the fluorochrome excited are accessible at the Semrock Searchlight website, https://goo.gl/dnYoMc.
After the first submission of this manuscript, we became aware that two commercial sources (Chroma Technology Corporation, Rockingam, VT; Semrock) have produced a filter set tailored for BV480 (excitation 436 or 438 nm, emission 478 or 483 nm, dichroic 458 nm) and which may be also used for AF acquisition.
Stained slides to be acquired were labeled with a two-dimensional (2D) barcode for automatic file name acquisition (TEC-IT Datenverarbeitung GmbH; Steyr, Austria), the bottom surface rinsed in distilled water to remove traces of sucrose and scanned in batch mode with predefined parameters. The barcode content is the sample identifier and the biomarker sequence, each biomarker flanked by a short fluorescence channel acronym for automatic coupling of each image with the biomarker by an image analysis software. The file name was kept as short as possible (i.e., below 20 characters).
Histograms data of fluorescence images as 8bit gray levels (0–255) and of at least 2 mm × 2 mm were obtained with Fiji and exported in an Excel spreadsheet. Cumulative percentage pixel numbers of the total in each channel were accrued over 255 channels, and the channel position where 90% of the pixels are found was used as the measurement of the fluorescence intensity (Scalia et al.,29 Supplemental Fig. 2, ibid.).
AF
The emission of tissue AF was quantified by exposing dewaxed, antigen-retrieved reference tissues (placenta, kidney, skin, etc.) to multiple exposure times in each of the filter combinations available (Supplemental Fig. 2A).
AF was acquired either with a separate channel on four-color stained sections during the image acquisition or on antigen-retrieved unstained sections, mounted with DAPI and coverslipped, in each filter combination, before the first round of immunostaining. In this case, the images were acquired at submaximal intensity for AF-rich tissues such as kidney or liver.
The mean pixel AF data for each tissue, each fluorescence channel, and each time point were extracted with the histogram function of ImageJ from the 8bit grayscale images, then plotted as intensity over exposure time in an Excel spreadsheet.
Chemical Inactivation of AF
For endogenous fluorescence quenching, a 1:280 dilution of an NaBH4 stock solution (NaBH4 15.7 mM, NaOH 0.05 N) was added to either 95% ethanol or to a 0.05 M Tris-buffer pH 9. As a control, an identical dilution of a stock 14-M NaOH solution was used. Sections were exposed to the NaBH4 or control solution for 10 and 30 min, either after the 99% ethanol deparaffinization step, or before antigen retrieval. Autofluorescent tissue samples, exposed to NaBH4, were imaged with three filter combinations (420/10: AF; 480/17: FITC; 556/20: TRITC) and the autofluorescence (AF) values imported in an Excel sheet.
Digital Subtraction of AF
AF was subtracted essentially as published by Pang et al.36,37 and Van DeLest et al.38
AF in some tissues (placenta, lymphoid tissue) had values of the same scale of intensity in each channel when measured and compared pixel by pixel (Supplemental Fig. 2C and 2D). In these cases, the linear equations for fluorescence in the AF filter and in the other filter were used to calculate an accommodation factor, to equalize the image obtained with the AF filter with the exposure and fluorescent response of the specific image, before subtraction.
As an example, the slope of the regression line for AF excited by the 420 nm filter is 0.61 × x + 2.7 = y, the slope for the 488 nm excitation is 0.26 × x + 0.31 = y.
If AF were acquired on placenta at 112 ms and the FITC image at 80 ms, then the factor to be used would be calculated as follows: (0.61 × 112 + 2.7) / (0.26 × 80 + 0.31) = 0.29, the first term being the value for AF, the second term the value for FITC, and 0.29 the factor to apply to the AF image to equal the AF background in the FITC image.
After importing in Fiji with the BioFormat Plug-in all the .ndpi files and saving them as .tiff files, the AF image was adjusted for the numerical factor of accommodation and subtracted from the stained image by using the Image Calculator function.
The procedure worked also in tissues with channel-specific AF values, although manual adjustment of the accommodation factor may be required. Alternatively, prestaining AF in each specific channel was acquired and then aligned and subtracted.
An example of AF subtraction is shown in Supplemental Fig. 2B.
AF subtracted images were used throughout for quantitative analysis and for imaging.
Registration
Removing and replacing the very same slide on the scanner stage entails microscopic translations and rotations, which misalign subsequently acquired images.
To realign the images (register), one image of the nuclei in the tissue (DAPI) was set as reference; then each other DAPI image acquired with a subsequent scan was aligned with the TurboReg Fiji plug-in.39 The coordinates of the registration were recorded as landmark (a .txt file with coordinates) and applied to the entire stacks made up of the images of the same round with the Multi StackReg Plug-in.40
Care was taken to produce whole slide images (WSI) as close to each other as possible; the registration process is considerably more difficult with images of different sizes, textures, and content.
Image Analysis
The amount of positive pixels, expressed as percentage of the area analyzed, was measured on each grayscale IF staining whole image, after application of a uniform threshold algorithm (Huang), for each staining cycle for that marker. WSI from routine IHC stain on serial sections, stained with DAB and counterstained with hematoxylin, were deconvoluted as published,30 inverted, and the pixel area quantified with the very same algorithm used for the IF images. Three separate fields per stain were analyzed.
Results
Antibody Removal by 2ME/SDS
A method based on strong reducing agent (beta mercaptoethanol; 2ME) and a detergent (sodium dodecyl phosphate; SDS) was previously published.19
We applied this method to sequential staining and stripping over 10 cycles with nine different antibodies representing membrane, cytoplasmic, and nuclear proteins and quantified the staining results. Variations for each subsequent staining were comprised within 10% above or below the initial result at time 0 (Fig. 1 and Supplemental Fig. 3A). We confirmed the reduced intensity for Ki-67 staining with 2ME/SDS incubation, however, the change was about 5% of the initial value and did not change after the first step (Supplemental Fig. 3B). CD44 was similarly mildly affected by this stripping method.
Figure 1.

Variability for nine markers over 10 staining and stripping 2ME/SDS cycles. One single section for every three markers was stained (time 0) and sequentially stripped and restained for the same markers 10 times. Variation in staining intensity is expressed as a fraction of the 256 8bit channels over the initial staining intensity. Primary Ab incubation time: 1 hr, 2nd Ab: 30 min, single indirect IF. Data are from four independent 10-cycle experiments. Abbreviations: 2ME/SDS, beta-mercaptoethanol/sodium dodecyl sulfate; Ab = antibody; IF, immunofluorescence.
An enhancing effect of the 2ME/SDS treatment, previously described,19 for example, on keratins, was not apparent or reduced when the ARx30 initial treatment was applied. ARx maximizes the immunoreactivity and overcomes the requirement for a tailored pH for AR (ibid.). Most of the nine antigens are expressed homogeneously on the cell, therefore, we digitally threshold the images and measured the amount of pixel, as a measure of the effect of the staining variations on the detection of the target. There was little variation in the amount of positive cells detected over the cycles (Supplemental Fig. 3C).
Antibody Removal by Chaotropic Salts
The 2ME/SDS stripping method uses hazardous chemicals with a strong odor; therefore, we investigated alternative methods.
Initial experiments of antibody removal with antichaotropic (saturated ammonium sulfate) and chaotropic compounds (GnHCl) showed that saturated ammonium sulfate was ineffective (not shown) but GnHCL at 5.3 and 6 M efficiently removed primary and secondary antibodies from sections (Fig. 2). The process was more efficient at pH 433 (not shown). Differently from 2ME/SDS, very dense spatially arranged proteins (e.g., pentameric cytoplasmic IgM, but not dimeric IgA or IgG) could not be completely stripped by GnHCl of bound primary and secondary antibodies (Fig. 2). This did not happen with 2ME/SDS.
Figure 2.
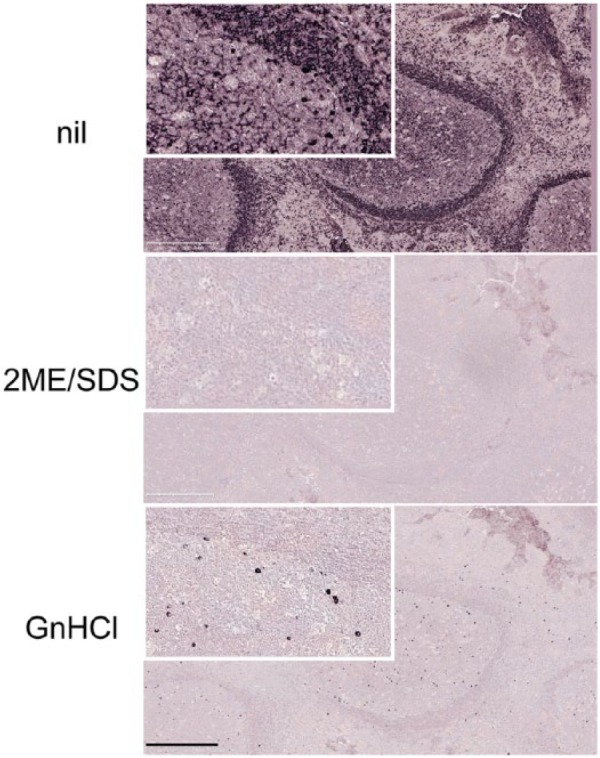
Comparison of antibody removal efficiency of 2ME/SDS and GnHCl. Tonsil sections were incubated with a FITC-conjugated anti-human IgM, stripped with either 2ME/SDS or GnHCl, counterstained with an AP-conjugated anti-FITC antibody and developed in NBT-BCIP. 2ME/SDS stripping (middle) leaves no stainable primary antibody. Stripping by GnHCl (bottom) leaves IgM+ plasma cells. Identical results are obtained with an anti-Rabbit AP-conjugated (not shown). High magnification in the insets. Scale bar: 500 µm. Abbreviations: 2ME/SDS, beta-mercaptoethanol/sodium dodecyl sulfate; GnHCl, guanidinium hydrochloride; FITC, Fluorescein Isothiocyanate; AP, alkaline phosphatase; NBT-BCIP, 5-bromo-4-chloro-3′-indolyl phosphate.
The treatment with GnHCl alone reduced the antigen availability33: This negative effect could be fully reversed by transferring the sections to a 6-M urea solution, followed by a 3-M urea solutions,33 both containing sucrose as a protein refolding agent (ibid.) and/or by a brief 1 min AR step (not shown). The combination of both 6 and 3 M urea followed by a short AR treatment in 10 mM EDTA in Tris-buffer pH 8 was found the most effective.
Variations for each subsequent staining over 10 stripping cycles were comprised between −5% and +30% of the initial result at time 0 (Fig. 3). The enhanced staining obtained with some antigens after one or more stripping cycles is due to the short AR step, as the same antigens showed a reduced detection (+5% to −15% variations over five cycles) when the AR step was not included (Supplemental Fig. 4). However, longer AR after a GnHCL-6-M urea treatment may mildly decrease some antigens (e.g., CD20, CD44; not shown). A broader variation in the amount of cell target detected was observed (Supplemental Fig. 3C) compared with the data obtained with 2ME/SDS.
Figure 3.

Variability for nine markers over 10 staining and stripping GnHCl, 6-M urea cycles. One single section for every three markers was stained (time 0) and sequentially stripped and restained for the same markers 10 times. Variation in staining intensity is expressed as a fraction of the 256 8bit channels over the initial staining intensity. Primary Ab incubation time: 1 hr, 2nd Ab: 30 min, single indirect IF. Abbreviations: Ab = antibody; IF, immunofluorescence.
Antibody Removal by NaBH4
NaBH4 has proteolytic properties,41 acting on peptide bonds mildly and selectively,42,43 particularly for serine, threonine, and asparagine, but not others. NaBH4 can cleave peptide linkages and reduce disulfide bonds.42 In addition, it suppresses fluorochrome fluorescence.44 Thus, it is an attractive chemical to remove bound antibodies.
We tested NaBH4 15 mM on bound antibodies of different species and mouse isotypes and found it most effective on rabbit and goat Ig, less on mouse IgG1 and IgG2a (Fig. 4). NaBH4 at lower molarity was much less effective (not shown). The addition of 0.5% SDS improved the removal of mouse IgG2a and partially of rabbit and goat Ig, at the cost of tissue damage with repeated applications (not shown).
Figure 4.
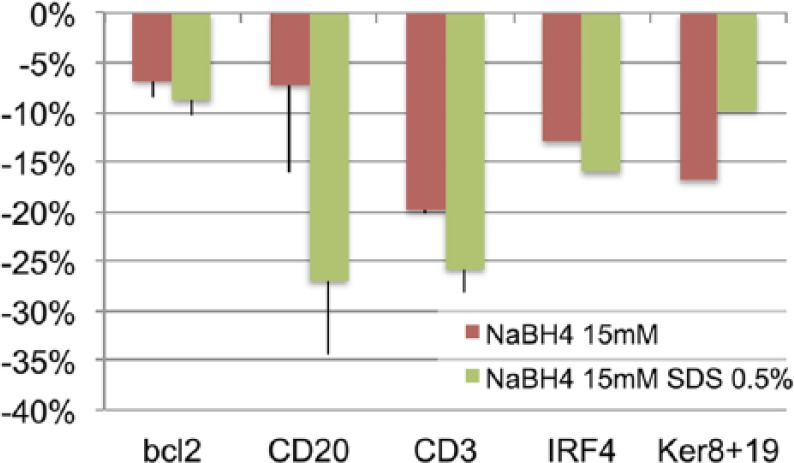
Effect of NaBH4 15 mM alone or in combination with SDS on bound primary antibodies. Stripping is enhanced by SDS and the effect is proportional to the starting abundance of the target. Bcl-2, mouse IgG1; CD20, mouse IgG2a; CD3, rabbit Ig; IRF4, goat Ig; Ker8+19, pooled rabbit Ig anti-keratin 8 and 19. Abbreviations: NaBH4, sodium borohydride; SDS, sodium dodecyl phosphate.
The application of 1 mg/ml (26 mM) NaBH4 in buffer at RT for longer than 20 min results in loss of the tissue (Supplemental Fig. 5A and 5B). However, NaBH4 treatment for 10 min at a concentration between 0.6 and 15 mM does not affect tissue antigens, even after repeated applications (Fig. 5). Because of its similarities with 2ME in reducing disulfide bonds, we tested NaBH4 as a 2ME substitute in the ARx method,30 and found it ineffective (not shown).
Figure 5.
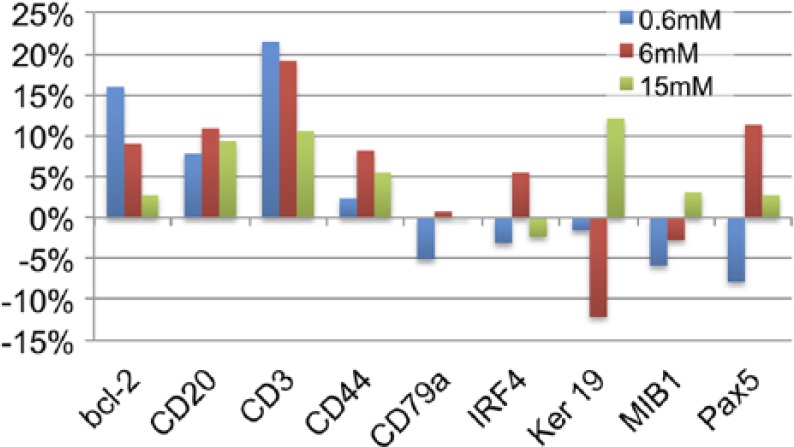
Effect of NaBH4 0.6, 6, and 15 mM on antigens. Sections have been treated with 10 cycles of 10 min NaBH4 pH 9, followed by pH 4 buffer. Channel intensity variation over 256 channels is shown. Abbreviation: NaBH4, sodium borohydride.
Effect of Antibody Removal on Target Detection and AF
As shown in Figs. 1 and 3 and Supplemental Figs. 3 and 4, the variation in staining over repeated cycles of staining and stripping is low. As published before,19 a decrease in staining affects only some antigens, and, here, we show that it is limited to the very first cycles (Supplemental Fig. 3B).
Moreover, by alternating positive antibody and negative control staining over 10 cycles, the controls remain below the lower detection limit of a positive stain across the cycles (Supplemental Fig. 3A).
To quantitatively assess the extent of antibody removal by 2ME/SD and GnHCl, we first addressed how the two stripping methods affect the fluorochromes, to distinguish between antibody removal and fluorochrome quenching. Stained sections were extensively cross-linked with formalin and subject to 2ME/SD or GnHCl stripping; differently from NaBH4, no effect on fluorescence was observed after both stripping methods (not shown). Formalin fixation did not affect fluorescence (not shown). Based on this result, any change in staining must be due to antibody removal.
We then stained in double indirect IF the sections for abundant proteins with repetitive motifs (i.e., keratins and vimentin), measured the staining intensity, stripped, restained the stripped sections with negative control Abs and fluorochrome-conjugated antibodies (Fig. 6 and Supplemental Fig. 6). Not only the antibody removal, both primary and secondary, was almost total, but restaining with the same powerful double indirect IF failed to detect a significant amount of leftovers.
Figure 6.
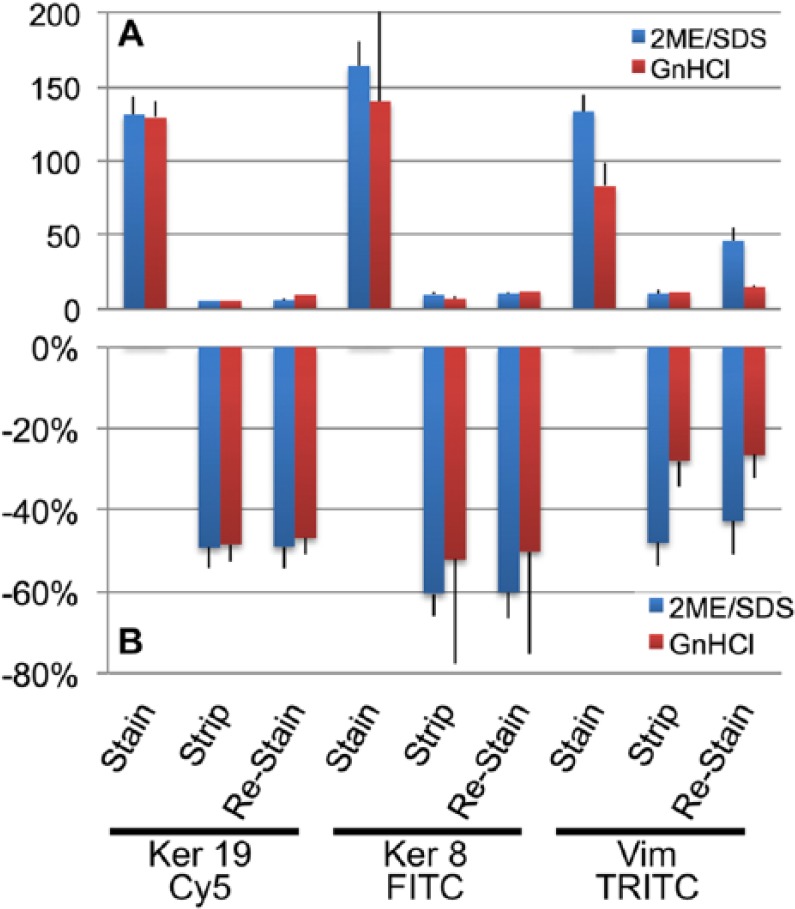
Effect of stripping and restaining on abundant antigens. Sections in duplicate were stained in double indirect IF for the indicated abundant antigens (intermediate filaments). The channel value (A) or the % changes from the time 0 stain over 256 channels (B) ± SD are plotted before (Stain), after stripping (Strip), and after restaining with species- and isotype-matched nonimmune control Abs (Restain). Primary Ab incubation time: O/N, 2nd Ab: 30 min, double indirect IF. Abbreviations: Ab = antibody; IF, immunofluorescence.
We succeeded in sequentially staining and stripping in excess of 20 cycles (i.e., detection of 63 antigens) a single FFPE section, using the 2ME/SD method.
AF obtained before and after AR was qualitatively different, as measured by pixel-by-pixel comparison (Supplemental Fig. 2E).
For most tissues, where AF is scarce, there was little variation over several staining and stripping cycles; however, AF-rich tissues such as kidney displayed a bimodal loss of AF over the cycles (Fig. 7). Pixel-by-pixel comparison at time 0 and after the last stripping showed a heterogeneous loss of AF, not seen in an immune infiltrate in the same section (Supplemental Fig. 2F), suggesting selective inactivation or extraction of highly fluorescent tissue components.
Figure 7.
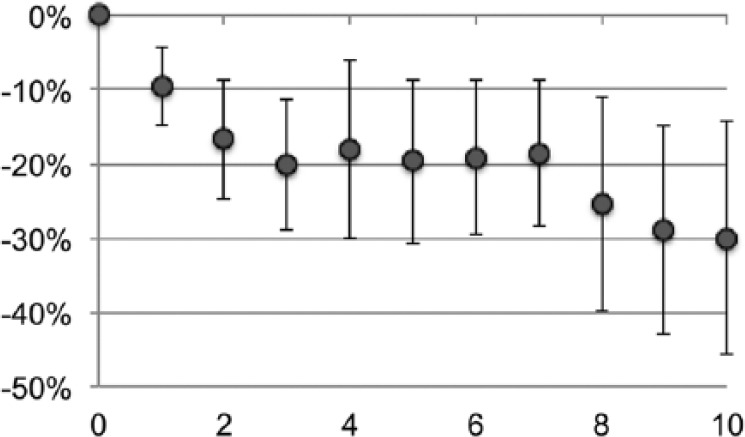
Cycle-dependent decrease of kidney tissue AF. Variation over time of the kidney tubules AF (em 520 nm), expressed as % channel changes from time 0, over 10 staining and 2ME/SDS stripping cycles. Eleven different areas from three samples have been imaged, and the average intensity change over the starting AF is expressed as % change over 256 channels ± SD. Abbreviations: AF, autofluorescence; 2ME/SDS, beta-mercaptoethanol/sodium dodecyl sulfate; SD, standard deviation.
AF Chemical Subtraction
Exposure of highly autofluorescent tissues to NaBH4 15 mM in 95% EtOH for 10 min reduced the tissue AF in the three channels by 6–16% below the control values (Supplemental Fig. 5). The reduction was specifically due to NaBH4, because the vehicle, NaOH 0.05 N, produced a modest increase in AF. The treatment of dewaxed sections, before the AR step, for 10 min with 15 mM NaBH4, caused a 19–24% increase of AF and a significant reduction of antigenicity (not shown) and, if contacted for 30 min or more, partial or total loss of the tissue.
In multiple experiments on tonsil and kidney tissue, no significant AF reduction was obtained (not shown), as previously published.45
Discussion
A dewaxed, antigen-retrieved FFPE section is essentially identical to a Western blot sheet,46 which can be stained and stripped multiple times of previously deposited antibodies. In a Western blot, the proteins are linearized and separated by molecular weight. The FFPE tissue, however, contains individual proteins cross-linked in situ with unknown bystanders whose epitopes are rescued from the processing-associated masking and reexposed. As for the proteins immobilized on a membrane, tissue epitopes, once reexposed, need to maintain the minimum amount of water to preserve the shape in an immuno-recognizable fashion.28 This essential requirement has been totally overlooked in routine IHC or IF, because there has never been the need to reuse a stained section, until it became a prominent issue for reproducibility when performing multiple sequential staining rounds. By moving the concept of a tissue section closer to a blot membrane and by examining the effect of every component on the antigens at each step of the multiplexing process, we have achieved optimal reproducibility and a low coefficient of variation upon repeated staining.
Diverse Molecular Mechanism of Antibody Removal Yields an Identical Outcome
The two methods shown here for multiplexing work with two quite different mechanisms.
The 2ME/SDS method is chemically altering the structure of the primary and secondary antibodies. Disulfide bonds are in a thermodynamic equilibrium for each given type of antibody47: the strong reducing agent, 2ME, tilts the balance toward an unbound form. The detergent SDS favors the dissociation of the Ig heavy chains,48 and the combined effect overcomes the bond between epitope and paratope, even for high-energy affinity.19 The heavily cross-linked tissue is resistant to the combined action of 2ME and SDS, at least for repeated exposure but at a temperature below the one required for tissue solubilization.30
The decrease of tissue AF with stripping cycles is in contrast with the little variation shown in antigen detection. Data for the smallest protein tested, bcl-2 weighing 26 kDa, point to a stable presence over repeated cycles for proteins of that molecular weight or larger, cross-linked in the tissue. Either repeated application of 2ME and SDS are quenching the AF, or small soluble AF compounds, not firmly cross-linked in the tissue, may be progressively removed from the section. A similar decrease of AF with cycle repetition was observed with a fluorochrome-quenching technique49; differently from this method, we did not observe cell loss.
The method based on GnHCL produces a rapid conformational change of the epitope and of the paratope,50 in part, altering the water structure that holds the epitope in shape. The detachment of the bound antibody is the effect sought, but the amount of the available epitopes is significantly diminished, probably because of incorrect refolding of epitopes after GnHCl.33 To correctly refold any available epitopes exposed after AR, the section is transferred to high molar urea,33 then to a diluted urea solution, in the presence of a folding enhancer, sucrose.51 Epitope-refolding is further enhanced with a brief exposure to high temperature, analogously to AR.
The interaction of GnHCl with the protein-associated water molecules in the first phase of denaturation50 may account for the inability to remove antibodies bound in high amount in packed molecular structures, possibly resulting in precipitation of the antibody at the antigen site and insolubility, because of the removal of critical water molecules.
The use of a chaotropic agent to reversibly modify the epitope conformation in a controlled fashion, to detach a bound antibody, further emphasizes the metastable nature of reexposed epitopes in FFPE material, as previously published.28 The efforts aimed at preserving the epitope conformation during each and every step of the multiplexing process, including antibody incubation and coverslipping, represent the other novelty of the present method: Reproducibility over multiple staining cycles has been obtained by using buffers and chemicals tested on purpose by quantitative IF for epitope stability. Differently from other multiplexing methods,26 removal of antibodies with 2ME/SDS does not require additional treatments such as AR between cycles, leaving tissue antigens unmodified for the whole sequence. Because of that, no staining prioritization is necessary.
Comparison of 2ME/SDS Versus GnHCl
Despite the identical outcome of the two protocols, there are specific characteristics for each one, which suggest a preferential use.
GnHCl is a nontoxic, safe method that works best where a short cycling sequence is required and sparsely represented proteins are involved. The enhancing effect of the short AR step may be exploited to prioritize the staining sequence. The inability to entirely remove dense antibody deposits such as IgM in plasma cells is compensated by the poor immune reactivity of the aggregated leftovers that are eventually left (Fig. 2).
The 2ME/SDS method has a thorough antibody removal ability and, combined with disaccharide protection, minimal or no effect on the antigens over extensive staining and stripping cycles. However, it depends on an optimal antigen retrieval from the very beginning, because no additional AR steps are involved. It requires a waterbath with controlled temperature and confinement of a chemical with a strong odor.
The extent of primary and secondary antibody removal was tested in multiple ways, quantitatively and qualitatively, exemplified in Figs. 2 and 6, and Supplemental Figs. 3A and 6. First, we excluded a bleaching effect of both 2ME/SD and GnHCl on the fluorochromes used. Next, we acquired the tissue images poststripping with the very same fluorescence setting. Finally, we restained stripped sections with the same protocol used to get the initial stain, with the substitution of a negative, isotype-matched irrelevant antibody as the first step (Fig. 6, and Supplemental Figs. 3A and 6) and imaged again. To test for the consistency of stripping, sections were consecutively stained with a positive and a negative antibody every other cycle (Supplemental Fig. 3A for 2ME/SDS and not shown for GnHCl). To test for stripping completeness in IHC (Fig. 2), we used a small molecule (FITC) as a robust hapten, resistant to modifications28 and a sensitive IHC development. The aggregate results show that residual antibodies after stripping, if ever present, are below detectability, within the staining method employed.
During this investigation, we came across multiple interesting characteristics of NaBH4, a compound used in biochemistry, fluorescent microscopy, and tissue fixation. The ability to quench the fluorochrome emission and the proteolytic activity suggested its use for multiplexing, although we found that in several previous reference papers, the pH of the diluent used shortened the half-life and, therefore, its action to mere seconds.52,53 Differently from proteases, occasionally used for multiplexing9,23 and known to affect tissue antigens, NaBH4 by itself proved to possess some ability to remove antibodies from tissues without damage (Figs. 4 and 5). However, the addition of SDS causes substantial damage to the tissue with treatment repetitions, and NaBH4 was not further pursued.
Antibody Removal-Based Multiplexing Is a Powerful Technique of Widespread Use
There are several advantages for the antibody stripping method we describe.
All reagents and instruments are common, commercially available, and low cost, compared with other techniques employing directly conjugated primary antibodies, amplification kits, complex software, and spectral microscopes.
One single section of routine FFPE material is required, opening a wealth of clinical specimens for study. Because all the stains are eventually combined in one multiplexed stack of images, the system can be optimized by selecting antibodies that differ by species and isotypes, filling each staining round with three stains.
Steric hindrance caused by interacting or heterodimerizing proteins can be circumvented by spacing the stainings in two separate staining cycles.
In addition, mistakes in staining can be mended by restaining for the same antibody.
Despite the fact that the filter setting we used allows the detection of seven fluorochromes and up without cross talk (DAPI/BV421, FITC, TRITC, Cy5, Pacific Orange/BV480, BV605, PerCp-Cy5.5, BV711; see https://goo.gl/scE3sd), the limit is the simultaneous detection in indirect IF of two antibodies of the same kind with two separate fluorochromes, an area we are investigating.
During this investigation, we found it convenient to allocate antigens in the various channels according to abundance (lowest to highest) in the following order: TRITC, Cy5, FITC. This is because of the dye brightness with our filter set (Rhodamine RedX: 1.55 × 10−6 mW; Alexa 488: 2.89 × 10−7 mW; Alexa 647: 3.96 × 10−7 mW), the differential sensitivity of the sensor to light (maximal between 520 and 640 nm), the absence of AF in the Cy5 channel, and the digital removal of AF.
The combination of (1) a careful choice of validated antibodies used at a concentration not exceeding 1µg/ml; (2) an enhancement of the sensitivity (superior AR, double indirect IF); (3) the choice of buffers and mounting media; (4) the acquisition with a sensitive, linear detector; and (5) the removal of the noise caused by AF, all concur to deliver a superior signal over multiple staining and stripping cycles.
A limitation of some popular multiplexing methods consists in the size limit of the area that can be digitally acquired, either because of how the software has been designed or because of long acquisition time or the image format. By acquiring whole slide images in a format that can be read by many platforms, the areas of interest need not to be known upfront, and additional regions of interest can be investigated later. Care should be taken to increase the computing random-access memory (RAM) to at least 32 Gb and to limit each single whole slide image below 6 Gb.
Multiplexing as a Discovery Tool
Multiplex IF, as we describe here, is a powerful discovery tool. By applying as little as a dozen markers on the very same tissue section, we could dissect the phenotype of rare and scattered immune cells such as interfollicular dendritic cells or germinal center follicular helper T cells, highlighting the power of multiplexing to investigate polymorphous reactive immunopathology.
As shown in Fig. 8, S-100+ cells of dendritic appearance can be simultaneously assessed for activation, proliferation, Class II major histocompatibility complex (MHC) molecules, and transcription factors expression, monocyte markers, and geographic localization in the tissue.
Figure 8.
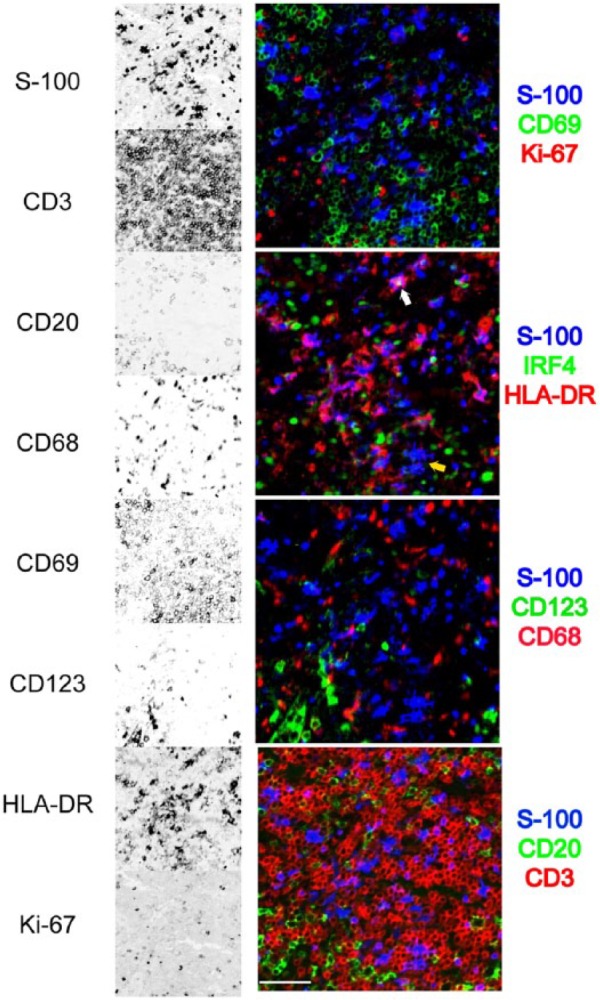
Detail of a multiplexed interfollicular tonsil area. Eight stains out of a 32-antibody multiplex are selected from a tonsil interfollicular area and shown as single stains (inverted gray scale) on the left, or combined into four three-color RGB composites. The area contains numerous S-100+ dendritic cells, HLA-DR+ with exceptions (yellow arrow), occasionally IRF4+ (white arrow), negative for Ki-67, CD69, CD68, and CD123. The dendritic cells are located in a CD3+ T-cell area, without contact with CD20+ B cells. GnHCl stripping method. Scale bar = 100 µm. Abbreviations: RGB, red green blue additive color; GnHCl, guanidinium hydrochloride.
By multiplex staining, we reassessed the expression of PD-1/CD274 on a subset of germinal center proliferating B cells expressing BCL6 (Fig. 9), a previously described subset.54 Interestingly, we were able to confirm the B cell expression by using the UMAB197 IgG2a but not the NAT105 IgG1 antibody, despite the full co-localization on follicular helper T cells (not shown).
Figure 9.
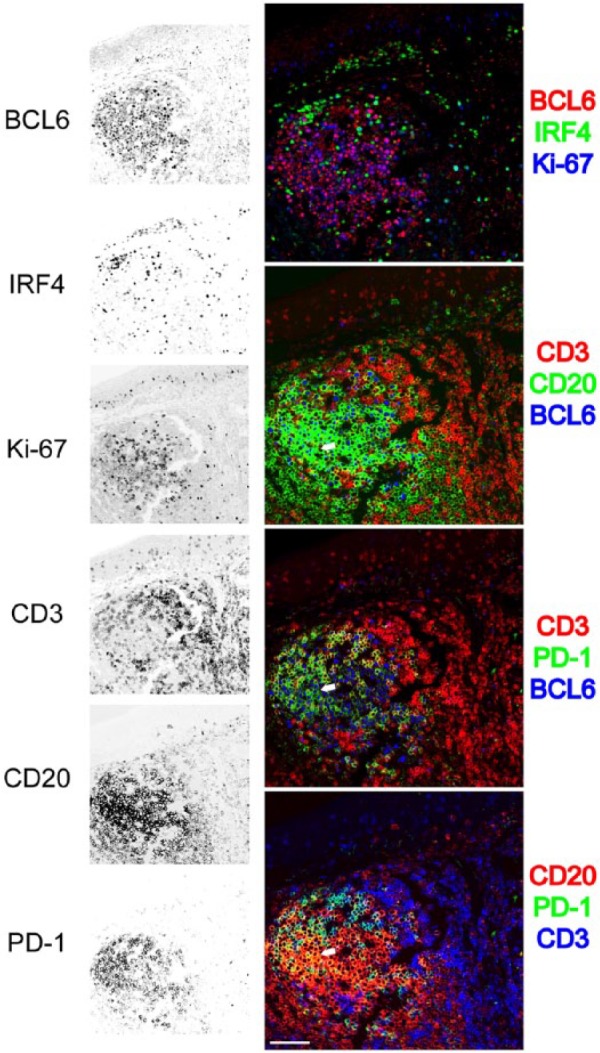
Detail of a multiplexed tonsil follicle. Six stains out of a 32-antibody multiplex are selected from a tonsil follicle and shown as single stains (inverted gray scale) on the left, or combined into four three-color RGB composites. Proliferating (Ki-67+) germinal center BCL6+ CD20+ B cells are mutually exclusive stained for IRF4. PD-1 stains CD3+ T cells in the germinal center as well as rare PD-1+, BCL6+, CD20+ centroblasts (white arrow). GnHCl stripping method. Scale bar = 100 µm. Abbreviations: RGB, red green blue additive color; GnHCl, guanidinium hydrochloride.
The comparison in cell number detection shown here (Supplemental Fig. 3C) between traditional single color IHC and multiplex IF opens the use of multiplexing for clinical investigation and use.
Supplementary Material
Acknowledgments
We wish to thank Dr. Franco Ferrario for continuous support, Emanuele Martella, Marco Cicuttin (Nikon, Italia), and Giulio Simonutti (Hamamatsu Italia) for expert advice; Linde DeSmedt (HistoGeneX NV, Antwerpen, Belgium) for suggestions; and Riccardo Tagliabue for expert technical help. The Hamamatsu S60 digital scanner was obtained as part of a clinical research project BEL114054 (HGS1006-C1121) of the University of Milano–Bicocca and GlaxoSmithKline, on which project Carla Rossana Scalia and Maddalena Maria Bolognesi are also supported.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: GC, CRS, and MMB equally designed the experiments. CRS performed immunofluorescent tests and acquired the digital preparations. MMB and MM devised the image analysis algorithms. FMB provided essential reagents. MF provided essential knowledge on fluorochrome characteristics, filter settings, and image analysis tools. FMB and SZ performed visual and digital image analysis. GC and MMB wrote the manuscript. All authors have read and approved the final manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by Departmental University of Milano–Bicocca funds. Carla Rossana Scalia and Maddalena Maria Bolognesi are employed by the Department of Medicine and Surgery of the University of Milano–Bicocca within a GlaxoSmithKline clinical research project BEL114054 (HGS1006-C1121). Francesca Maria Bosisio is funded by the MEL-PLEX research training program (“Exploiting MELanoma disease comPLEXity to address European research training needs in translational cancer systems biology and cancer systems medicine,” Grant Agreement No: 642295, MSCA-ITN-2014-ETN, Project Horizon 2020, in the framework of the MARIE SKŁODOWSKA-CURIE ACTIONS).
Literature Cited
- 1. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, Ishikawa Y, Wistuba I, Flieder DB, Franklin W, Gazdar A, Hasleton PS, Henderson DW, Kerr KM, Petersen I, Roggli V, Thunnissen E, Tsao M. Diagnosis of lung cancer in small biopsies and cytology: implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch Pathol Lab Med. 2013;137:668–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rimm DL. What brown cannot do for you. Nat Biotechnol. 2006;24:914–16. [DOI] [PubMed] [Google Scholar]
- 3. Ramos-Vara JA, Miller MA. When tissue antigens and antibodies get along: revisiting the technical aspects of immunohistochemistry—the red, brown, and blue technique. Vet Pathol. 2014;51:42–87. [DOI] [PubMed] [Google Scholar]
- 4. Mason DY, Micklem K, Jones M. Double immunofluorescence labelling of routinely processed paraffin sections. J Pathol. 2000;191:452–61. [DOI] [PubMed] [Google Scholar]
- 5. Mittag A, Lenz D, Gerstner AOH, Sack U, Steinbrecher M, Koksch M, Raffael A, Bocsi J, Tarnok A. Polychromatic (eight-color) slide-based cytometry for the phenotyping of leukocyte, NK, and NKT subsets. Cytometry A. 2005;65:103–15. [DOI] [PubMed] [Google Scholar]
- 6. Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Böckelmann R, Malykh Y, Gollnick H, Friedenberger M, Bode M, Dress AWM. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270–78. [DOI] [PubMed] [Google Scholar]
- 7. Adams DL, Alpaugh RK, Tsai S, Tang C-M, Stefansson S. Multi-phenotypic subtyping of circulating tumor cells using sequential fluorescent quenching and restaining. Sci Rep. 2016;6:33488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. PNAS. 2013;110:11982–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin J-R, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun. 2015;6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schubert W, Dress A, Ruonala M, Krusche A, Hillert R, Gieseler A, Walden P. Imaging cycler microscopy. PNAS. 2014;111:E215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clarke GM, Zubovits JT, Shaikh KA, Wang D, Dinn SR, Corwin AD, Santamaria-Pang A, Li Q, Nofech-Mozes S, Liu K, Pang Z, Filkins RJ, Yaffe MJ. A novel, automated technology for multiplex biomarker imaging and application to breast cancer. Histopathology. 2014;64:242–55. [DOI] [PubMed] [Google Scholar]
- 12. Remark R, Merghoub T, Grabe N, Litjens G, Damotte D, Wolchok JD, Merad M, Gnjatic S. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci Immunol. 2016;1:aaf6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lechago J, Sun NC, Weinstein WM. Simultaneous visualization of two antigens in the same tissue section by combining immunoperoxidase with immunofluorescence techniques. J Histochem Cytochem. 1979;27:1221–5. [DOI] [PubMed] [Google Scholar]
- 14. Pirici D, Mogoanta L, Kumar-Singh S, Pirici I, Margaritescu C, Simionescu C, Stanescu R. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J Histochem Cytochem. 2009;57:567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tóth ZE, Mezey E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J Histochem Cytochem. 2007;55:545–54. [DOI] [PubMed] [Google Scholar]
- 16. Negoescu A, Labat-Moleur F, Lorimier P, Lamarcq L, Guillermet C, Chambaz E, Brambilla E. F(ab) secondary antibodies: a general method for double immunolabeling with primary antisera from the same species. Efficiency control by chemiluminescence. J Histochem Cytochem. 1994;42:433–37. [DOI] [PubMed] [Google Scholar]
- 17. Nielsen B, Borup-Christensen P, Erb K, Jensenius JC, Husby S. A method for the blocking of endogenous immunoglobulin on frozen tissue sections in the screening of human hybridoma antibody in culture supernatants. Hybridoma. 1987;6:103–109. [DOI] [PubMed] [Google Scholar]
- 18. Lu QL, Partridge TA. A new blocking method for application of murine monoclonal antibody to mouse tissue sections. J Histochem Cytochem. 1998;46:977–84. [DOI] [PubMed] [Google Scholar]
- 19. Gendusa R, Scalia CR, Buscone S, Cattoretti G. Elution of high affinity (>10-9 KD) antibodies from tissue sections: clues to the molecular mechanism and use in sequential immunostaining. J Histochem Cytochem. 2014;62:519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glass G, Papin JA, Mandell JW. SIMPLE: a sequential immunoperoxidase labeling and erasing method. J Histochem Cytochem. 2009;57:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim M, Soontornniyomkij V, Ji B, Zhou X. System-wide immunohistochemical analysis of protein co-localization. PLoS ONE. 2012;7:e32043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tramu G, Pillez A, Leonardelli J. An efficient method of antibody elution for the successive or simultaneous localization of two antigens by immunocytochemistry. J Histochem Cytochem. 1978;26:322–24. [DOI] [PubMed] [Google Scholar]
- 23. Zrazhevskiy P, Gao X. Quantum dot imaging platform for single-cell molecular profiling. Nat Commun. 2013;4:1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tornehave D, Hougaard DM, Larsson L. Microwaving for double indirect immunofluorescence with primary antibodies from the same species and for staining of mouse tissues with mouse monoclonal antibodies. Histochem Cell Biol. 2000;113:19–23. [DOI] [PubMed] [Google Scholar]
- 25. Mansfield JR. Multispectral imaging: a review of its technical aspects and applications in anatomic pathology. Vet Pathol. 2014;51:185–210. [DOI] [PubMed] [Google Scholar]
- 26. Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods (San Diego, Calif). 2014;70:46–58. [DOI] [PubMed] [Google Scholar]
- 27. Rimm DL. Next-gen immunohistochemistry. Nat Methods. 2014;11:381–83. [DOI] [PubMed] [Google Scholar]
- 28. Boi G, Scalia CR, Gendusa R, Ronchi S, Cattoretti G. Disaccharides protect antigens from drying-induced damage in routinely processed tissue sections. J Histochem Cytochem. 2015;64:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scalia CR, Boi G, Bolognesi MM, Riva L, Manzoni M, DeSmedt L, Bosisio FM, Ronchi S, Leone BE, Cattoretti G. Antigen masking during fixation and embedding, dissected. J Histochem Cytochem. 2017;65:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scalia CR, Gendusa R, Cattoretti G. A 2-step Laemmli and antigen retrieval method improves immunodetection. Appl Immunohistochem Mol Morphol. 2015;24:436–46. [DOI] [PubMed] [Google Scholar]
- 31. Scalia CR, Gendusa R, Basciu M, Riva L, Tusa L, Musarò A, Veronese S, Formenti A, D’Angelo D, Ronzio AG, Cattoretti G, Bolognesi MM. Epitope recognition in the human-pig comparison model on fixed and embedded material. J Histochem Cytochem. 2015. a;63:805–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cattoretti G, Büttner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood. 2006;107:3967–75. [DOI] [PubMed] [Google Scholar]
- 33. Narhi LO, Caughey DJ, Horan T, Kita Y, Chang D, Arakawa T. Effect of three elution buffers on the recovery and structure of monoclonal antibodies. Anal Biochem. 1997;253:236–45. [DOI] [PubMed] [Google Scholar]
- 34. Narhi LO, Caughey DJ, Horan TP, Kita Y, Chang D, Arakawa T. Fractionation and characterization of polyclonal antibodies using three progressively more chaotropic solvents. Anal Biochem. 1997;253:246–52. [DOI] [PubMed] [Google Scholar]
- 35. Yakulis V, Schmale J, Costea N, Hellerp Production of Fc fragments of IgM. J Immunol. 1968;100:525–9. [PubMed] [Google Scholar]
- 36. Pang Z, Barash E, Santamaria-Pang A, Sevinsky C, Li Q, Ginty F. Autofluorescence removal using a customized filter set. Microsc Res Tech. 2013;76:1007–1015. [DOI] [PubMed] [Google Scholar]
- 37. Pang Z, Laplante NE, Filkins RJ. Dark pixel intensity determination and its applications in normalizing different exposure time and autofluorescence removal. J Microsc. 2012;246:1–10. [DOI] [PubMed] [Google Scholar]
- 38. Van de Lest CH, Versteeg EM, Veerkamp JH, Van Kuppevelt TH. Elimination of autofluorescence in immunofluorescence microscopy with digital image processing. J Histochem Cytochem. 1995;43:727–30. [DOI] [PubMed] [Google Scholar]
- 39. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thevenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7:27–41. [DOI] [PubMed] [Google Scholar]
- 41. Crestfield AM, Moore S, Stein WH. The preparation and enzymatic hydrolysis of reduced and S-carboxymethylated proteins. J Biol Chem. 1963;238:622–7. [PubMed] [Google Scholar]
- 42. Jentoft N, Dearborn DG. Labeling of proteins by reductive methylation using sodium cyanoborohydride. J Biol Chem. 1979;254:4359–65. [PubMed] [Google Scholar]
- 43. Paz MA, Henson E, Rombauer R, Abrash L, Blumenfeld OO, Gallop PM. Alpha-amino alcohols as products of a reductive side reaction of denatured collagen with sodium borohydride. Biochemistry. 1970;9:2123–7. [DOI] [PubMed] [Google Scholar]
- 44. Vaughan JC, Jia S, Zhuang X. Ultrabright photoactivatable fluorophores created by reductive caging. Nat Methods. 2012;9:1181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Corrodi H, Hillarp NA, Jonsson G. Fluorescence methods for the histochemical demonstration of monoamines. 3. Sodium Borohydride reduction of the fluorescent compounds as a specificity test. J Histochem Cytochem. 1964;12:582–6. [DOI] [PubMed] [Google Scholar]
- 46. Kaufmann SH, Ewing CM, Shaper JH. The erasable Western blot. Anal Biochem. 1987;161:89–95. [DOI] [PubMed] [Google Scholar]
- 47. Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 2012;4:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gupta J, Hoque M, Zaman M, Khan RH, Saleemuddin M. A detergent-based procedure for the preparation of IgG-like bispecific antibodies in high yield. Sci Rep. 2016;6:39198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sood A, Miller AM, Brogi E, Sui Y, Armenia J, McDonough E, Santamaria-Pang A, Carlin S, Stamper A, Campos C, Pang Z, Li Q, Port E, Graeber TG, Schultz N, Ginty F, Larson SM, Mellinghoff IK. Multiplexed immunofluorescence delineates proteomic cancer cell states associated with metabolism. JCI Insight. 2016;1:e87030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jha SK, Marqusee S. Kinetic evidence for a two-stage mechanism of protein denaturation by guanidinium chloride. PNAS. 2014;111:4856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsumoto K, Ejima D, Kumagai I, Arakawa T. Practical considerations in refolding proteins from inclusion bodies. Protein Expr Purif. 2003;28:1–8. [DOI] [PubMed] [Google Scholar]
- 52. Baschong W, Suetterlin R, Laeng RH. Control of autofluorescence of archival formaldehyde-fixed, paraffin-embedded tissue in confocal laser scanning microscopy (CLSM). J Histochem Cytochem. 2001;49:1565–72. [DOI] [PubMed] [Google Scholar]
- 53. Davis AS, Richter A, Becker S, Moyer JE, Sandouk A, Skinner J, Taubenberger JK. Characterizing and diminishing autofluorescence in formalin-fixed paraffin-embedded human respiratory tissue. J Histochem Cytochem. 2014;62:405–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thibult M-L, Mamessier E, Gertner-Dardenne J, Pastor S, Just-Landi S, Xerri L, Chetaille B, Olive D. PD-1 is a novel regulator of human B-cell activation. Int Immunol. 2013;25:129–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.