

Abstract
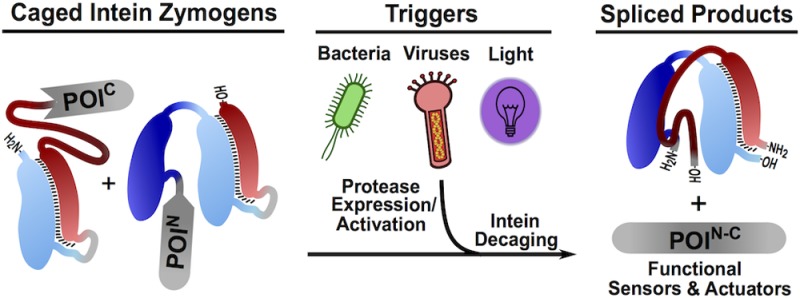
Naturally split inteins have found widespread use in chemical biology due to their ability to drive the ligation of separately expressed polypeptides through a process termed protein trans-splicing (PTS). In this study, we harness PTS by rendering association of split intein fragments conditional upon the presence of a user-defined protease. We show that these intein “zymogens” can be used to create protein sensors and actuators that respond to the presence of various stimuli, including bacterial pathogens, viral infections, and light. We also show that this design strategy is compatible with several orthogonal split intein pairs, thereby opening the way to the creation of multiplexed sensor systems.
Inteins are intervening protein domains that undergo a spontaneous post-translational process known as protein splicing. During this autoprocessing event, the intein excises itself from a larger precursor polypeptide, in the process ligating the flanking polypeptides (exteins) via a peptide bond (Figure S1a).1 Unlike the more common contiguous inteins, naturally split inteins such as DnaE from the cyanobacterium Nostoc punciforme PCC73102 (Npu) are translated separately and must first associate to initiate the splicing reaction, so-called “protein trans splicing” (PTS) Figure S1b). Many of these split intein pairs have extremely high affinity for one another (Npu, KD = 1.2 nM)2 and, upon complex formation, exhibit remarkably fast splicing rates3−6 (Npu splicing t1/2 < 1 min),4 making them attractive tools for a wide range of protein engineering applications.1
Due to the dramatic change in primary sequence that accompanies protein splicing, it has long been recognized that controlling intein activity offers a means to regulate associated protein (i.e., extein) function at the post-translational level.7 Indeed, a number of so-called “conditional protein splicing” (CPS) systems have been reported in which the splicing event is triggered by external stimuli such as small molecules7−11 or light.12−16 Split inteins such as Npu are especially attractive starting points for the development of CPS methods since they support the extremely rapid ligation of two separately expressed polypeptides. While significant progress has been made in the development of CPS systems based on the naturally split inteins,12,14,17 none of these offer modularity with respect to the type of triggers employed. Here, we describe a general strategy for controlling the association and subsequent trans splicing of naturally split inteins. Guided by the mechanism of split intein association and folding,2 we designed genetically encoded caged split intein pairs that are activated by targeted proteolysis, in effect creating split intein zymogens. We show that this design paradigm can be applied to several orthogonal split intein pairs and used to create CPS sensors and actuators that respond to the presence of bacterial pathogens, viral infections, and light.
Previously, we found that complementation of the Npu split intein pair is initiated by electrostatic interactions between an unstructured cationic region in the C-terminal fragment, NpuC (residues 1–13), and an unstructured anionic region in the N-terminal fragment, NpuN (residues 51–102).2 This initial “capture” step leads to formation of a binding intermediate structure that subsequently undergoes a hydrophobic “collapse” to complete the native intein fold (Figure 1a). Based on this folding mechanism, we designed caged NpuN and NpuC constructs in which residues 51–102 of NpuN were fused to full-length NpuC (NpuCCage) and residues 1–13 of NpuC were fused to full-length NpuN (NpuNCage). We hypothesized that these cage sequences would participate in intramolecular interactions with the split intein fusion partner, effectively locking each half of the split intein in its binding intermediate structure and thus preventing fragment association and splicing (Figure 1b). As part of our design, we also included a protease cleavage sequence between each native split intein and its corresponding cage sequence. We imagined that proteolytic removal of the cage sequences would trigger split intein association and hence PTS (Figure 1b). Consequently, we refer to these as split intein zymogens, reflecting their potential to be enzymatically activated.
Figure 1.
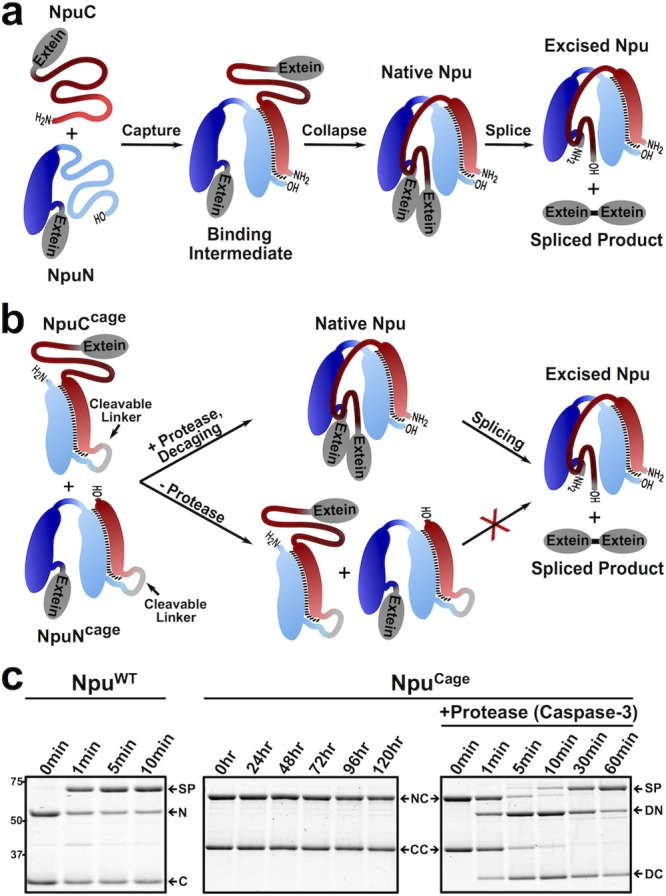
Design and characterization of caged intein zymogens: N = MBP-NpuN, C = NpuC-eGFP, NC = MBP-NpuNCage, CC = NpuCCage-eGFP, DN = decaged-NC, DC = decaged-CC, SP = splice product (MBP-eGFP). (a) Schematic depicting two-step mechanism by which the split fragments of Npu (NpuC and NpuN) assemble and splice. (b) Schematic depicting the design of NpuCCage and NpuNCage constructs, where addition of a protease cleaves or “decages” the inteins leading to protein splicing. (c) Coomassie-stained SDS-PAGE gels of splicing reactions (37 °C, 1 μM each intein fragment) monitored over time of either the wild-type (NpuWT) or zymogen (NpuCage) versions of the split Npu intein. Caspase-3 was added to a separate aliquot of NpuCage at time = 0 h.
To assess the efficacy of our CPS strategy, we used recombinant expression methods to generate the two caged split inteins, each fused to model exteins (Figure S2). The purified constructs were then individually incubated with the corresponding Npu wild-type (NpuWT) split intein fragment, also fused to a model extein (Figures S2 and S3). This revealed that the caging strategy was effective for NpuC, increasing the half-life of the Npu splicing reaction from less than 1 min to well over 1 h (Figure S3b). By contrast, attachment of the caging sequence to NpuN had no observable effect on the splicing rate (Figure S3d). Anticipating that our CPS system would ultimately need both intein fragments effectively inhibited, we carried out several rounds of optimization on the caged NpuN construct, ultimately arriving at a design which contained tandem copies of the cage unit, in addition to some point mutations at the binding interface (see Figures S2, S4, and S5 for details). Use of this optimized NpuNCage in PTS reactions with NpuCWT led to a substantial reduction in the rate of product formation, analogous to what was observed with the NpuCCage construct (Figure S4). Importantly, our two CPS constructs, NpuNCage and NpuCCage, exhibited no affinity for one another in pull-down assays, and, critically, no splicing activity was observed between them, even after 5 days of incubation (Figures S6 and 1c). We note that this represents a remarkable level of inhibition over the native Npu split intein, which splices in seconds.4 Consistent with our original design, addition of a protease (caspase-3) that is able to remove the cage sequence from these constructs leads to rapid onset of protein splicing (t1/2 ≈ 15 min) (Figure 1c).
Encouraged by these initial results, we next asked whether the same caging strategy used on Npu could be extended to create zymogens of other split inteins. We focused on the GP41-1, GP41-8, and NrdJ-1 split inteins, since they all exhibit ultrafast splicing rates comparable to Npu.6 Moreover, they are complementary to Npu in terms of the local extein environments they prefer to splice from (i.e., those residues immediately flanking the intein), thus, making them attractive candidates for expanding our CPS toolbox. We aligned each intein sequence to Npu and used NpuCCage and an early iteration of NpuNcage as templates to design corresponding IntCCage and IntNCage pairs (Figures S2 and S7). Similar to Npu, the caged versions of GP41-8, GP41-1, and NrdJ-1 were completely inactive until treated with a protease (caspase-3) that removes the cage sequences, whereupon efficient PTS ensued (Figures 2a and S8). The ease with which this caging strategy, originally developed for Npu, could be applied to other naturally split inteins reveals a remarkable level of robustness in our basic design and suggests a common folding mechanism between these separate families of split inteins.
Figure 2.

Splicing reactions of orthogonal intein zymogens: NC = MBP-IntNCage, CC = IntCCage-eGFP, DN = decaged-NC, DC = decaged-CC, SP = splice product (MBP-eGFP). (a) Coomassie-stained SDS-PAGE gels showing in vitro splicing reactions of caged versions of GP41-8, GP41-1, and NrdJ-1 split inteins in the absence and presence of protease trigger, caspase-3 (Cas3). Each intein zymogen contains a Cas3 cut site and for each reaction, Cas3 was added in a separate aliquot at time = 0 h. (b) Western blots showing in vitro splicing reactions of caged GP41-8, GP41-1, NrdJ-1, and Npu split inteins with indicated protease cleavage sequences in the absence and presence of protease triggers, carried out for 1 h at 25 °C and 1 μM of each indicated intein zymogen pair.
We next sought to test two key features of our CPS platform, namely the exchangeable nature of the triggering feature and the capacity to generate a functional protein as output. To explore the first of these parameters, we replaced the caspase cut site in the original Npu zymogen with a cleavage sequence for the NS3/4A protease from the hepatitis C virus (Figure S2). As in the original example, we observed protease-dependent PTS (Figure 2b). The exchangeability of the trigger feature was also confirmed in the context of the GP41-8, GP41-1, and NrdJ-1 inteins—these were converted into intein zymogens that could be activated by human rhinovirus 3C protease, tobacco etch virus (TEV) protease, and thrombin, respectively (Figures 2b and S2). The efficiency of splicing varied somewhat depending on the intein-protease pair used, for example, Npu splicing triggered by the NS3/4A protease was less efficient than that triggered by Cas3. Notably, by installing unique cleavage sites in each of the four orthogonal inteins, we could selectively activate a desired PTS reaction in a split intein mixture (Figure S9). This illustrates the orthogonality of the inteins and highlights the potential of this system for multiplexed type applications.
Turning to the question of functional output, we generated an Npu intein zymogen pair designed to sense the bacterial pathogen Staphylococcus aureus and generate a pathogen-specific antibacterial agent as a response. These constructs contained cleavage sequences for the S. aureus secreted protease ScpA and were fused to a peptidoglycan degrading domain (Lys16) and a cell wall binding domain (SH3B), which possess potent anti-staphylococcal activity when linked18 (Figures S2 and S10a). In initial studies, we confirmed that PTS activity was dependent on the presence of log phase S. aureus cultures (Figure S10b). Gratifyingly, the splicing reaction between the exogenously added caged constructs was directly coupled to a 99.9% reduction in bacterial colony forming units, confirming the ability of the CPS system to act as an actuator (Figure S10c).
Next, we examined the functionality of the CPS system in mammalian cells. We designed intein zymogens that could be utilized to generate a splicing-dependent enhanced green fluorescent protein (eGFP) (Figures S11 and S14). This allowed us to test whether our CPS system can be used as a sensor for viral infection (Figure 3a). Initial studies revealed that NpuNCage was poorly expressed in HEK-293T cells, a problem that was ultimately traced to two residues that had been mutated during the initial optimization process (E7K and E61K)—fortunately reverting these to their native glutamates largely rescued expression to WT levels (Figures S12 and S14). The TEV cleavage sequence was embedded within NpuNCage and NpuCCage and the resulting split intein zymogen pair fused to the respective N- and C-terminal fragments of an eGFP reporter (Figures 3a and S14). As expected, no protein splicing was observed when these CPS constructs were co-expressed in HEK-293T cells. By contrast, exposure of these cells to a baculovirus strain encoding mCherry-TEV resulted in robust generation of eGFP splice product (Figure 3b,c). This result demonstrates that our system can sense the presence of virally encoded proteases and, by extension, viral infection.
Figure 3.

Fluorescent response to viral infection or photo-activation. (a,d) Schematics depicting TEV-activated split eGFP intein zymogen system in mammalian cells with either viral infection (a–c) or photolysis (d–f) acting as CPS triggers. Note that, for clarity, additional domains fused to eGFP fragments are not shown. For more details on constructs used, see Supporting Information. (b) Images of HEK 293T cells expressing the TEV-activated split eGFP intein zymogen constructs either with (bottom) or without (top) baculoviral infection. Images were taken following an overnight incubation with baculovirus. (c) Top: Western blot analysis (anti-HA) of cells from images in (b). SP = splice product, NC = HA-eGFP(1–64)-NpuNCage, DN = decaged-NC. Bottom: anti-β-actin loading control. (e) Images of HEK 293T cells expressing split eGFP intein zymogen system and photo-caged TEV with (bottom) or without (top) 1 min UV (365 nM) irradiation. Images were taken 6 h post irradiation. (f) Western (anti-HA) analysis of cells from images in (e). NC-F = HA-eGFP(1–64)-NpuNCage-Tev-(C151TAG) full translation product, NC-T = truncated translation product of NC-F, SP = splice product, DN = decaged-NC.
Inspired by previous TEV activation technologies,19 we next sought to add an additional layer of control to our system by utilizing amber codon suppression to incorporate a photo-caged cysteine into the protease active site (Figures 3d and S13). In principle, this would enable photo-control of fragment association in the context of the TEV-activated intein zymogen system. Such a system would complement an existing optogenetic tool based on incorporation, by amber suppression, of a caged cysteine residue directly into an artificially fused version of Npu.16 With this in mind, we designed a construct in which a mutant TEV protease was fused to the C-terminus of NpuNCage (NpuNCage-TEV, Figure S14). HEK-293T cells harboring the requisite orthogonal tRNA/tRNA synthetase pair for the unnatural amino acid S-[1-(4′,5′-(methylenedioxy)-2′-nitrophenyl)ethyl]cysteine19 were transfected with NpuCCage and NpuNCage-TEV constructs fused to the split eGFP pair as N- and C-exteins (Figures 3d and S14). Consistent with our design, we observed irradiation-dependent generation of spliced eGFP in the presence of the unnatural amino acid (Figure 3e,f). The success of this light-activated split intein zymogen system is expected to expand the range of protein targets amenable to optogenetic control, due to the known promiscuity of the Npu split intein.20 Moreover, we note that placing the photo-caged residue in the protease, rather than the intein itself, could help mitigate one of the drawbacks sometimes associated with amber suppression, namely lower expression yields caused by premature chain termination. The amplification effect inherent to the enzymatic trigger, in principle, means that a small amount of the full-length protease should be sufficient to support efficient photo-uncaging.
Despite the extraordinarily high binding affinity and rapid splicing kinetics of ultrafast split inteins, the CPS strategy reported herein effectively cages the association of the split intein fragments while allowing for a diverse set of proteases to trigger the splicing reaction. Applications of these intein zymogens, such as the conditional killing of S. aureus and sensing of viral infection in mammalian cells, illustrate this system’s ability to detect and respond to biological processes involving proteases. Moreover, coupling the caged constructs to a photo-activatable TEV protease adds an additional layer of spatiotemporal control. We envision that the development of orthogonally caged systems, each able to respond to a unique trigger, will expand the range of targets amenable to CPS and enable one-pot reactions where multiple separate splicing reactions can be controlled.
Acknowledgments
The authors thank Anahita Z. Mostafavi, Olivia S. Walker, Glen P. Liszczak, and Robert E. Thompson for valuable discussions. This work was supported by the U.S. National Institutes of Health (NIH grant R37-GM086868.)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02618.
Full methods and experimental data, including Figures S1–S14 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shah N. H.; Muir T. W. Chem. Sci. 2014, 5, 446. 10.1039/C3SC52951G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. H.; Muir T. W.; et al. J. Am. Chem. Soc. 2013, 135, 18673. 10.1021/ja4104364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai H.; Züger S.; Jin J.; Tam P. H. FEBS Lett. 2006, 580, 1853. 10.1016/j.febslet.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Zettler J.; Schütz V.; Mootz H. D. FEBS Lett. 2009, 583, 909. 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Shah N. H.; Dann G. P.; Vila-Perello M.; Liu Z.; Muir T. W. J. Am. Chem. Soc. 2012, 134, 11338. 10.1021/ja303226x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vallejos P.; Pallisse R.; Mootz H. D.; Schmidt S. R. J. Biol. Chem. 2012, 287, 28686. 10.1074/jbc.M112.372680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootz H. D.; Muir T. W. J. Am. Chem. Soc. 2002, 124, 9044. 10.1021/ja026769o. [DOI] [PubMed] [Google Scholar]
- Buskirk A. R.; Ong Y. C.; Gartner Z. J.; Liu D. R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 10505. 10.1073/pnas.0402762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen C. M.; Rodda S. J.; Vokes S. A.; McMahon A. P.; Liu D. R. J. Am. Chem. Soc. 2006, 128, 8939. 10.1021/ja062980e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. C.; Saez L.; Young M. W.; Muir T. W. Nat. Chem. Biol. 2007, 3, 50. 10.1038/nchembio832. [DOI] [PubMed] [Google Scholar]
- Peck S. H.; Chen I.; Liu D. R. Chem. Biol. 2011, 18, 619. 10.1016/j.chembiol.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Perelló M.; Hori Y.; Ribó M.; Muir T. W. Angew. Chem., Int. Ed. 2008, 47, 7764. 10.1002/anie.200802502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyszkiewicz A. B.; Muir T. W. Nat. Methods 2008, 5, 303. 10.1038/nmeth.1189. [DOI] [PubMed] [Google Scholar]
- Böcker J. K.; Friedel K.; Matern J. C.; Bachmann A. L.; Mootz H. D. Angew. Chem., Int. Ed. 2015, 54, 2116. 10.1002/anie.201409848. [DOI] [PubMed] [Google Scholar]
- Jung D.; Sato K.; Min K.; Shigenaga A.; Jung J.; Otaka A.; Kwon Y. Chem. Commun. 2015, 51, 9670. 10.1039/C5CC01067E. [DOI] [PubMed] [Google Scholar]
- Ren W.; Ji A.; Ai H. W. J. Am. Chem. Soc. 2015, 137, 2155. 10.1021/ja508597d. [DOI] [PubMed] [Google Scholar]
- Wong S.; Mosabbir A. A.; Truong K. PLoS One 2015, 10, e0135965. 10.1371/journal.pone.0135965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul V. D. A novel bacteriophage Tail-Associated Muralytic Enzyme (TAME) from Phage K and its development into a potent antistaphylococcal protein. BMC Microbiol. 2011, 11, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. P.; Mahesh M.; Elsässer S. J.; Hancock S. M.; Uttamapinant C.; Chin J. W. J. Am. Chem. Soc. 2014, 136, 2240. 10.1021/ja412191m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan M.; Pedamallu C. S.; Tori K.; Perler F. J. Biol. Chem. 2013, 288, 6202. 10.1074/jbc.M112.433094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.