

Abstract
Neuroretina and retinal pigment epithelium (RPE) are differentiated from the progenitors in optic vesicles, but it is unclear when and how the two lineages are segregated. Manipulation of chick embryos reveals that the early anteroventral optic vesicle is crucial for neuroretinal development, but the molecular mechanism is unclear. Homeodomain transcription factor Six3 is required for neuroretinal specification and is dispensable for RPE formation, but the cell fates of Six3-deficient progenitors and the origins of remnant RPE are unknown. Here, we performed lineage tracing of Six3-Cre positive cells in wild-type and Six3-deficient mouse embryos. Six3-Cre positive progenies were found in a population of progenitors in the anteroventral optic pits/vesicles starting at E8.5, and were found in neuroretina, optic stalk, ventral forebrain, but not RPE, at E10.5. Six3-deletion in the small population of progenitors at E8.5 was sufficient to cause rostral expansion of Wnt8b and drastic reduction of Fgf8/MAPK signaling, ablating neuroretinal specification without affecting RPE. Lineage tracing revealed Six3-deficient progenitors at E8.5 were eventually lost and the remnant RPE was derived from Six3-Cre negative cells. Thus, Six3 in a small population of progenitors expressing Six3-Cre at E8.5 is required for neuroretinal specification via regulating cell signaling and survival in mice.
Keywords: Retina, Cell specification, Lineage tracing, Six3, Wnt8b, Pituitary
1. Introduction
Morphogenesis of the vertebrate eyes is a multistep process starting with eye field formation in the anterior neural plate. In mice, optic pits are visible in the anterior neural plate at 5- to 7-somite stages (E8.0) (Kaufman, 1992). Fate mapping in mouse embryos identifies the origins of eyes in a rostral territory inside the anterior neural plate (Inoue et al., 2000). The optic pits evaginate to form optic vesicles, which then invaginate to form double-layered optic cups at E10.5 when neuroretina (NR) and retinal pigmented epithelium (RPE) are partitioned into the inner and outer layers of optic cups, respectively (Fuhrmann, 2010). Meanwhile, surface ectoderm overlaying the optic vesicles forms lens placodes, which later form lens vesicles (Cvekl and Ashery-Padan, 2014). Live imaging of medaka, zebrafish, and chick embryos reveals complex cell movements coordinated between the prospective NR, RPE and lens (Ivanovitch et al., 2013; Kwan et al., 2012; Rembold et al., 2006). Pinwheel movement and anterior rotation in 8- to 9-somite chick embryos make the cells at the anterior margin of optic vesicles move laterally and ventrally whereas the cells at the lateral margin move medially and dorsally (Kwan et al., 2012). In chick embryos, removal of ventral or anterior optic vesicles at 9- to 14-somite stages ablates NR, leading to pigmented vesicles that express RPE marker Mitf at the cost of NR markers Rax and Vsx2 (Hirashima et al., 2008). On the other hand, the anteroventral quadrant of optic vesicles develops whole eye after the other three quadrants are surgically removed, indicating that the anteroventral quadrant of early optic vesicles has the full potential for ocular development (Hirashima et al., 2008). In addition, transplantation of an anteroventral quadrant of the optic vesicle and the surrounding anterior cephalon from quail embryos into the same stage (9- to 11-somite) chick embryos generates a second eye (Kobayashi et al., 2009). Despite these findings, it is unclear why anteroventrally localized activity in the early optic vesicles are crucial for NR specification in chicks.
Transcription factors and cell signaling molecules coordinately regulate eye morphogenesis. In E8.0 mouse embryos, the eye field expresses a group of transcription factors, including Six3, Rax, Pax6, Lhx2, and Otx2 (Liu et al., 2010; Zuber et al., 2003). Rostral to the eye field, an Fgf8-expressing signaling center at the anterior neural ridge is required for the expression of telencephalon and rostral retina marker Foxg1 (Hebert and McConnell, 2000; Shimamura and Rubenstein, 1997). Inactivation of Shp2-mediated Fgf/MAPK signaling converts parts of the NR into the cells expressing RPE marker Mitf (Cai et al., 2010). Caudal to the eye field, a territory expresses Wnt8b, and overexpression of Wnt8b suppresses NR specification (Liu, 2012; Liu et al., 2010). In the eye field and optic vesicles, the markers are initially expressed in the whole optic vesicles, and the optic vesicle neuroepithelium is bipotential: the prospective NR and RPE can trans-differentiate under certain experimental conditions (Fuhrmann, 2010). In late stage optic vesicles (E9.5-E10.0), prospective NR and RPE become delineated by the expression of homeodomain transcription factor Vsx2 and bHLH transcription factor Mitf at ventral and dorsal optic vesicles, respectively. At E10.5, Vsx2 and Mitf are expressed in retinal progenitor cells (RPCs) in NR and the progenitors in RPE, respectively (Fuhrmann, 2010; Nguyen and Arnheiter, 2000). In retinal organoids derived from human embryonic stem cells, VSX2 and MITF positive cells are spontaneously partitioned into two adjacent domains (Lowe et al., 2016; Nakano et al., 2012). Still, it is unclear when and how NR and RPE lineages are segregated.
The homeobox gene Six3 is dynamically expressed during forebrain and visual system development (Oliver et al., 1995). Germ line inactivation of Six3 in mice causes truncation of forebrain, loss of the eye field, and rostral expansion of Wnt1 (Lagutin et al., 2003). Haploinsufficiency of Six3 fails to activate Shh expression in the ventral forebrain and causes holoprosencephaly (Geng et al., 2008). Conditional inactivation of Six3 using Rx-Cre ablates NR specification, but does not affect RPE formation (Liu et al., 2010). Direct transcriptional repression of Wnt8b by Six3 is required for NR specification (Liu et al., 2010). Six3 overexpression and knockdown into neonatal mouse retinae via electroporation causes small quantitative changes in cell proportions (Rapicavoli et al., 2011). Collectively, Six3 is required for NR specification and is dispensable for RPE formation, but the cell fates of Six3-deficient progenitors and the origins of remnant RPE are unclear.
Lineage tracing is a powerful tool to understand cell-fate decisions in tissue morphogenesis (Kretzschmar and Watt, 2012). We sought to determine the cell fate of Six3-deficient progenitors and the origins of remnant RPE in mice. Compared with Rx-Cre, Six3-Cre positive progenies are restricted to NR lineage (Furuta et al., 2000; Liu et al., 2010; Swindell et al., 2006), making it useful for tracking NR lineage. Here, we performed lineage tracing of Six3-Cre positive cells in wild-type and Six3-deficient mouse embryos and found that Six3 in a small population of progenitors in the anteroventral optic pits/vesicles at E8.5 regulates cell survival and signaling in NR specification.
2. Material and methods
2.1. Mice
Six3F/F and Six3+/Δ (Liu et al., 2006), Six6+/− (Li et al., 2002), Six3-Cre (Furuta et al., 2000), CAGG-CreER (Hayashi and McMahon, 2002), and Pax6 α-Cre mice (Marquardt et al., 2001) were maintained in the NMRI background and genotyped as previously reported. Six3 F/Δ;Six3-Cre embryos were generated by crossing female Six3F/F mice with male Six3+/Δ;Six3-Cre mice. Six3 F/Δ;α-Cre mice were generated by crossing female Six3F/F mice with male Six3+/Δ;α-Cre mice. Six3 F/Δ;Six3-Cre;Six6−/− embryos were generated by crossing female Six3F/F;Six6+/− mice with male Six3+/Δ;Six3-Cre;Six6+/− mice. Six3 F/Δ;CAGGCreER;Six6−/− embryos were generated by crossing female Six3F/F;Six6+/− mice with male Six3+/Δ;CAGGCreER;Six6+/− mice. Tamoxifen (TM) (2.25 mg/40 g body weight) was intraperitoneally injected into pregnant dams for early and late phases of deletion: at E8.0 and E8.5 for early phase deletion, and at E9.25 and E10.0 for late deletion. Animal experiments were approved by the Animal Care and Use Committees in St. Jude Children's Research Hospital and Albert Einstein College of Medicine.
2.2. Lineage tracing
For lineage tracing in wild-type condition, embryos and pups from the breeding between female Six3-Cre mice and male homozygous R26R mice (also known as Gtrosa26tm1Sor, Jackson lab, Stock No: 003309) were harvested for X-gal staining as described previously (Liu et al., 2006). For lineage tracing in Six3 F/Δ;Six3-Cre embryos, embryos from the breeding between female Six3F/F;R26R/R26R mice and male Six3+/Δ;Six3-Cre mice were harvested for X-gal staining.
2.3. Immunohistochemistry and mRNA in situ hybridization
Standard protocols were followed (Liu et al., 2010, 2006). The following antibodies were used: anti-Pou4f2 (also known as Brn3b, Santa Cruz #SC-6026, 1:100), anti-Calb2 (also known as Calretinin, Chemicon #AB149, 1:2000), anti-Calb1 (also known as Calbindin D28K, Sigma #C9848 clone CB-955, 1:2000), anti-Glul (also known as glutamine synthetase, BD Bioscience #610517, 1:500), anti-pH3 (Upstate #05806, 1:2000), anti-Isl1 (T. Jessell, 1:1000), anti-Ki67 (NeoMarkers #RM-9106-51, 1:500), Lhx3 (T. Jessell, 1:4000), activated MAP Kinase (also known as diphosphorylated ERK-1 & 2, Sigma #M8159, 1:200, with TSA signal amplification (PerkinElmer #NEL701A001KT)), anti-Mitf (H. Arnheiter, 1:2000), anti-Pax6 (Covance #PRB-160P, 1:500), anti-Prkca (also known as PKCα, Upstate #05-154, 1:300), anti-Rax (also known as Rx, Abcam #ab86210, 1:1000), anti-Rcvrn (K.W. Koch, 1:2000), anti-Rho (B. Molday, 1:500), anti-Six3 (G. Oliver, 1:500), anti-Sox2 (Chemicon #AB5770, 1:800), anti-Tubb3 (BabCO #MMS435P clone Tuj1, 1:500), anti-Vsx2 (Abcam #AB9016, 1:200). The following in situ hybridization probes were used: Wnt8b (XbaI/T3), Fgf8 (PstI/T7), Shh (HindIII/T3), Bmp4 (AccI/T7), Six6 (XbaI/T7), and Nkx2.1 (XbaI/T3).
2.4. TUNEL assay
TUNEL assay was performed on frozen sections (10 μm) or whole mount embryos by using the ApoTag Kit (Chemicon) according to the manufacturer's instructions.
3. Results
3.1. Lineage tracing reveals that Six3-Cre positive progenitors at E8.5-9.5 are fated to NR, optic stalk, and ventral forebrain at E10.5
We sought to specifically delete Six3 in NR lineage using Six3-Cre mouse line for phenotype analysis. Six3-Cre mice carry transgenic Cre under the control of a Six3 promoter/enhancer, and R26R reporter for Six3-Cre was previously found in retina and ventral forebrain starting at E9.0–9.5 (Furuta et al., 2000). In our studies, β-gal (R26R reporter) was detected in the anteroventral optic pits/vesicles as early as at 8-somite stage (E8.0–8.5) with variable onset (Fig. 1A-E, n = 3/8). On sections, β-gal positive cells intermingled with β-gal negative cells in optic vesicle epithelium at 11-somite stage (E8.5) (Fig. 1E). At E9.0 and E9.5, β-gal was widely found in ventral optic vesicles and ventral forebrain, but not in dorsal optic vesicles (Fig. 1F-I). At E9.75, NR marker Rax became restricted to ventral optic vesicles, whereas RPE marker Mitf was expressed in dorsal optic vesicles, delineating prospective NR domain and prospective RPE domain at the ventral and dorsal optic vesicles, respectively (Fig. 1J,K). The β-gal positive domain overlapped with the prospective NR domain, but not with the prospective RPE (Fig. 1I-K). At E10.5, β-gal positive cells were found at NR, optic stalk and ventral forebrain, but not at RPE (Fig. 1L). Only one or two Six3-Cre positive progenies were found in RPE at E10.5. Collectively, Six3-Cre positive progenies were found in a small population of progenitors in the anteroventral optic pits/vesicles starting at E8.5, and sequentially accumulated progenies were fated to NR, optic stalk, and ventral forebrain at E10.5.
Fig. 1.
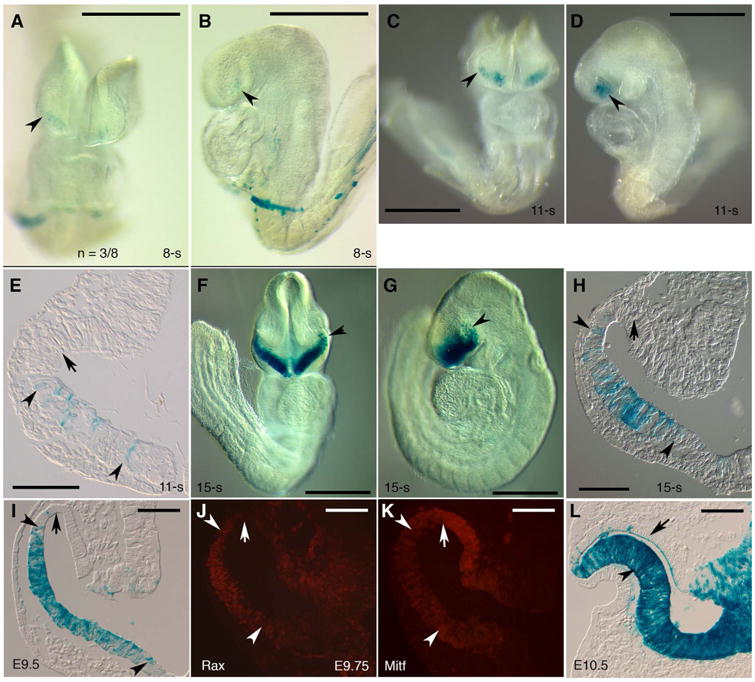
Six3-Cre positive progenitors at E8.5–9.5 are fated to NR, optic stalk, and ventral forebrain at E10.5. (A-E) In the anteroventral optic pits/vesicles as early as at 8-somite stage (E8.0–8.5), weak β-gal expression was detected (arrowheads in A, B, n = 3 (β-gal positive embryos) /8 (embryos with a genotype positive for Six3-Cre and R26R). In the same area at 11-somite stage (E8.5), β-gal expression became stronger (arrowheads in C, D, n = 2/4). On sections, β-gal positive cells intermingled with negative cells in the ventral optic vesicles, but were not found in the dorsal optic vesicles (arrowheads and arrow in E). (F-K) At 15-somite stage, β-gal positive cells were found in areas comprising the ventral optic vesicles, ventral optic stalks and ventral forebrain, but were not detected in the dorsal optic vesicles (arrowheads and arrow in F-H, n = 3/3). At E9.5, β-gal positive cells were located at the entire ventral vesicles, but were not found in the dorsal optic vesicles (arrowheads and arrow in I, n = 3/3). At E9.75, NR marker Rax and RPE marker Mitf were restricted to the ventral and dorsal optic vesicles, respectively (arrowheads and arrows in J, K). The β-gal positive domain overlapped with the Rax positive domain, but not with the Mitf positive domain (I-K). (L) At E10.5, β-gal positive cells were found in the NR, optic stalk, and ventral forebrain (arrowhead in L, n = 3/3, a whole mount view in Fig. 5). β-gal positive cells were generally not found in RPE at E10.5 (arrow in L) except for one or two mysterious cells. Scale bars, 500 μm (A-D, F, G), 100 μm (E, H, I-L).
3.2. Inactivation of Six3 using Six3-Cre causes two major retinal phenotypes
Compared with Rx-Cre, Six3-Cre is restricted to NR lineage with later onset, allowing functional studies of Six3 in a more specific cell population during NR specification. Conditional inactivation of Six3 in Six3 F/Δ;Six3-Cre mutant embryos (referred to as Six3Six3-Cre KO mice hereafter) by crossing female Six3F/F mice (Liu et al., 2006) with male Six3+/Δ;Six3-Cre mice led to two major retinal phenotypes at E10.5–11.5, a stage when NR has just been specified and pigmentation in RPE becomes visible (Fig. 2). In type I mutant embryos (n=12/26), optic cups formed normally or were slightly reduced in size, and NR was present. In type II mutant embryos (n = 12/26), optic cups did not form, NR was absent, but RPE remained (Fig. 2A-C,D-F). In addition, the size of forebrain was significantly reduced in type II mutant embryos, but not in type I mutant embryos. A few Six3Six3-Cre KO mutant embryos only had optic stalks (n=2/26). In sum, Six3Six3-Cre KO mutant embryos displayed two major retinal phenotypes.
Fig. 2.
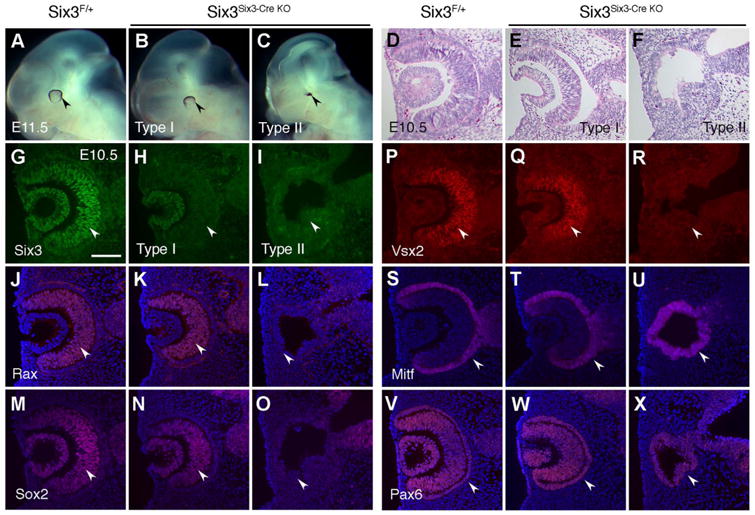
Inactivation of Six3 using Six3-Cre causes two major retinal phenotypes. Six3Six3-Cre KO and littermate control embryos at E10.5–11.5 were used. Morphology examination at E11.5 and molecular characterization at E10.5 identified type I (grossly normal, n = 12/26) and type II (severely affected, n = 12/26) Six3Six3-Cre KO embryos. (A-C) At E11.5, a dorsal rim of pigmentation became visible in the control eyes. Compared with the controls, type I Six3Six3-Cre KO eyes were grossly normal, whereas type II Six3Six3-Cre KO eyes were severely affected and displayed as pigmentation dots (arrowheads in A-C). In addition, the forebrain was grossly normal in type I Six3Six3-Cre KO embryos, but was severely reduced in size in type II Six3Six3-Cre KO embryos. (D-F) On H & E stained sections at E10.5, the control and type I Six3Six3-Cre KO embryos exhibited double-layered optic cups, but type II Six3Six3-Cre KO embryos did not have optic cups and exhibited defective vesicles. (G-R) At E10.5, Six3 was absent in both type I and type II Six3Six3-Cre KO mutant retinae (arrowheads in G-I). However, NR markers Rax, Sox2, and Vsx2 were normal in type I Six3Six3-Cre KO retinae, but were absent in type II Six3Six3-Cre KO retinae (arrowheads in J-R). (S-T) RPE marker Mitf was found at the outer layer of optic cups in the control and type I Six3Six3-Cre KO embryos, but was in the whole defective vesicles in type II Six3Six3-Cre KO embryos (arrowheads in S-T). (V-X) Pax6 was expressed in both NR and RPE in the control and type I Six3Six3-Cre KO embryos, but was found in the whole defective vesicles in type II Six3Six3-Cre KO embryos (arrowheads in V-X). In addition to retinal phenotypes, lens vesicles were normal in type I Six3Six3-Cre KO embryos, but were absent in type II Six3Six3-Cre KO embryos, as indicated by both altered morphology and the absence of lens vesicle markers Six3, Sox2, and Pax6 (D-F, G-I, M-O, V-X). Scale bar, 100 μm.
3.3. Six3-deficiency disrupts NR specification and lens vesicle formation in type II Six3Six3-Cre KO mutant embryos
Although Six3 was absent in both type I and II mutant retinae (Fig. 2G-I), RPC markers Rax (Mathers et al., 1997), Sox2 (Taranova et al., 2006), and Vsx2 (Nguyen and Arnheiter, 2000) appeared normal in type I mutant embryos, but were absent in type II mutant embryos (Fig. 2J-O,P-R). RPE marker Mitf (Nguyen and Arnheiter, 2000) was normal in type I mutant embryos, but was expressed in whole remnant vesicles in type II mutant embryos (Fig. 2S-U). In the control and type I mutant embryos, Pax6 was expressed in both NR and RPE. In type II mutant embryos, Pax6 was expressed in whole mutant vesicles, reflecting Pax6 expression in RPE (Fig. 2V-X). In addition, lens vesicle markers Sox2 and Pax6 were absent in the defective surface ectoderm, indicating the lack of lens placodes/vesicles in type II mutant embryos (Fig. 2M-O,V-X). The retinal phenotypes in type II Six3Six3-Cre KO mutant embryos mimic the phenotypes in Six3Rx-Cre KO embryos driven by Rx-Cre (Liu et al., 2010), confirming our previous findings. Thus, inactivation of Six3 using Six3-Cre disrupted NR specification and lens vesicle formation in type II Six3Six3-Cre KO mutant embryos.
3.4. Retinal cell fate determination and proliferation of RPCs are grossly normal in type I Six3Six3-Cre KO mutant NR
We sought to determine the retinal phenotypes in type I Six3Six3-Cre KO retinae at later stages. Although Six3 was absent in mutant NR at E13.5–14.5 and mutant retinae were slightly smaller (Fig. S1A,B), the expression of retinal ganglion cell markers Tubb3 and Pou4f2 (also known as Brn3b) (Gan et al., 1996) was mostly normal (Fig. S1C-F, n=3/3), except for occasional axon pathfinding errors (arrow in Fig. S1D, n=1/3). The axon pathfinding errors are consistent with the retinal phenotypes previously described in compound six3a;six3b mutant zebrafish (Samuel et al., 2016). The reduction in retinal size could be due to smaller optic cups at E10.5 (see Fig. S6). In type I Six3Six3-Cre KO retinae, RPCs were unaffected, as indicated by normal Pax6 and Vsx2 expression (Fig. S1G-J). In addition, there were no overt change in proliferation and cell survival at E13.5–14.5 (Fig. S1K-P). At postnatal stages, Six3Six3-Cre KO mutant pups grew much slower compared with the controls, and most of the mutant pups did not survive beyond postnatal day (P) 5. One mutant pup survived to P11 and was used for retinal phenotype analysis. In the mutant NR, although Six3 was absent, markers for the major retinal cell types were present in the normally stratified NR (Fig. 3). The tested markers include ganglion cell markers (Pou4f2, Isl1), amacrine cell markers (Pax6, Calb2 (also known as calretinin)), horizontal cell marker (Calb1, also known Calbindin D28), bipolar cell markers (Isl1, Vsx2, Prkca (also known as PKCα)), Müller glia marker (Glul, also known as glutamine synthetase), and photoreceptor markers (Rcvrn, Rho). The reduced thickness of the mutant NR could be due to growth retardation caused by pituitary defects (see Fig. 7), smaller optic cups at E10.5 (see Fig. S6), or minor changes in proliferation and apoptosis. To overcome the lethality in postnatal Six3Six3-Cre KO mutant pups, we also conditionally inactivated distal retinae using Pax6 α-Cre mice (Marquardt et al., 2001) and found that retinal cell fate determination was normal (Fig. S2), confirming the findings in Six3Six3-Cre KO mutant retina. Taken together, our findings indicate that retinal cell fate determination and proliferation of RPCs were grossly normal in type I Six3Six3-Cre KO mutant NR.
Fig. 3.
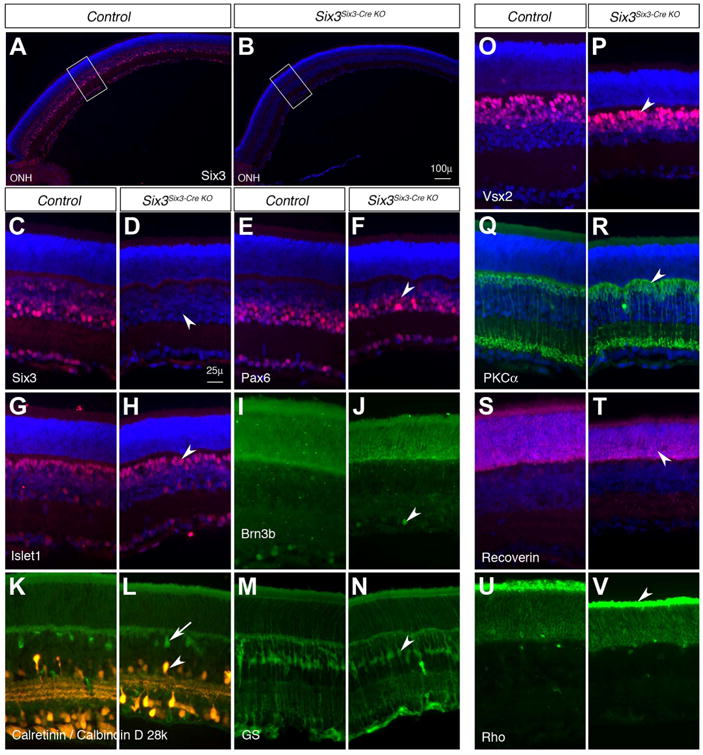
Retinal cell fate determination is grossly normal in type I Six3Six3-Cre KO retinae on postnatal day 11. The results represent the only survival pup in three litters on postnatal day (P) 11. (A-D) Six3 was efficiently deleted in type I Six3Six3-Cre KO retinae (arrowhead in D). Panels C and D show higher magnifications of the rectangle areas in A and B, respectively. (E-V) Type I Six3Six3-Cre KO retinae on P11 contained all major retinal cell types as revealed by immunostaining of the markers: Pax6 for amacrine cells (arrowhead in F), Isl1 for ganglion and bipolar cells (arrowhead in H), Pou4f2 for ganglion cells (arrowhead in J), Calb1 for horizontal cells (arrow in L), Calb2 for amacrine cells (arrowhead in L), Glul for Müller glia (arrowhead in N), Vsx2 and Prkca for bipolar cells (arrowheads in P, R), Rcvrn and Rho for photoreceptors (arrowheads in T, V). The green signals in photoreceptor layers in I and J were artifacts from red channel that was used to identify Rcvrn. ONH, optic nerve head. Scale bars, 100 μm (B), 25 μm (D).
Fig. 7.
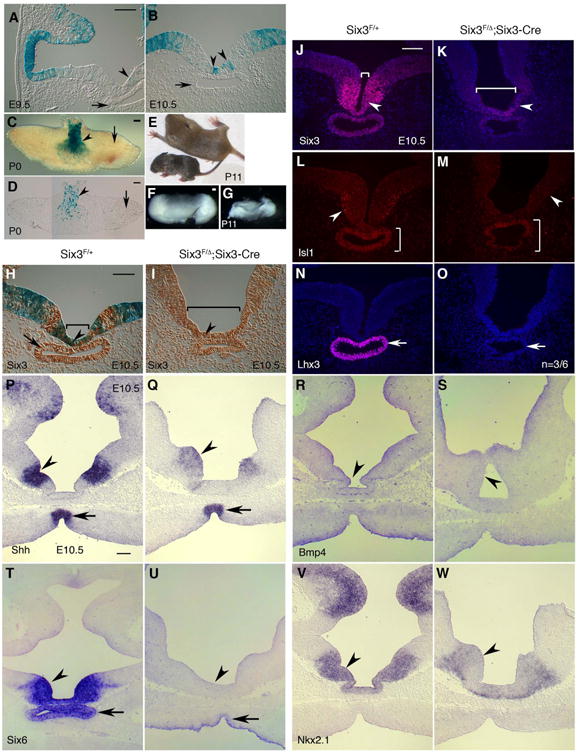
Six3-deficiency in the ventral forebrain leads to the loss of Six3-Cre positive progenies and the defects in the infundibulum and Rathke's pouch. (A-D) Six3-Cre positive progenies were found in the infundibulum and ventral forebrain at E9.5 and E10.5 (arrowheads in A-B, n =3/3) and in the anterior pituitary on postnatal day 0 (arrowheads in C-D, n = 3/3), but were not in the Rathke's pouch and the posterior pituitary (arrows in A-D). (E-F) One survival pup on postnatal day 11 was significantly smaller than its littermate (E) and displayed pituitary hypoplasia (F, G). (H-I) Six3-Cre positive progenies in the infundibulum were lost upon Six3 deficiency. In the control embryos, ß-gal positive cells were only found in the infundibulum and ventral forebrain, although endogenous Six3 was expressed in the infundibulum, ventral forebrain, and Rathke's pouch (brown in H). In Six3Six3-Cre KO mutant
3.5. Six3-deficiency is compensated by Six6 in NR specification intype I Six3Six3-Cre KO mutant embryos
The lack of overt phenotypes in type I Six3Six3-Cre KO mutant retinae could be due to compensation by Six6, the closely related gene family member that is expressed in E9.5 ventral optic vesicles (Jean et al., 1999). Previous studies show that inactivation of Six6 in mice results in variable retinal and pituitary hypoplasia (Li et al., 2002). In the 129sv background, RPC proliferation is moderately reduced, but all retinal cell types are present in Six6-null adult retinae (Li et al., 2002). We closely compared Six3 and Six6 expression in mouse retinal development. In the mouse eye field at 6-somite stage (E8.0), Six3 was strongly expressed, whereas Six6 was undetectable (Fig. S3A,F). At 12-somite stage (E8.5), Six3 was strongly expressed in the optic vesicles, whereas Six6 was weakly expressed in the distal ventral optic vesicles (Fig. S3B,C,G,H). At E9.5, Six3 was expressed in the whole optic vesicles, whereas Six6 was expressed in the ventral optic vesicles (Fig. S3D,I). At E10.5, both Six3 and Six6 were widely expressed in the NR (Fig. S3E,J). Thus, in mouse retinal development, Six3 expression precedes Six6, and Six3 and Six6 are later co-expressed in NR lineage.
To evaluate potential redundant functions between Six3 and Six6 in NR development, we performed phenotypic analysis of Six3Six3-Cre KO;Six6−/− compound mutant embryos. At E11.0, Six6-null embryos did not show any overt ocular defects. However, in all Six3Six3-Cre KO;Six6−/− compound mutant embryos, type II retinal phenotypes (NR was absent but RPE vesicles remained) was observed, and no type I retinal phenotype was found (Fig. 4, n =6/6). This result indicates that inactivation of Six6 in type I Six3Six3-Cre KO embryos led to type II retinal phenotypes, and thus Six6 compensated Six3-deficiency in type I Six3Six3-Cre KO embryos when Six6 was expressed in optic vesicles.
Fig. 4.
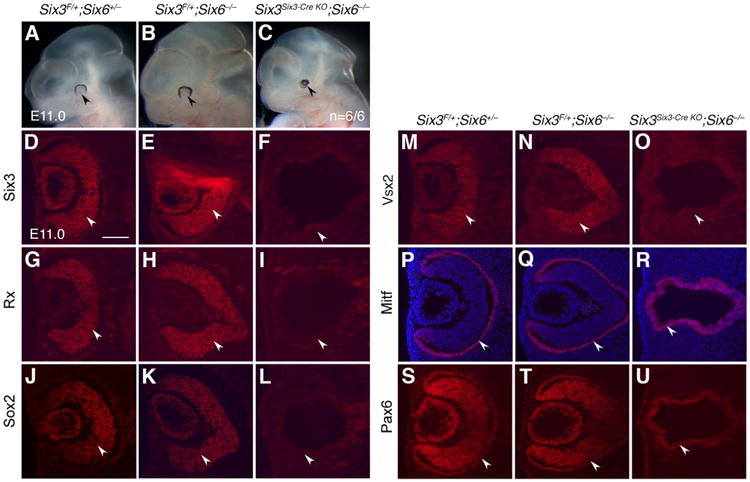
Six3Six3-Cre KO;Six6−/− embryos displayed type II retinal phenotypes. The results represent six out of six mutant embryos. (A-C) At E11.0, the control (Six3F/+;Six6+/−) and Six6-null (Six3F/+;Six6−/−) eyes were grossly indistinguishable (arrowheads in A, B). In contrast, Six3Six3-Cre KO;Six6−/− eyes were severely affected and displayed as pigmentation dots (arrowhead in C, n = 6/6). (D-U) At E11.0, Six3 was expressed in the control and Six6-null NR, but was absent in Six3Six3-Cre KO;Six6−/− embryos (arrowheads in D-F). In Six6-null embryos, optic cups formed and NR markers Rax, Sox2, Vsx2, and RPE marker Mitf were expressed indistinguishably compared with the controls. In Six3Six3-Cre KO;Six6−/− embryos, optic cups did not form, NR markers Rax, Sox2 and Vsx2 were absent, and RPE marker Mitf was expressed in the whole defective vesicles (arrowheads in G-R). In the control and Six6-null eyes, Pax6 was expressed in both NR and RPE. In Six3Six3-Cre KO;Six6−/− eyes, Pax6 was expressed in the whole defective vesicles (arrowheads in S-U). Scale bar = 100 μm.
3.6. Six3 expression at E8.0-8.5 is required for NR specification
Type II retinal phenotypes in Six3Six3-Cre KO embryos could be due to Six3 deletion at E8.0–8.5, a stage before Six6 is expressed. To test this hypothesis, we temporally deleted Six3 in Six3F/Δ;CAGG-CreER embryos (referred to as Six3CAGGCreER KO hereafter) at early and late phases by Tamoxifen (TM) administration at E8.0–8.5 and E9.25–10.0, respectively. Six3-deletion at E8.0–8.5 led to type II retinal phenotypes, whereas Six3-deletion at E9.25–10.0 resulted in type I retinal phenotypes (Fig. S4, n=3/3). Consistent with the phenotypes caused by temporal Six3-deletion, Six3-deletion using Rx-Cre, which becomes active much earlier and wider than Six3-Cre, also led to type II retinal phenotypes (Liu et al., 2010). These findings indicate that early (E8.0–8.5) and late (E9.25–10.0) Six3-deficiency cause type II and type I retinal phenotypes, respectively. Thus, the essential functions of Six3 in NR specification have temporal dependence, and Six3-deletion at E8.0–8.5 disrupts NR specification.
3.7. Six3-Cre positive progenies are lost and the remnant RPE cellsare derived from Six3-Cre negative cells in type II Six3Six3-Cre KO mutant embryos
In type II Six3Six3-Cre KO mutant embryos, NR was absent, but RPE remained. To clarify whether the progenitors fated to NR were directly converted into RPE cells upon Six3-deletion, we performed lineage tracing using R26R reporter in Six3Six3-Cre KO mutant embryos. As early as 14-somite stage (E8.5), the number of β-gal positive cells in type II Six3Six3-Cre KO mutant optic vesicles became reduced compared that in the controls (Fig. 5A-D). Drastic reduction of β-gal positive cells in type II Six3Six3-Cre KO mutant optic vesicles was continuously observed at 18-somite stage and E9.5 (Fig. 5E-L). At E10.5, β-gal positive cells were undetectable in type II Six3Six3-Cre KO mutant optic vesicles that exclusively expressed RPE marker Mitf (Fig. 5M-P). In contrast to Six3-Cre, R26R reporter expression for Rx-Cre (Liu et al., 2010; Swindell et al., 2006) was found in both NR and RPE in E10.5 control embryos (arrowhead and arrow in Fig. S5A). When Six3 was conditionally deleted using Rx-Cre, NR was absent, but RPE remained. Importantly, R26R reporter expression for Rx-Cre remained in the RPE in Six3Rx-Cre KO mutant embryos (arrow in Fig. S5F), indicating that gene expression in R26R locus was active in both wild type and Six3-null RPE and does not requires Six3 functions. Thus, the lack of β-gal positive cells in type II Six3Six3-Cre KO mutant optic vesicles indicates that the progenies of Six3-Cre positive cells were lost upon Six3-deficiency and the remnant RPE cells were differentiated from Six3-Cre negative progenitors.
Fig. 5.
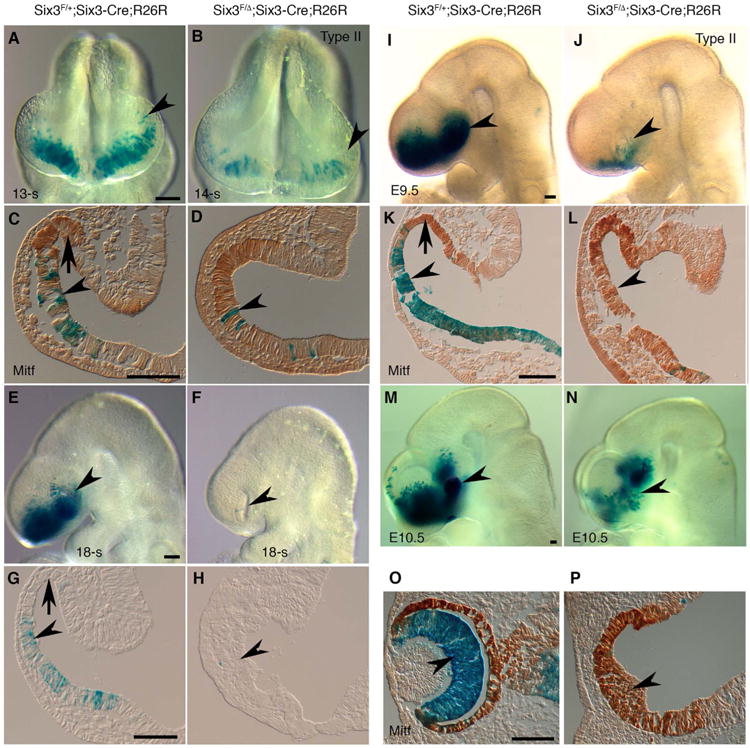
Six3-Cre positive progenies are eventually lost in type II Six3Six3-Cre KO (Six3F/Δ;Six3-Cre;R26R) optic vesicles. Control and type II mutant embryos (defined by altered morphology in the eyes, n = 3 for each stage) were sectioned and immunostained with a Mitf antibody (C, D, K, L, O, P). (A-L) At 14-somite to E9.5 stages, ß-gal positive cells were located in the ventral but not the dorsal optic vesicles in the control embryos (arrowheads and arrows in C, G, K). In type II Six3Six3-Cre KO embryos, the number of ß-gal positive cells gradually reduced in the defective optic vesicles (arrowheads in A-L). In E9.5 mutant optic vesicles, ß-gal positive cells were absent, and Mitf expression extended ventrally (K, L). (M-N) In E10.5 type II Six3Six3-Cre KO embryos, optic cups did not form, ß-gal positive cells were absent, and Mitf was expressed in the entire defective vesicles (arrowheads in M-P). Scale bars = 500 μm (A, E, I, M), 100 μm (C, G, K, O).
In contrast to type II Six3Six3-Cre KO mutant retinae, β-gal positive cells remained in type I Six3Six3-Cre KO mutant retinae in which optic cups formed and NR specification took place (Fig. S6). Thus, progenies of Six3-Cre positive cells behave differently in the two types of Six3Six3-Cre KO embryos and the loss of Six3-Cre positive progenies coincides with the ablation of NR.
3.8. Six3-deficiency in a small population of progenitors expressing Six3-Cre at E8.0–8.5 is sufficient to disrupt paracrine signaling and cell survival
We sought to determine whether Six3-deletion in the small population of progenitors expressing Six3-Cre at E8.5 is sufficient to cause type II retinal phenotypes. Consistent with the reporter assay, immunofluorescence demonstrated that Six3 was only deleted in a small cell population in optic pits at 8-somite stage (E8.0) (arrowhead in Fig. 6A,B, n = 2/4). In the mutant embryos, one Six3 allele is conditional and the other Six3 allele is null, and thus Six3 expression was reduced even in the cells in which Six3-Cre was negative. Importantly, Six3 heterozygotes were routinely maintained in our mouse colonies and never displayed type II retinal phenotypes (data not shown). To obtain an overview of Six3-deletion in whole embryos, we performed immunostaining of Six3 on serial coronal sections of 7-somite embryos. In the control embryos, Six3 expression displayed a gradient along anteroposterior axis and ventrodorsal axis, with high levels at the anteroventral eye field / optic pit (Fig. S7 A,D,G,J,M). In Six3Six3-Cre KO embryos, Six3-deletion was found in a small population of progenitors in the eye field / optic pit, and gross reduction of Six3 expression was striking at the anteroventral eye field / optic pit (Fig. S7 B,C,E,F,H,I, K,L,N,O), which pattern is consistent with that of R26R reporter for Six3-Cre at 8- to 11-somite stages (Fig. 1). Interestingly, Six3-deficient cells appeared to be disintegrated from retinal epithelium (arrowheads in Fig. S7 B,C,H,I,K,L,N,O), indicating that these cells were dying. Upon Six3-deletion, Rax expression was reduced in corresponding areas (Fig. 6C,D), and so was Lhx2 (Fig. S8). It was reported that non-parallel recombination exists between R26R reporter and targeted gene deletion, and the differences in the chromosomal location, the distance spanning LoxP sites, the sequences around the LoxP sites and the level of Cre activity in the cells probably contribute to the non-parallel recombination (Liu et al., 2013). In our hand, Six3-deletion and β-gal reporter expression are mostly exclusive and we did not see complete Six3-deletion in E8.5 Six3 mutant embryos in which drastic phenotypes were obvious, no matter Six3-deletion was driven by Six3-Cre (Fig. S9) or Rx-Cre (Fig. S10). Despite partial Six3-deletion in Six3Six3-Cre KO mutant embryos, Wnt8b expression was strikingly expanded rostrally to the anterior neural ridge at 7-somite stage (Fig. 6E-H, n = 2/4), and Fgf8 expression was significantly reduced at the anterior neural ridge at 8-somite stage (Fig. 6I-L, n=2/4). In addition, activated MAP Kinase (also known as diphosphorylated ERK-1 and ERK-2), downstream mediators of FGF8 signaling (Harada et al., 2016), was found in the anteroventral optic pits at 6- to 8-somite (E8.0) stages in control embryos, but was absent in type II Six3Six3-Cre KO mutant embryos (Fig. 6M-P, n=3/4, Fig. S11). In control embryos, a number of physiological apoptotic cells were found at the anterior neural ridge at 8-somite stage. In type II Six3Six3-Cre KO mutant embryos, the physiological apoptosis at the anterior neural ridge was significantly reduced (arrows in Fig. 6Q-T), and ectopic apoptosis were found in the optic pits (arrowhead in Fig. 6S,T). On sections, ectopic apoptosis was found in Six3Six3-Cre KO mutant eye field / optic pit at 8-somite stage and became rare at 12-somite stage (arrowheads in Fig. S12), but was significant in the dorsal optic vesicles and dorsal periocular mesenchyme at 12-somite stage (arrows in Fig. S12), whereas physiological apoptosis was significantly reduced in the ventral optic stalks and the anteroventral neural ridge (double-headed arrows in Fig. S12). In addition, anterior neuropore closure was delayed in Six3Six3-Cre KO mutant embryos (Fig. S12). The apoptosis profiles in Six3Six3-Cre KO mutant optic vesicles at 12-somite stage are consistent with our previous findings in Six3Rx-Cre KO mutant embryos (Liu et al., 2010). At E9.5, a significant increase of apoptosis in the periocular mesenchyme was found in Six3Six3-Cre KO mutant embryos (Fig. S13). Taken together, these results indicate that Six3-deficiency in a small population of progenitors expressing Six3-Cre at E8.0–8.5 is sufficient to disrupt paracrine signaling and cell survival, ablating NR specification.
Fig. 6.
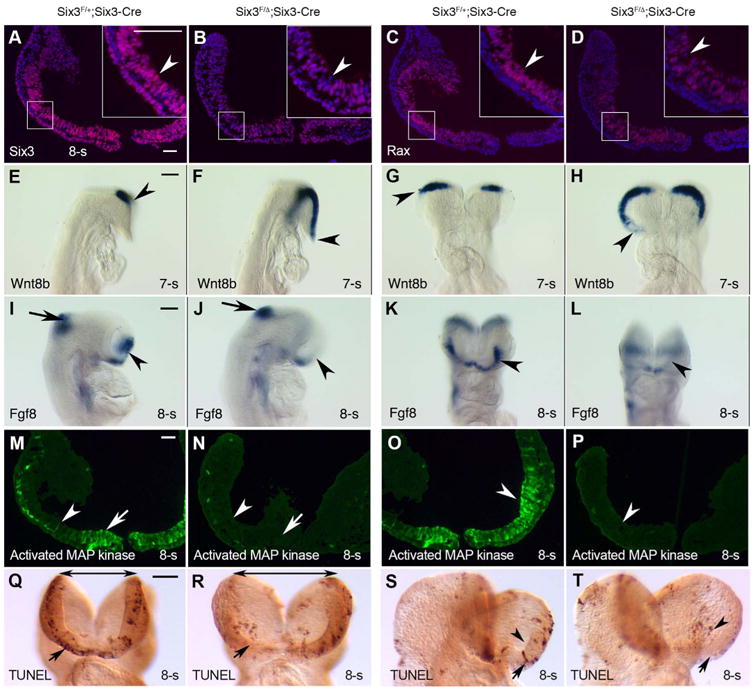
Six3-deficiency in a small population of progenitors in the anteroventral optic pits/vesicles at E8.5 is sufficient to disrupt paracrine signaling and cell survival. (A, B) Six3 was deleted in a small population of progenitors in the optic pits/vesicles in 8-somite stage Six3Six3-Cre KO embryos (arrowheads in A, B, n = 2/4). In the mutant embryos, one Six3 allele is conditional and the other Six3 allele is null, and thus Six3 expression was widely reduced. (C, D) Rax expression was reduced (arrowheads in C, D, n = 2/4). (E, L) In Six3Six3-Cre KO embryos at 7- to 8-somite stages, Wnt8b was expanded rostrally and Fgf8 was downregulated drastically at the anterior neural ridge (arrowheads in E-L, n = 2/4). Unaffected Fgf8 expression at midbrain-hindbrain boundary served as an internal control. (M-P) Severe downregulation of activated MAP kinase (also known as diphosphorylated ERK-1 & 2) in Six3Six3-Cre KO embryos (arrows and arrowheads, n = 2/4). Coronal sections at two levels were displayed. (Q-T) Drastic changes in apoptosis in Six3Six3-Cre KO embryos at 8-somite stage: ectopic apoptosis was found in the optic pits/vesicles (arrowheads in S, T, n = 2/4), but physiological apoptosis at the anterior neural ridge was reduced (arrows in Q-T. In addition, the folding of the anterior neural plate and evagination of optic pits/vesicles in Six3Six3-Cre KO embryos were defective (double-headed arrows in Q, R, n = 2/4). Scale bars = 100 μm (A, M), 500 μm (E, I, Q).
3.9. Six3-deficiency in the ventral forebrain leads to the loss of Six3-Cre positive progenies and the defects in the infundibulum and Rathke's pouch
To determine the mechanisms of growth retardation in Six3Six3-Cre KO mutant pups, we examined R26R reporter for Six3-Cre in pituitary development. β-gal positive cells were found in a population of cells in the infundibulum and ventral forebrain at E9.5–10.5, but never in Rathke's pouch (Fig. 7A,B). Later, β-gal positive cells were located in the posterior pituitary, but not in the anterior pituitary at postnatal day 0 (Fig. 7C,D).
Six3Six3-Cre KO mutant pups at P11 displayed severe growth retardation and pituitary hypoplasia (Fig. 7E-G). Defective infundibulum was obvious as early as at E10.5.
In E10.5 control embryos, Six3 was expressed in the infundibulum, ventral forebrain and Rathke's pouch (Fig. 7H), whereas β-gal positive cells (Six3-Cre positive progenies) were only found in the infundibulum and ventral forebrain. In E10.5 Six3Six3-Cre KO mutant embryos, β-gal positive cells were absent in these areas, and the infundibulum was broader (Fig. 7H,I), indicating that Six3-Cre positive progenies were lost in the defective infundibulum upon Six3-deficiency. Meanwhile, Six3 expression in the mutant infundibulum and Rathke's pouch was gradually reduced (Fig. 7H-K). It is worth noting that the reduction of Six3 expression in these areas reflected overall cell fate changes rather than Cre recombinase-mediated gene deletion, because these cells were derived from Six3-Cre negative cells (Fig. 7I) and a number of other key markers also showed defective expression in the mutant embryos: Isl1 (Rizzoti and Lovell-Badge, 2005) at the infundibulum was reduced (Fig. 7L,M), Lhx3 (Zhao et al., 2006) in Rathke's pouch was absent (Fig. 7N,O, n = 3/6), Shh and Bmp4 at ventral forebrain (Rizzoti and Lovell-Badge, 2005) was reduced (Fig. 7P-S), Six6 (Li et al., 2002) was absent or significantly reduced (Fig. 7T, U), and Nkx2.1 (Kimura et al., 1996) was moderately reduced (Fig. 7V,W). We conclude that Six3-deficiency at the ventral forebrain causes the loss of Six3-Cre positive progenies and defective cell signaling, leading to defects in the infundibulum and Rathke's pouch.
4. Discussion
Using lineage tracing and conditional Six3-deletion in mice, we demonstrate that Six3 in a small population of progenitors expressing Six3-Cre at E8.5 is required for neuroretinal specification. Six3-deletion driven by Six3-Cre at E8.5 is sufficient to cause rostral expansion of Wnt8b and drastic reduction of Fgf8/MAPK signaling, disrupting NR specification and leading to the loss of Six3-Cre positive progenies. Similarly, Six3-deficiency in the ventral forebrain driven by Six3-Cre causes defective paracrine signaling and the loss of Six3-Cre positive progenies, leading to defects in the infundibulum and Rathke's pouch. Thus, Six3 is essential for neuroretinal and pituitary development through regulating paracrine signaling and cell survival.
Our findings provide novel insight into the molecular and cellular mechanisms of NR specification. Previously, we demonstrated that inactivation of Six3 using Rx-Cre abolishes NR specification without affecting RPE (Liu et al., 2010). Six3-Cre positive progenies are restricted to NR lineage and is later-onset compared with Rx-Cre (Liu et al., 2010; Swindell et al., 2006). Inactivation of Six3 using Six3-Cre causes the same retinal phenotypes as those using Rx-Cre, confirming our previous findings. Importantly, our current studies narrow down the cell population in which Six3 exerts its essential roles in NR specification, since Six3-deletion in a small population of progenitors expressing Six3-Cre at E8.0–8.5 is sufficient to cause rostral expansion of Wnt8b and drastic downregulation of Fgf8/MAPK signaling, disrupting NR specification. Similar expression changes in Wnt8b and Fgf8 are also found in Six3Rx-Cre KO embryos (Liu et al., 2010). The contrast between the restricted Six3-deletion in E8.5 Six3Six3-Cre KO mutant embryos and the wide-range changes in Wnt8b and Fgf8/MAPK signaling indicate that, besides the direct transcriptional repression of Wnt8b identified previously (Liu et al., 2010), non-cell-autonomous regulation of Wnt8b and Fgf8/MAPK signaling by Six3 also exists. We previously demonstrated that ectopic Wnt8b expression is sufficient to abolish NR specification (Liu et al., 2010). Cai et al. (2010) demonstrated that inactivation of Shp2-mediated Fgf/MAPK signaling at an early stage, but not at a late stage, converts parts of the NR into the cells expressing RPE marker Mitf. Thus, we conclude that Six3 in a small population of progenitors expressing Six3 at E8.5 is required for suppressing Wnt8b and maintaining Fgf8/MAPK signaling in NR specification.
Lineage tracing reveals lineage relationships during NR specification. Furuta et al. (2000) previously described that β-gal reporter for Six3-Cre were restricted to prospective NR, but not prospective RPE, at E9.5 (27-somite); at E12.5, staining was also found in RPE. The staining in RPE at E12.5 described by Furuta et al. (2006) is questionable, since Six3-Cre failed to delete β-catenin or α-catenin genes in RPE in mouse embryos (Chen et al., 2012; Fu et al., 2006). Consistent with Chen et al. and Fu et al.'s findings and Furuta et al.'s findings at E9.5 (27-somite), we found that β-gal reporter for Six3-Cre was initially found in the anteroventral optic pits at E8.5 and then was restricted to NR but not RPE at E10.5. The findings that RPE marker Mitf expands to the entire defective optic vesicles at the cost of NR markers Rax, Sox2 and Vsx2 in both Six3Six3-Cre KO and Six3Rx-Cre KO mutant embryos at E10.5 easily lead to the speculation that prospective NR progenitors are directly converted to RPE progenitors upon Six3-deficiency. However, lineage tracing of Six3-Cre positive cells in Six3Six3-Cre KO embryos refutes this speculation, because β-gal reporter for Six3-Cre was absent in the remnant RPE in type II Six3Six3-Cre KO embryos in which NR was absent. Importantly, gene expression in R26R locus is active in RPE and does not require Six3 functions, since R26R reporter for Rx-Cre is present in the remnant RPE in Six3Rx-Cre KO mutant optic vesicles. Although non-parallel recombination between R26R reporter and targeted gene deletion was found to an extent (Liu et al., 2013), there is still a very high correlation between R26R reporter and Six3-deletion. Accordingly, the lack of β-gal reporter in the remnant RPE cells in type II Six3Six3-Cre KO embryos indicates that Six3-Cre positive progenies were eventually lost upon Six3-deletion and the remaining RPE cells were derived from Six3-Cre negative cells in the optic vesicles. Consistent to the findings revealed by lineage tracing, ectopic apoptosis was found in Six3Six3-Cre KO mutant optic pits/vesicles at E8.5. It is unclear at the moment whether the ectopic apoptosis is directly or indirectly caused by Six3-deletion. Interestingly, mutations in so, the Six3 homolog in Drosophila, result in massive cell death anterior to the morphogenetic furrow in eye discs (Cheyette et al., 1994). The lack of drastic ectopic apoptosis in Six3Six3-Cre KO ventral optic vesicles at later stages indicates that Six3-Cre negative cells in the ventral optic vesicles remained to be Six3-Cre negative upon non-cell-autonomous Six3-deficiency rather than switched on Six3-Cre expression and then went on apoptosis. We conclude that 1) the progenitors at the ventral optic vesicles sequentially switch on Six3-Cre starting at E8.5, and Six3-Cre expression in the optic vesicles indicates a change of cell state towards prospective NR fate, because Six3-Cre positive progenies are found in NR but not in RPE at E10.5; therefore, lineage segregation between neuroretina and RPE starts at E8.5 and is a progressive process; 2) upon Six3-deficiency at E8.5, Wnt8b is rostrally expanded and Fgf8/MAPK signaling is drastically reduced, Six3-Cre positive progenies are eventually lost, leading to the phenotypes that Six3-Cre negative cells in the ventral and dorsal optic vesicles remain to be Six3-Cre negative and are specified as RPE; 3) Six3-Cre negative cells in E8.5 ventral optic vesicles eventually express Six3-Cre at later stages fating to NR in wild-type embryos, but remain to be Six3-Cre negative fating to RPE upon non-cell-autonomous Six3-deficiency in Six3Six3-Cre KO mutant embryos, and thus are bipotential.
The population of Six3-Cre positive progenies in E8.5 mouse embryos is interesting. It is located at the anteroventral optic pits/vesicles and is close to the anterior neural ridge, and is reminiscent of the anteroventral quadrant of optic vesicles that plays crucial roles in early eye development in chick embryos (Hirashima et al., 2008; Kobayashi et al., 2009). Importantly, Six3-deficiency in this cell population in mice leads to retinal phenotypes very similar to the phenotypes generated by surgical removal of ventral or anterior optic vesicles in chick embryos at 9- to 14-somite stages (Hirashima et al., 2008). In chick and zebrafish embryos, pinwheel movement and anterior rotation make the cells at the anterior and ventral margins of the optic vesicles to take part in retinal morphogenesis (Kwan et al., 2012), and such cell movements could also exist in mouse embryos. Fate mapping of mouse embryos reveals the origins of eyes at a rostral territory inside the anterior neural plate (Inoue et al., 2000). These findings support that the cell population where Six3-Cre positive progenies are located in E8.5 mouse embryos are important in early eye development. It is worth noting that Six3-Cre positive progenies in E8.5 mouse embryos are only a part of the cell population in which endogenous Six3 is expressed and it is unclear that why Six3-Cre positive progenies display such an pattern. Nevertheless, Six3 in the small population of progenitors expressing Six3-Cre in E8.5 mouse embryos is essential for proper paracrine signaling at the anterior neural ridge during NR specification, providing a molecular mechanism why anteroventrally localized activity in the early optic vesicles are crucial for NR specification.
The essential roles of Six3 in NR specification have temporal dependence. Analysis of temporal Six3-deficient embryos driven by CAGG-CreER indicates that Six3-deletion at early and late phases causes type II and type I retinal phenotypes, respectively. In the temporal Six3-deficient embryos, Six3 is deleted ubiquitously, and thus Six3-deletion in surface ectoderm could also contribute to defective NR specification (Liu et al., 2006). However, Six3-deficiency in neural lineage at E8.5 driven by Six3-Cre causes rostral expansion of Wnt8b and drastic reduction of Fgf8/MAPK signaling, indicating that Six3 expression in the neural lineage at E8.5 is essential for NR specification. The two major phenotypes in Six3Six3-Cre KO mutant embryos are caused by variable onset of Six3-Cre activity, as variable onset of R26R reporter for Six3-Cre is found for an unknown reason. In compound Six3Six3-Cre KO;Six6−/− mutant embryos, only type II retinal phenotypes are found, indicating that inactivation of Six6 in type I Six3Six3-Cre KO embryos converts type I retinal phenotypes into type II retinal phenotypes, and thus Six6 compensates Six3-deficiency when Six6 is expressed in the optic vesicles. The functional redundancy between Six3 and Six6 in NR specification is further supported by their identical homeodomain sequences and largely co-localized expression in the optic vesicles. We conclude that Six3 expression at E8.5 in a small population of progenitors expressing Six3-Cre is required for NR specification, and Six3 and Six6 jointly regulate NR specification when Six6 is expressed in the optic vesicles.
Six3 plays essential roles in early pituitary development. Although Six3 is expressed in the infundibulum of ventral forebrain and the Rathke's pouch, Six3-Cre positive progenies are only found in a cell population in the infundibulum and ventral forebrain, but not in the Rathke's pouch. Similar to what happens in NR specification, Six3-deletion driven by Six3-Cre is sufficient to disrupt proper cell signaling in the infundibulum of ventral forebrain, leading to defects in the infundibulum and Rathke's pouch and the loss of Six3-Cre positive progenies in the defective infundibulum. Thus, Six3 regulates cell signaling and survival in neuroretinal specification as well as in early pituitary development.
Supplementary Material
Acknowledgments
We thank Drs. G. Oliver for generous support and critical comments, from the Editor and anonymous reviewers for constructive comments, M.G. Rosenfeld and X. Li for the Six6 mice, and H. Arnheiter, B. Molday, T. Jessell, and A. McMahon for probes and antibodies. We thank R.G. Harris and A. Lowe for editing. This work was partially supported by the National Institutes of Health grants (EY022645 to W. Liu, EY012200 to A. Cvekl).
Footnotes
Appendix A. Supporting information: Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ydbio.2017.05.026.
References
- Cai Z, Feng GS, Zhang X. Temporal requirement of the protein tyrosine phosphatase Shp2 in establishing the neuronal fate in early retinal development. J Neurosci. 2010;30:4110–4119. doi: 10.1523/JNEUROSCI.4364-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lewis B, Moran A, Xie T. Cadherin-mediated cell adhesion is critical for the closing of the mouse optic fissure. PLoS One. 2012;7:e51705. doi: 10.1371/journal.pone.0051705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheyette BN, Green PJ, Martin K, Garren H, Hartenstein V, Zipursky SL. The Drosophila sine oculis locus encodes a homeodomain-containing protein required for the development of the entire visual system. Neuron. 1994;12:977–996. doi: 10.1016/0896-6273(94)90308-5. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Ashery-Padan R. The cellular and molecular mechanisms of vertebrate lens development. Development. 2014;141:4432–4447. doi: 10.1242/dev.107953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Sun H, Klein WH, Mu X. Beta-catenin is essential for lamination but not neurogenesis in mouse retinal development. Dev Biol. 2006;299:424–437. doi: 10.1016/j.ydbio.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann S. Eye morphogenesis and patterning of the optic vesicle. Curr Top Dev Biol. 2010;93:61–84. doi: 10.1016/B978-0-12-385044-7.00003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26:130–132. [PubMed] [Google Scholar]
- Gan L, Xiang M, Zhou L, Wagner DS, Klein WH, Nathans J. POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc Natl Acad Sci USA. 1996;93:3920–3925. doi: 10.1073/pnas.93.9.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X, Speirs C, Lagutin O, Inbal A, Liu W, Solnica-Krezel L, Jeong Y, Epstein DJ, Oliver G. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev Cell. 2008;15:236–247. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Sato T, Nakamura H. Fgf8 signaling for development of the midbrain and hindbrain. Dev Growth Differ. 2016;58:437–445. doi: 10.1111/dgd.12293. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Hirashima M, Kobayashi T, Uchikawa M, Kondoh H, Araki M. Anteroventrally localized activity in the optic vesicle plays a crucial role in the optic development. Dev Biol. 2008;317:620–631. doi: 10.1016/j.ydbio.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Inoue T, Nakamura S, Osumi N. Fate mapping of the mouse prosencephalic neural plate. Dev Biol. 2000;219:373–383. doi: 10.1006/dbio.2000.9616. [DOI] [PubMed] [Google Scholar]
- Ivanovitch K, Cavodeassi F, Wilson SW. Precocious acquisition of neuroepithelial character in the eye field underlies the onset of eye morphogenesis. Dev Cell. 2013;27:293–305. doi: 10.1016/j.devcel.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean D, Bernier G, Gruss P. Six6 (Optx2) is a novel murine Six3-related homeobox gene that demarcates the presumptive pituitary/hypothalamic axis and the ventral optic stalk. Mech Dev. 1999;84:31–40. doi: 10.1016/s0925-4773(99)00068-4. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. The Atlas of Mouse Development, Revised edition 1995 ed. Accadmic Press; London, UK: 1992. [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yasuda K, Araki M. Generation of a second eye by embryonic transplantation of the antero-ventral hemicephalon. Dev Growth Differ. 2009;51:723–733. doi: 10.1111/j.1440-169X.2009.01132.x. [DOI] [PubMed] [Google Scholar]
- Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Otsuna H, Kidokoro H, Carney KR, Saijoh Y, Chien CB. A complex choreography of cell movements shapes the vertebrate eye. Development. 2012;139:359–372. doi: 10.1242/dev.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L, Russell HR, McKinnon PJ, Solnica-Krezel L, Oliver G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003;17:368–379. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Perissi V, Liu F, Rose DW, Rosenfeld MG. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science. 2002;297:1180–1183. doi: 10.1126/science.1073263. [DOI] [PubMed] [Google Scholar]
- Liu J, Willet SG, Bankaitis ED, Xu Y, Wright CV, Gu G. Non-parallel recombination limits Cre-LoxP-based reporters as precise indicators of conditional genetic manipulation. Genesis. 2013;51:436–442. doi: 10.1002/dvg.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. Focus on molecules: Wnt8b: a suppressor of early eye and retinal progenitor formation. Exp Eye Res. 2012;101:113–114. doi: 10.1016/j.exer.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Lagutin OV, Mende M, Streit A, Oliver G. Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. Embo J. 2006;25:5383–5395. doi: 10.1038/sj.emboj.7601398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Lagutin O, Swindell E, Jamrich M, Oliver G. Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J Clin Investig. 2010;120:3568–3577. doi: 10.1172/JCI43219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe A, Harris R, Bhansali P, Cvekl A, Liu W. Intercellular adhesion-dependent cell survival and ROCK-regulated actomyosin-driven forces mediate self-formation of a retinal organoid. Stem Cell Rep. 2016;6:743–756. doi: 10.1016/j.stemcr.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Mathers PH, Grinberg A, Mahon KA, Jamrich M. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387:603–607. doi: 10.1038/42475. [DOI] [PubMed] [Google Scholar]
- Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–785. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Arnheiter H. Signaling and transcriptional regulation in early mammalian eye development: a link between FGF and MITF. Development. 2000;127:3581–3591. doi: 10.1242/dev.127.16.3581. [DOI] [PubMed] [Google Scholar]
- Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. 1995;121:4045–4055. doi: 10.1242/dev.121.12.4045. [DOI] [PubMed] [Google Scholar]
- Rapicavoli NA, Poth EM, Zhu H, Blackshaw S. The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev. 2011;6:32. doi: 10.1186/1749-8104-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold M, Loosli F, Adams RJ, Wittbrodt J. Individual cell migration serves as the driving force for optic vesicle evagination. Science. 2006;313:1130–1134. doi: 10.1126/science.1127144. [DOI] [PubMed] [Google Scholar]
- Rizzoti K, Lovell-Badge R. Early development of the pituitary gland: induction and shaping of Rathke's pouch. Rev Endocr Metab Disord. 2005;6:161–172. doi: 10.1007/s11154-005-3047-7. [DOI] [PubMed] [Google Scholar]
- Samuel A, Rubinstein AM, Azar TT, Ben-Moshe Livne Z, Kim SH, Inbal A. Six3 regulates optic nerve development via multiple mechanisms. Sci Rep. 2016;6:20267. doi: 10.1038/srep20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- Swindell EC, Bailey TJ, Loosli F, Liu C, Amaya-Manzanares F, Mahon KA, Wittbrodt J, Jamrich M. Rx-Cre, a tool for inactivation of gene expression in the developing retina. Genesis. 2006;44:361–363. doi: 10.1002/dvg.20225. [DOI] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006;20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Morales DC, Hermesz E, Lee WK, Pfaff SL, Westphal H. Reduced expression of the LIM-homeobox gene Lhx3 impairs growth and differentiation of Rathke's pouch and increases cell apoptosis during mouse pituitary development. Mech Dev. 2006;123:605–613. doi: 10.1016/j.mod.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Zuber ME, Gestri G, Viczian AS, Barsacchi G, Harris WA. Specification of the vertebrate eye by a network of eye field transcription factors. Development. 2003;130:5155–5167. doi: 10.1242/dev.00723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.